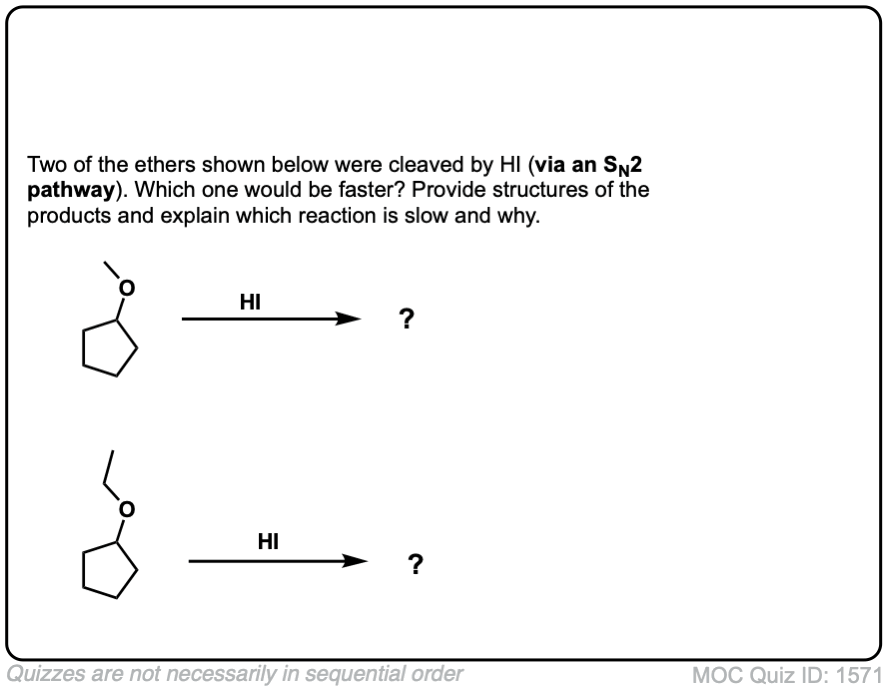
Acidic cleavage of ethers (SN2)
Description: When ethers are treated with strong acid in the presence of a nucleophile, they can be cleaved to give alcohols and alkyl halides. If the ether is on a primary carbon this may occur through an SN2 pathway.
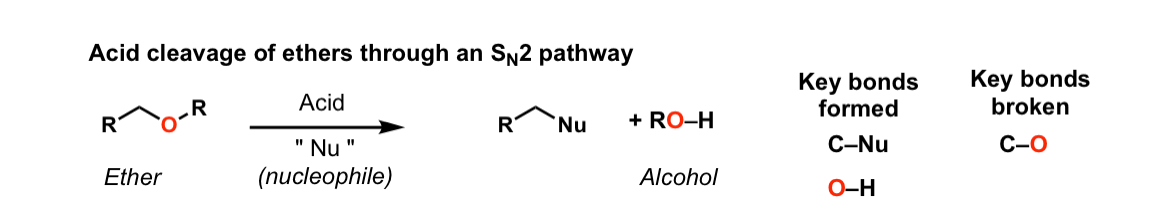
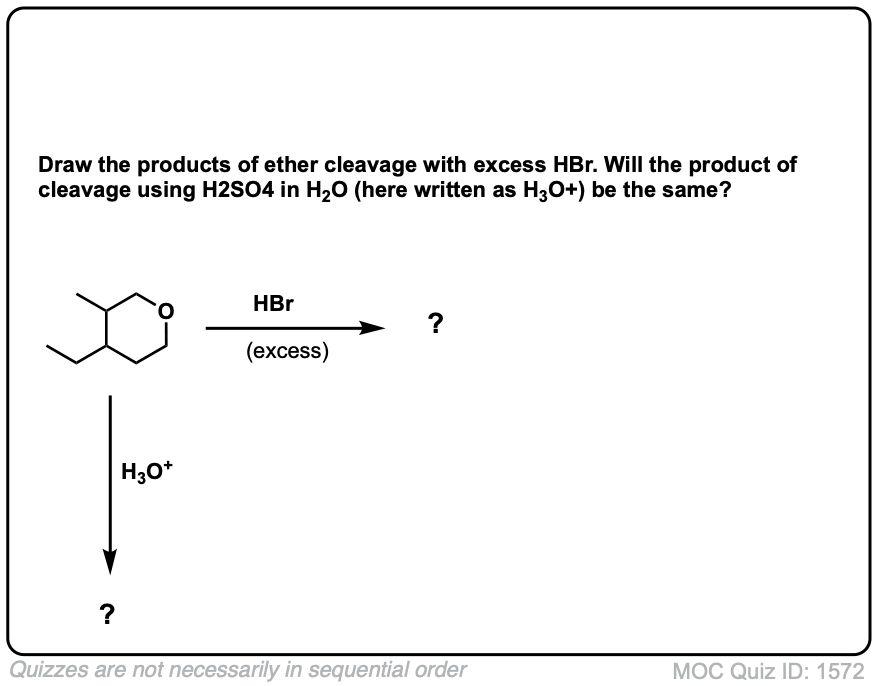
Notes: Common acids for this purpose are HI and other hydrogen halides, as well as H2SO4 in the presence of H2O.
