Addition of HBr to Alkenes
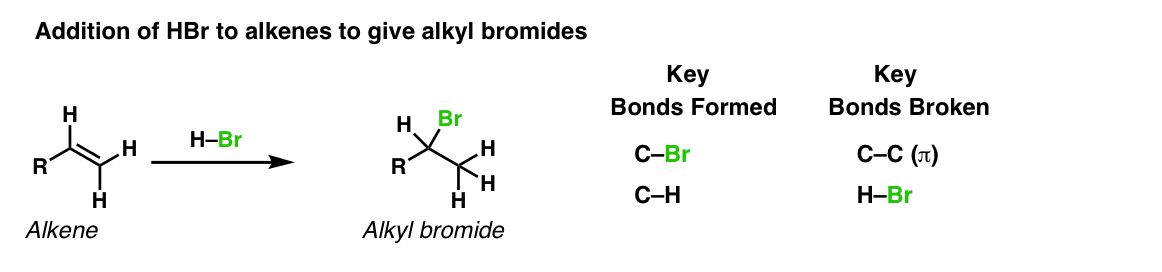
Description: Treatment of alkenes with hydrobromic acid will result in the formation of alkyl bromides.
Notes: This is an addition reaction. Note that the bromine always ends up at the more substituted carbon of the alkene (Markovnikoff-selectivity)
Examples:
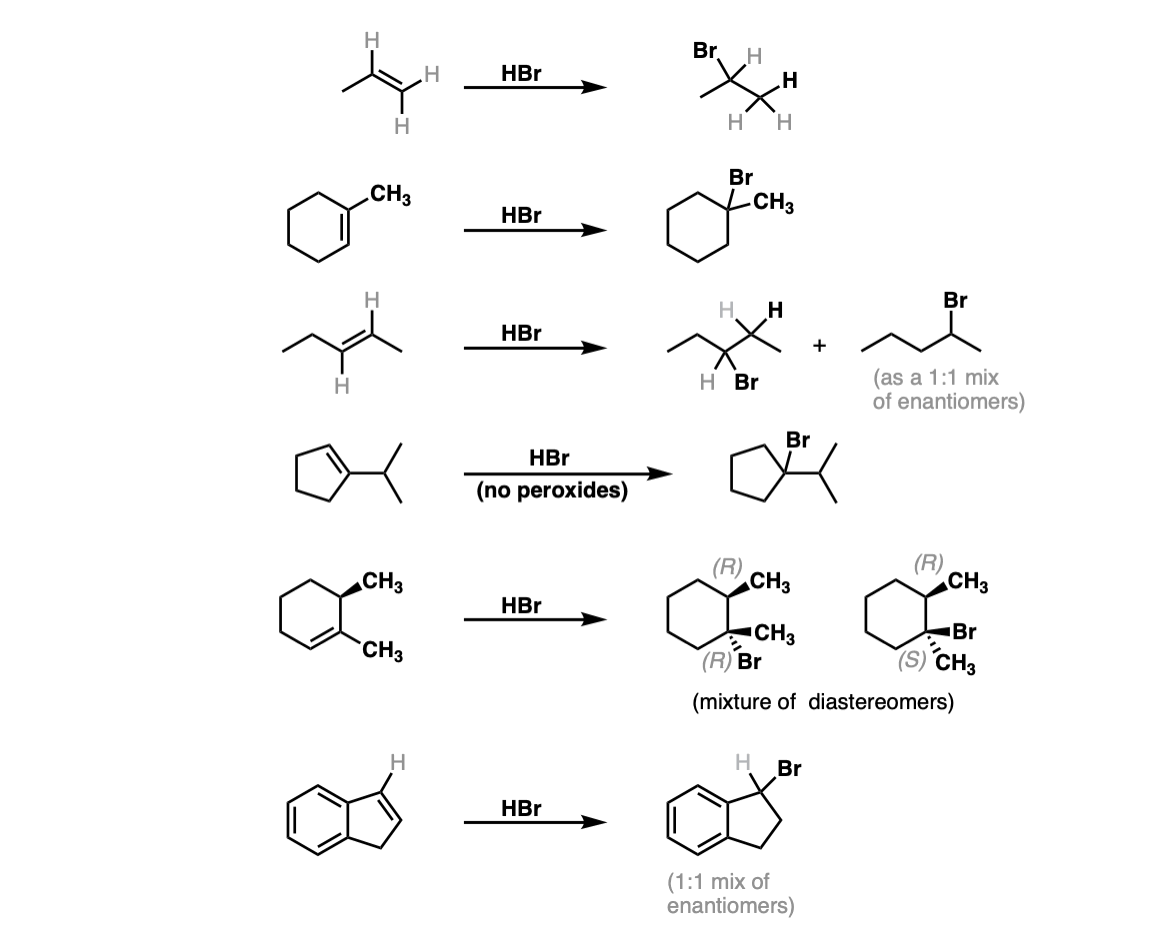
Notes: The first two examples show a simple addition of HBr to give the Markovnikov product. In the third example Markovnikov’s rule gives no clear preference, so a mixture is obtained. The fifth example gives a mixture of diastereomers, since addition of HBr across the alkene will not affect the initial (R) stereochemistry. The final (sixth) example will give a dominant product, despite identical substitution, because formation of the resonance-stabilized carbocation will be favored!