In the case where the ether being cleaved is secondary and has a stereocenter, there will be inversion of configuration.
Examples:
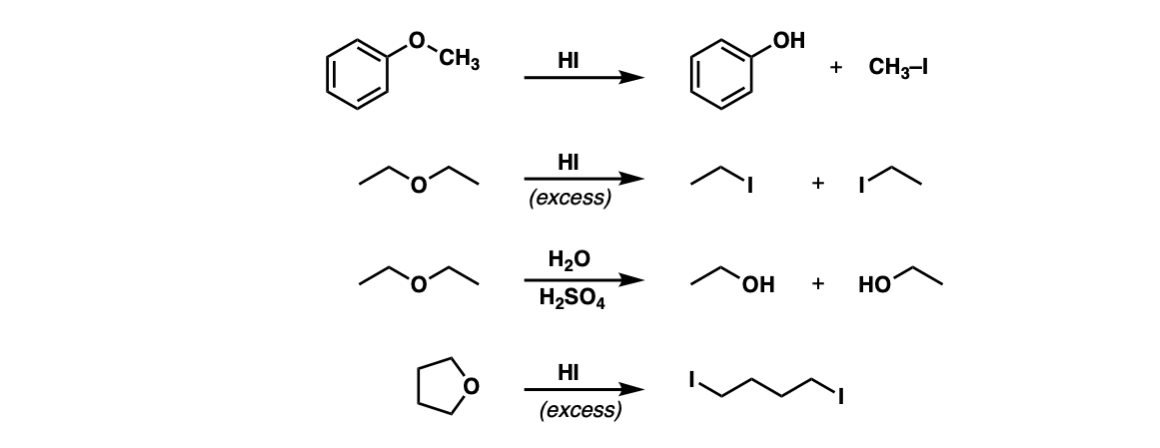
Notes: The third example could also be written “H3O+” . Note that excess HI will convert primary alcohols to alkyl halides via SN2, but not phenol (C6H5OH) since sp2 hybridized carbons do not undergo SN1 or SN2 reactions.
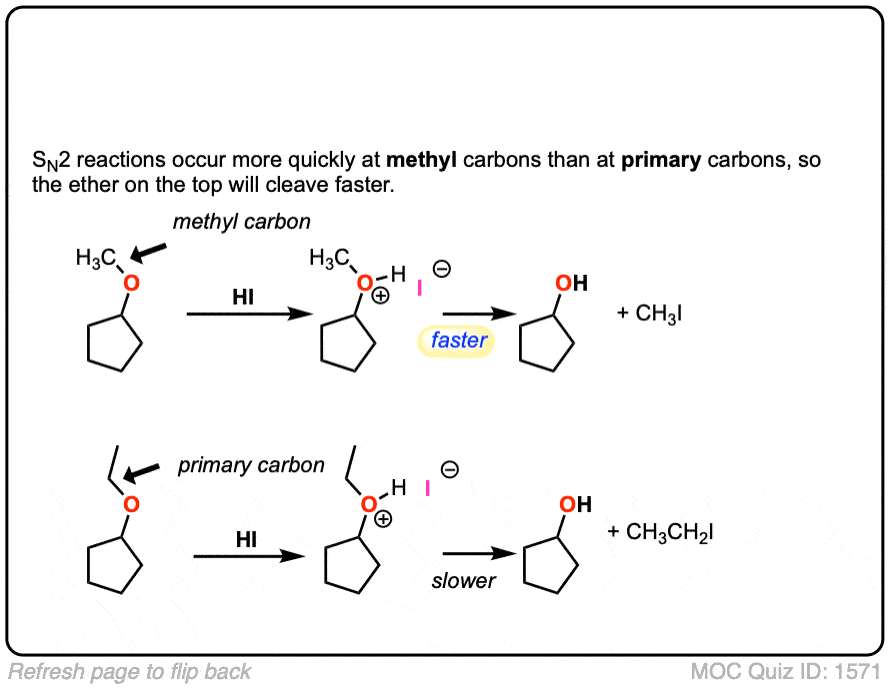
Mechanism: Strong acid (HI) protonates the ether oxygen, which turns it into a better leaving group (Step 1, arrows A and B). Next, the iodide ion attacks the carbon in an SN2 reaction (Step 2, arrows C and D) to give the alcohol and methyl iodide.