When a secondary carbocation is formed adjacent to a tertiary or quaternary carbon, rearrangements are possible. See these pages for more examples:
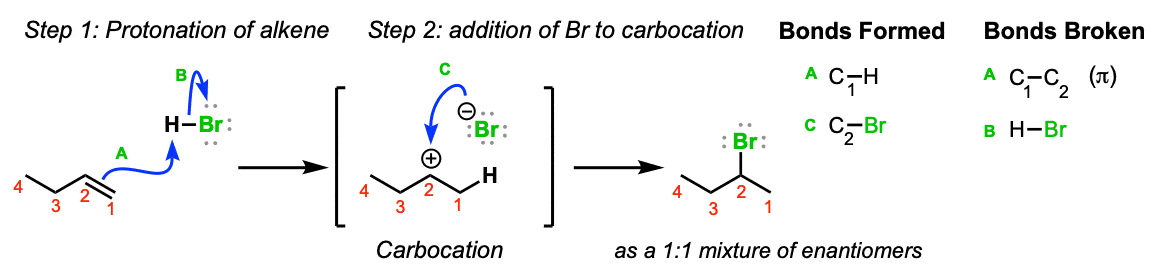
Mechanism: Electrons from the C1-C2 π bond attack the hydrogen of HBr, expelling the bromide anion and leading to the formation of a carbocation (Step 1, arrows A and B). Note here that the carbocation preferentially forms on C2 (secondary) and not C1 (primary) since secondary carbocations are more stable. The bromide anion then attacks the carbocation, leading to formation of the alkyl bromide (Step 2, arrow C)
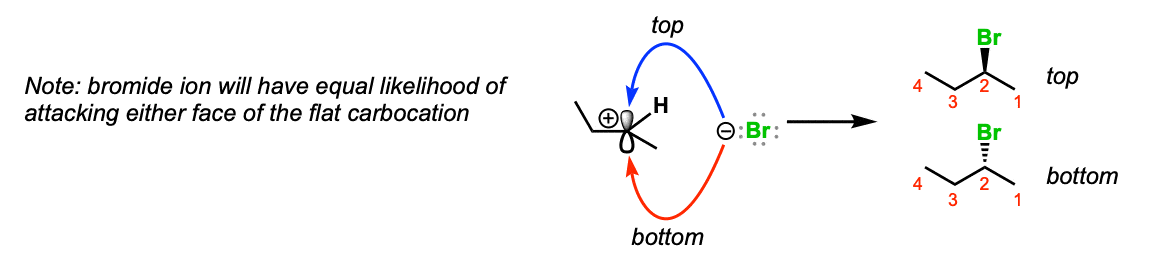
Notes: Since the carbocation is planar (flat) there is no preferred direction of attack of the bromide ion. If it’s possible to form a stereocenter, a mixture of stereoisomers will be obtained.
Rearrangements: When secondary (or primary) carbocations are formed adjacent to a more substituted carbon, adjacent hydrogen atoms or alkyl groups can shift, resulting in formation of a more stable carbocation.
The first step of the reaction below is formation of a carbocation. Here, the formation of a carbocation via attack of the alkene upon H-Br is shown. (Step 1, arrows A and B). Since we have a secondary carbocation adjacent to a tertiary carbon, shift of a hydrogen to the secondary carbocation will result in a (more stable) tertiary carbocation (Step 2, arrow C). The tertiary carbocation is then trapped, in this case, by Br(–) (Step 3, arrows D). The rearrangement step goes through a transition state such as the one pictured.
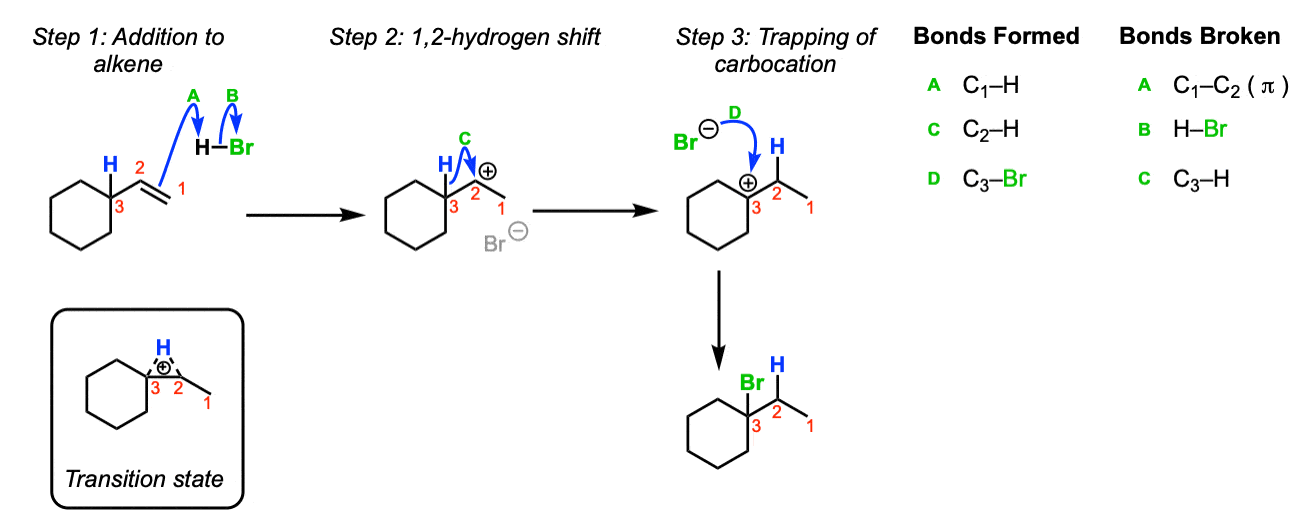
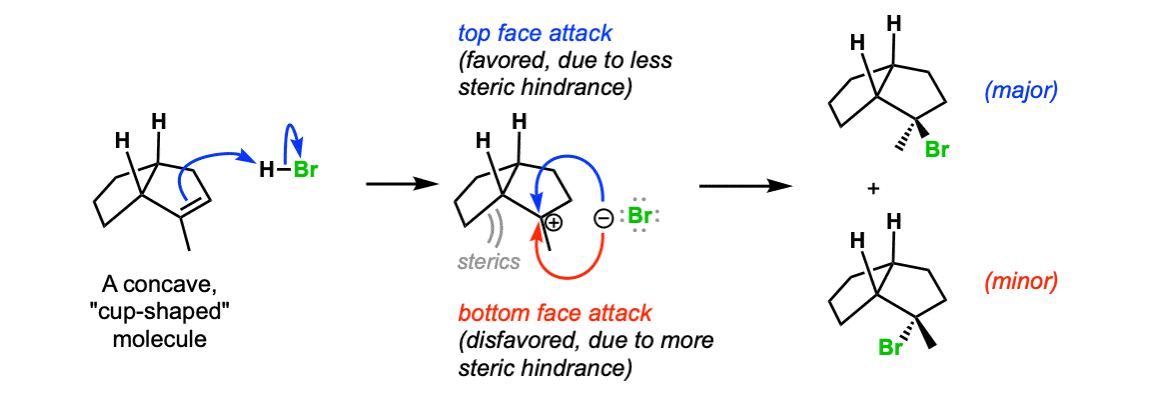
Additional example (advanced) : If the molecule contains adjacent stereocenters, the two directions of attack on the carbocation will no longer be of equal energy and a mixture of diastereomers will be obtained. In this example, attack of the bromide ion on the less hindered “top face” of the carbocation is favored, and a mixture of products (diastereomers) will be obtained.
Rearrangements can also occur. See Additions To Alkenes Accompanied By 1,2-Hydride Shifts
Quiz Yourself!