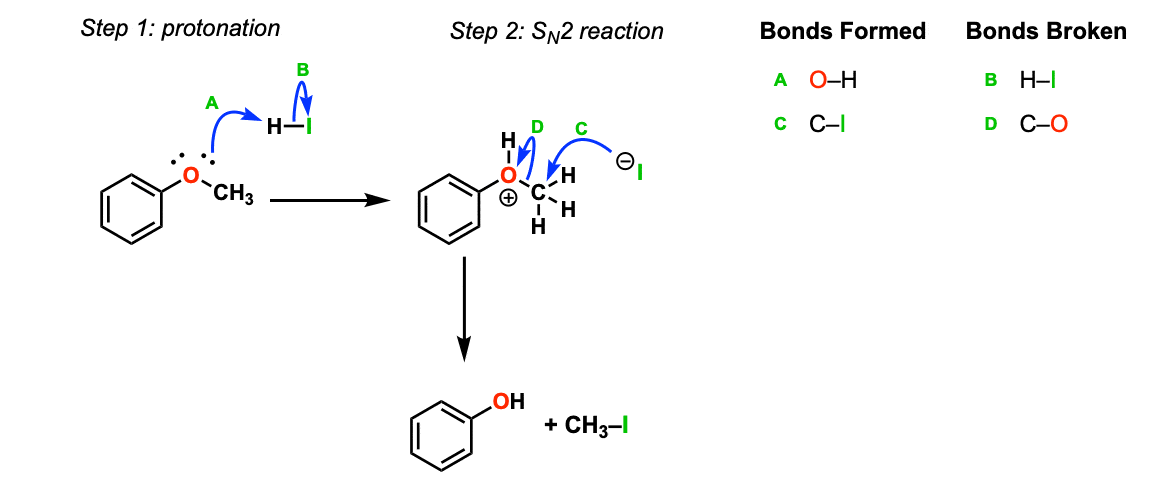
Notes: In cases where the ether being cleaved is secondary and has a stereocenter, there will be inversion of configuration.
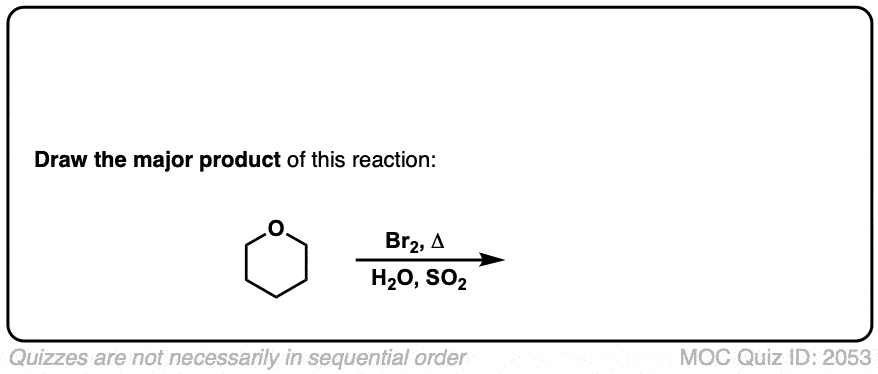
Another example: Opening of tetrahydrofuran (THF) with aqueous acid:
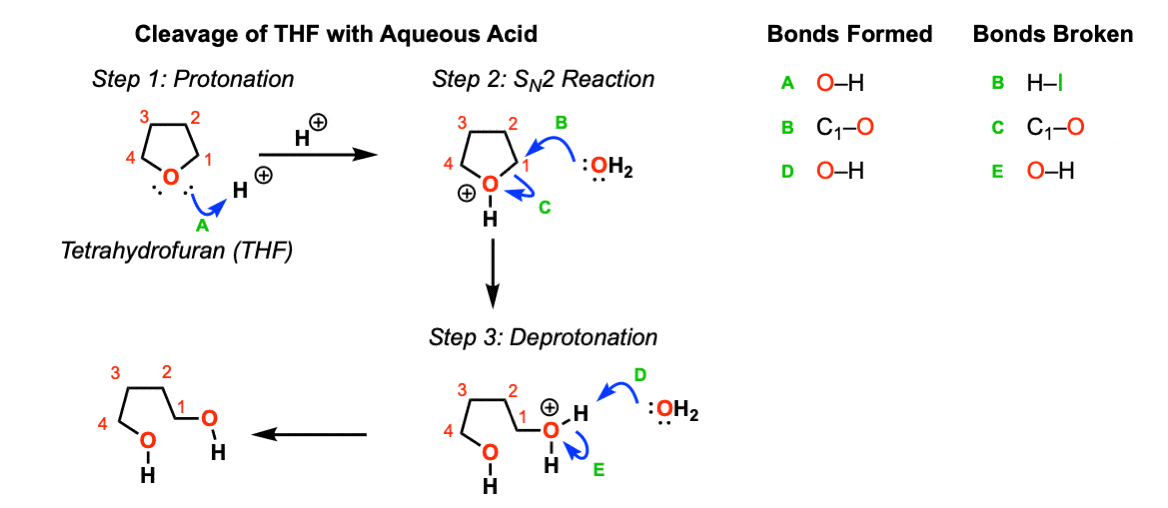
Test Yourself!
 Click to Flip
Click to Flip
 Click to Flip
Click to Flip
 Click to Flip
Click to Flip
(Advanced) References and Further Reading
Ethers are widely inert to a lot of conditions, and thus find common use as solvents (e.g. diethyl ether, THF (tetrahydrofuran), dioxane, glyme, and others). Ether cleavage generally requires strong acid and heat, which are forcing conditions. Alternatively, silane reagents can be used, which are reactive at room temperature.
- A NEW METHOD FOR THE PREPARATION OF ORGANIC IODIDES
HERMAN STONE and HAROLD SHECHTER
The Journal of Organic Chemistry 1950, 15 (3), 491-495
DOI: 10.1021/jo01149a008
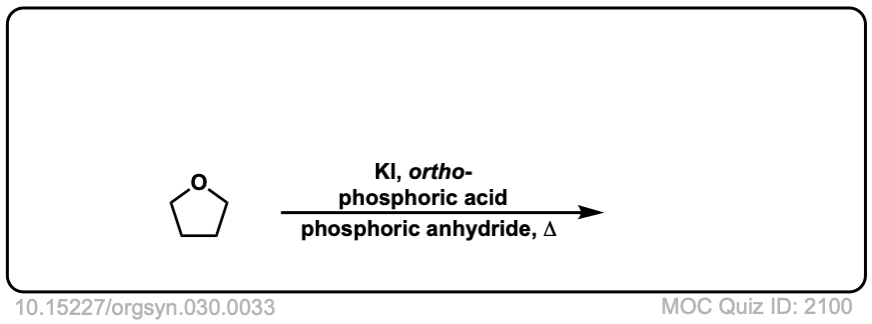
Instead of using HI, which is expensive (and an inconvenient gas, to boot), one can use the combination of phosphoric acid + KI for ether cleavage, which generates HI in situ. - 1,4-DIIODOBUTANE
Herman Stone and Harold Shechter
Org. Synth. 1950, 30, 33
DOI: 10.15227/orgsyn.030.0033
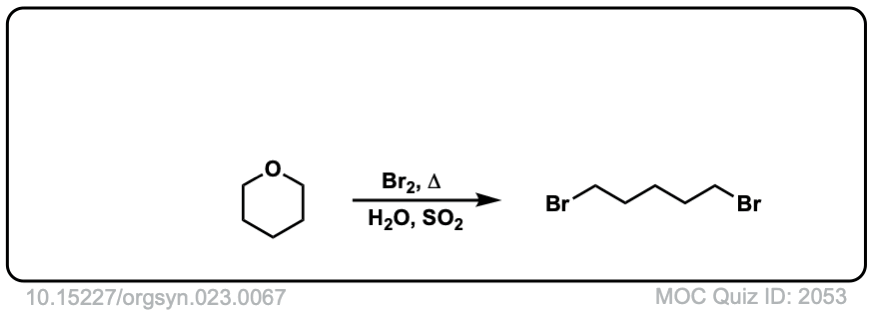
This procedure from Organic Syntheses, a reliable source of independently tested synthetic organic laboratory procedures, demonstrates the cleavage of THF with refluxing strong acid.Below are a variety of papers using silane-based reagents for ether cleavage. The Nobel Laureate late Prof. George Olah did a lot of work in this area in the middle of his career. - Cleavage of Esters and Ethers with Iodotrimethylsilane
Tse‐Lok Ho Prof. Dr. George A. Olah
Angew. Chem. Int. Ed. 1976, 15 (12), 774-775
DOI: 10.1002/anie.197607741 - Mild cleavage of methoxymethyl (MOM) ethers with trimethylsilyl bromide
Stephen Hanessian, Daniel Delorme, Yves Dufresne
Lett. 1984, 25 (24), 2515-2518
DOI: 10.1016/S0040-4039(01)81219-3
MOM ethers are commonly used as protecting groups for -OH in organic synthesis, and so strategies for selective deprotection of MOM ethers under mild cleavage are invaluable.
Real-Life Examples:
Org. Synth. 1943, 23, 67
DOI Link: 10.15227/orgsyn.023.0067
 Click to Flip
Click to Flip
Org. Synth. 1950, 30, 33
DOI Link: 10.15227/orgsyn.030.0033
 Click to Flip
Click to Flip
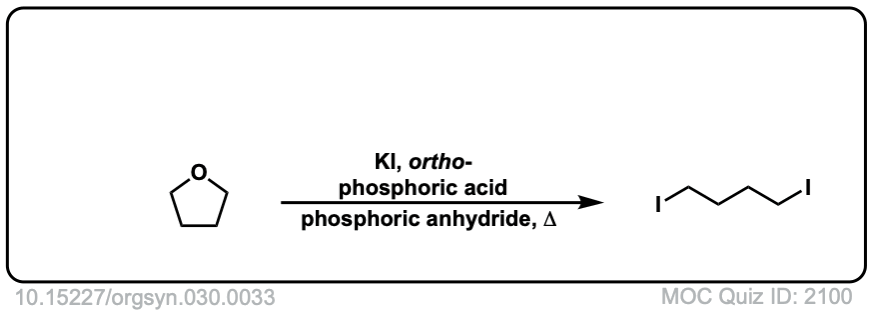
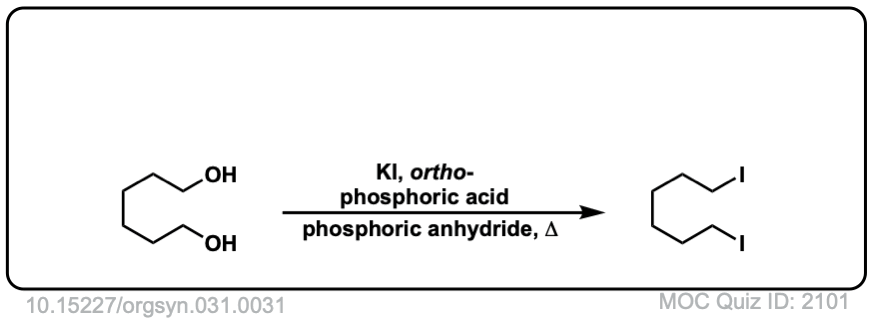
Org. Synth. 1951, 31, 31
DOI Link: 10.15227/orgsyn.031.0031
 Click to Flip
Click to Flip
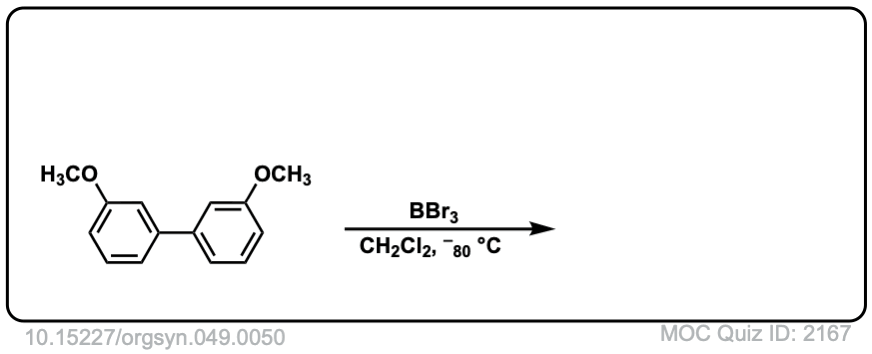
Org. Synth. 1969, 49, 50
DOI Link: 10.15227/orgsyn.049.0050
 Click to Flip
Click to Flip
What are the uses of CF3CO2H? Can it be used to cleave ethers? I have been asked to suggest what could happen with just this reagent, in the presence of heat, to a compound that contains a ether group attached to a benzene ring, a ketone group, an alkene group and a phenyl ring.
Many thanks.
Hi. I dont know if this site is still active.
What will happen when isopropyl methyl ether reacts with HI?
If you have excess HI, you will get a combination of CH3-I and isopropyl iodide.
Hi, just one detail I missed: if we want ro ckeave Ph-O-R (anisole) using H2SO4 with H2O as the solvent, what teperature does this have to be done at? And for how long do we have to continue the reaction to completeness? Thank you!
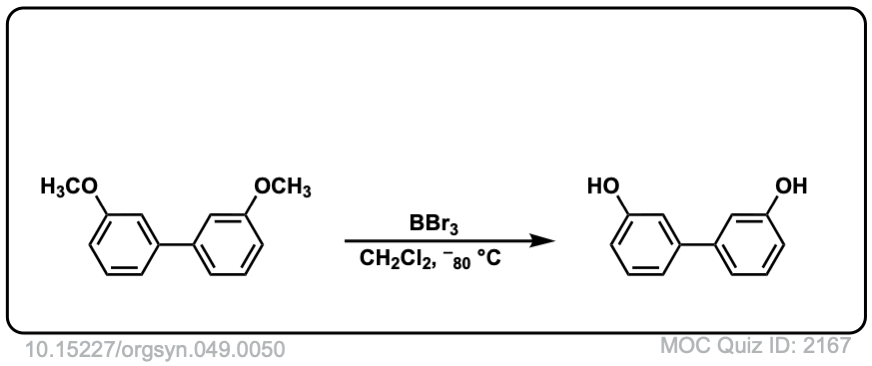
With anisole, R = CH3, the usual approach is to use HI (iodide ion will act as nucleophile to attach CH3 group) not H2SO4, since the HSO4(-) ion is a very poor nucleophile. You might have to wait awhile for that methyl group to cleave. An alternative approach is to use a very strong Lewis acid such as BBr3, as shown in this Organic Syntheses example.
http://www.orgsyn.org/demo.aspx?prep=CV5P0412
Will the nucleophile go to the smaller alkyl group and break the c-o bond there in the first R-c-o-R reaction
What is an excellent question. In this case, since we are discussing SN2, the answer is yes, since the SN2 proceeds most quickly on small alkyl groups.
What happens when allyl iodide reacts in excess of HI ?
There will likely be addition of HI across the pi bond of allyl iodide.
Sir,
Please explain the case of CH3-O-tBut in presence of HI {not anhydrous}?
Thanks.
The first step is protonation of oxygen. After that, there are two competing possibilities as to what could happen first: attack of I- on the methyl group to give CH3I and tBuOH, or ionization to give the t-butyl cation and CH3OH. I am not sure at the moment which path would initially dominate, but in the excess of HI, both will be converted into alkyl iodides.
And thank you sir for such a great resource…….it’s really helpful.
I have a question about how to decide where the nucleophile will attack in case of asymmetric ethers ?…..some where it’s said that it attacks primary carbon(less hindered) producing 1° alkyl halides..and in another instance it’s said that it produces 3° alkyl halide…..can u help me out here…Thnx.
If any one side of the -O- bond has a three degree carbon.The mechanism proceeds through an Sn1 type mechanism.This results in the attack on carbocation with more stability.So it reaults in a three degree alkyl halide.
In other instances.The mechanism is Sn2.Hence a primary alkyl halide is formed.
1-ethoxy-2-(1-methoxyethyl)benzene + HI —> ??
Is this a reduction reaction?
–
No, it is a substitution.
Rxn btwn C6H5CH(CH3)OCH3 with HI
After protonation with HI, the likely first step is ionization to give the secondary benzylic carbocation, liberating methanol. Attack of HI on the carbocation then gives an alkyl iodide. But in the excess of HI, even methanol will be converted to CH3I.
Is cyclic ether less reactive than aliphatic ether towards proton
According to the Evans pKa table the pKa of protonated dimethyl ether is -3.8 and the pKa of protonated THF is -2.0. Since the stronger the acid, the weaker the conjugate base, it’s fair to say that THF is a slightly stronger base than dimethyl ether.
diisopropyl ether +bbr3 HOW IS THIS WORk??
Boron coordinates to oxygen first, making an ion pair. At that point, there is either SN2 attack by bromine on the secondary carbon of one of the isopropyl groups, or ionization to give a secondary carbon followed by attack of bromine.
Hi James
Is HOOC-C6H5-O-CH2-C6H5 benzyl aryl ether that results from protection of phenol with benzyl chlorid, Is this compound stable if l do estirification reaction with using chloropropanol
Chloropropanol? that’s not stable
How we will do alkaline hydrolysis of ethers
It’s generally very difficult because your leaving group has to be an alkodie, which tends to be quite basic.
When does this reaction follow SN1 and when does it follow SN2. Because my textbook says it depends on the stability of the carbocation that the oxonium ion forms. If its stable then SN1 otherwise SN2. Pls Help
Well, that’s absolutely true. It does depend on the stability of the carbocation. This specific page covers the SN2 example. One unambiguous situation is the case of phenyl methyl ether where SN1 cleavage is impossible due to the instability of both the phenyl cation and the methyl cation.
In the 3rd example there is two -OH.
Is that the correct result or does it need to be corrected? I have a assignment( a conversion) => CH3-O-CH2CH3 ==> CH3-CH2-OH
In the presence of excess HI this would be converted to CH3CH2-I
What if the substance is like a THF but with a double bond. Would hcl react with the double bond first?
Yes. Assuming the double bond is adjacent to the oxygen, you’d have an enol ether. Draw some resonance forms of this species and you will see one with a negative charge on carbon. That’s the position which will be protonated.
Sir James when we r using the THF as a solvent for reacion there is no need to add the additives specially in organometalic reaction but in presence of ether their need to add additives. why?
? You’ll have to provide a specific example.
What would happen in the case of a cyclic diether? Would both ethers be cleaved or just one?
Not sure what you mean by “diether”. Are you referring to an actual di-ether, or an acetal?
CH3-O-CH2-CH3 +HI gives CH3I and CH3CH2OH and why not CH3OH and CH3CH2I??
Attack on methyl group will be considerably faster than on primary carbon. Note however, that if excess HI is added, all alcohols will be converted to alkyl iodides.
Always remember this thumb rule
If you have ROR’ type group and both R and R’ are 1 degree then first H+ will attach on O and in second step I- will do Sn2 attack on less hindered C ( if R is CH3 and R’ is C2H5 then R is less hindered) .
If any of the two R groups one is sec or tert group then first C+ will form and then I- will attach on it ( C+ will form where it is more stable)
Note that in SN2 reactions the steric hindrance plays a big role. To obtaine CH3CH2I, the nucleophile has to attack a more sterically hindered carbon when compared with just CH3, so preferably the nucleophile reach for the less hindered CH3 for the attack giving CH3I and CH3CH2OH rather than CH3OH and CH3CH2I.
Hi, do the cyclic ethers (not epoxides) too follow these type of reactions?
Yes, they do. For example the cyclic ether tetrahydrofuran (THF) can be opened with strong acid in the presence of a nucleophile. The Lewis acid titanium tetrachloride also does a particularly good job….
Will this reaction work with HCl? For example: ethanol + hcl = chloroethane?
Yes, it’s possible.
Hi, will secondary and tertiary cleavages be SN1?
Tertiary cleavages will certainly be SN1.
Hello James,
can you also do this cleaving of ethers quantitative to make an analysis for determining monomeric components in a poly-ether?
In theory yes, but I imagine that it would be very difficult on a practical level.
What happens when you put a reagent with THF? How does it react?
If the reaction, say for a tertiary ether, takes place at high temperature in an acidic medium, can an alkene be formed via elimination (provided the anion is a weak nucleophile) ?
will you please go over the reaction between thf and h3o+?
Great question. Added it as an “addendum”, see above.
Hi James,
could you give any ideas on the reaction conditions for the aequeous cleavage of THF? Since water is a rather bad nucleophile the reaction would take ages while giving unsatisfying yields. Except for the case one choses right temperatures and ratio between sulfuric acid / water. Maybe there also exists a usable catalyst ?
Why do you want to do aqueous cleavage of THF?
If there were a tertiary alkyl group among them, would the mechanism have been SN1 since the carbocation can be stablised now by +I effect ?
It’s hard to generalize. This specific post is about SN2 cleavage.
For the first example with the clevage of C6H5OCH3, how come C6H5I and CH3OH don’t form ?
Ah! This is an SN2 reaction…. and SN2 reactions require a backside attack. A backside attack would have to occur from inside the aromatic ring which is impossible (very crowded). Also, SN2 reactions don’t work on sp2 hybridized carbons (like alkenyl halides)
Does this method work for complex molecules like LIGNIN which contain a lot of ether linkages?
In regards to reactivity, are more substituted carbon centers less reactive because during the backside attack there is more steric hinderance?
Yes, exactly! (for SN2 reactions, anyway)
Hey, I have a question about keeping this reaction from happening. If I were so to say have a hydrogen halide in conditions that favor radical formation (H2O2, UV) would I be able to control this reaction, or cause the radical substitution to occur in higher likelihood than ether cleavage?
Depends on the ether, but most ethers are relatively stable towards HBr. Ethers don’t just fall off, you need to hit them with a hammer.
This was so helpful! Thank you so much! Do you have anything on epoxide ring opening?
Suppose I have a polymeric structure, with multiple ether linkages, say for instance, an ether of polyethlene glycol. What is the probability of the cleavage affecting only the terminal linkage and not the other ether bonds??? Thanx.
hi
this my qustion please answ??er
(Suppose I have a polymeric structure, with multiple ether linkages, say for instance, an ether of polyethlene glycol. What is the probability of the cleavage affecting only the terminal linkage and not the other ether bonds??? Thanx.)
Very low. If you have conditions for leaving off the terminal linkage, it will cleave off the internal linkages too. You need to modify the PEG somehow so that only the terminal residue can be removed.
Hi, in the mechanism is produced Ph-OH + CH3I. Why is not produced Ph-I + CH3-OH? Thx, P.
Because that would involve an SN2 of I- on an sp2 hybridized carbon, which is very slow – not to mention that “backside attack” would have to occur from the middle of the benzene ring!
incase of tert.ether and resonance stabilised ether we can use SN1 Mechanism
what about unsymmetrical ether cleavage???????
Is HI a stronger nucleophile than HCl? If asked to list all the hydrogen halides in terms of their cleavage speed with an ether without side chains, what order should you arrange them? I always thought HF is the strongest and as you go down it gets weaker. But that wasn’t the case right? Could you please explain to me why.
Thanks,
Hao
HI is a better nucleophile than HCl because the Iodine atom is more polarizable and hence more able to take place in SN2 reaction. As you go down the halide group, the electron shells get larger and farther away from the nucleus and therefore more polarizable. Thus they get more nucleophilic down gorup as well
You must be thinking that F being for electronegative stabilizes the negative charge on it the most. But remember thats not the only basis on which u decide the acidity. H-F bonds are hydrogen bonds(Very strong) so cleaving it would be the most difficult. While for the others hydration energy also plays a role. So the correct series is:
HI> HBr> HCl> HF
According to their acidic strength.
So I- acts as a very good nucleophile because it is a stable nucleophile.
But what about steric hindrance ? I would expect the chloride ion to be more reactive than the iodide due to its considerably smaller size. Also, that becomes even more important considering the fact that the reaction is Sn2.
Steric hindrance isn’t really an issue with halides because the size is generally inversely proportional to the length of the bond they form with carbon [i.e. small Cl, shorter bond length, large I, longer bond length] and these two factors largely cancel out. More important is the type of solvent. In polar protic solvents (solvents that can hydrogen bond) iodide is a better nucleophile than chloride. See this posthttps://www.masterorganicchemistry.com/2012/06/18/what-makes-a-good-nucleophile/
First of all we have to understand the bond distribution for take place in the any type of neucleophilic reaction #
H-F (least tendency to distribution of ions) due to smallest surface area
H-I (quick or good/fast tendency to distribution of ions) due to greatest surface area
Above in basis of reactivity order
F>CL>BR>I OR H-F<H-CL<H-Br<H-I
If you you look at the mechanism, the first step shows the Hydrogen-Iodine bond breaking. This occurs due to the fact that Iodine is a large atom and valence electrons are far from the nucleus, thus making it easy to cleave the bond to the small s orbital of the H. If you look at the conjugate base Iodine is the largest of the halides and is more polarizable than the rest thus it can stabilize a negative charge better. You can further expand this logic to the halides and observe the trend.
Yes, thanks for the comment Ali.