 Click to Flip
Click to Flip

 Click to Flip
Click to Flip
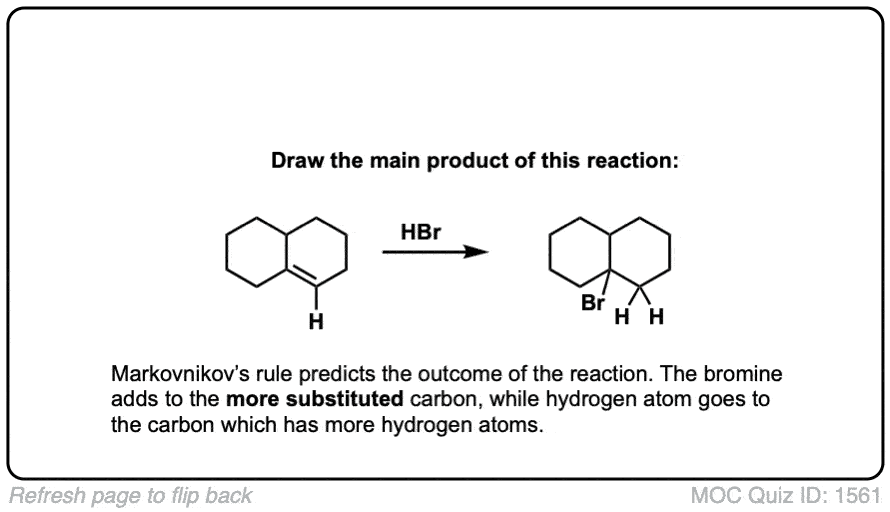
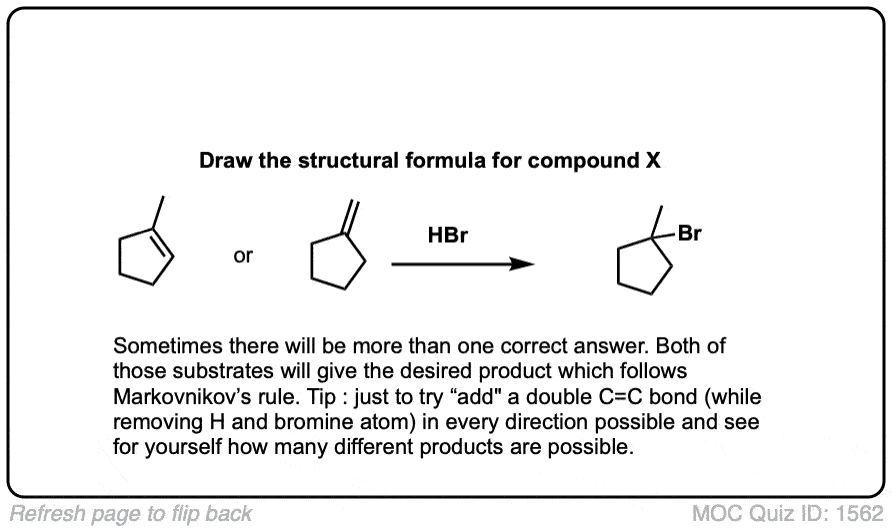
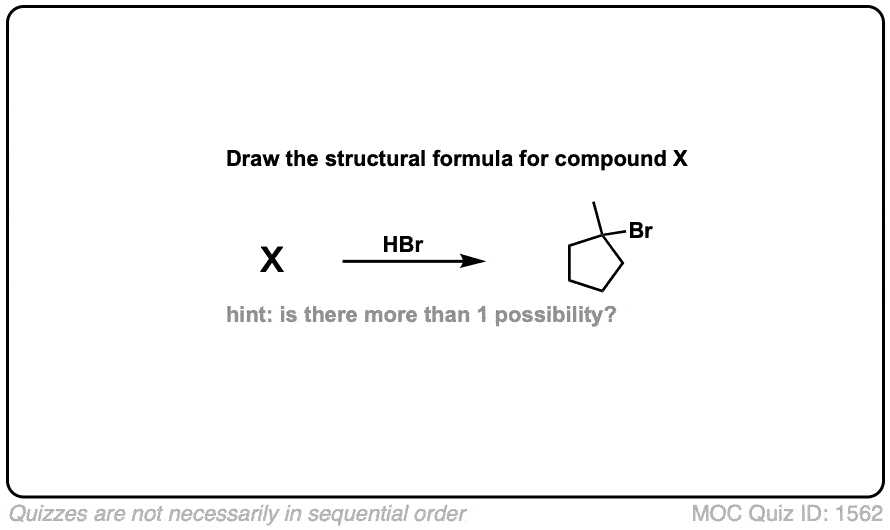 Click to Flip
Click to Flip
(Advanced) References And Further Reading:
- Early example
The Stereochemistry of the Addition of Hydrogen Bromide to 1,2-Dimethylcyclohexene
George S. Hammond and Thomas D. Nevitt
Journal of the American Chemical Society 1954 76 (16), 4121-4123
DOI: 10.1021/ja01645a020
Early paper from the 50’s by Prof. George Hammond (of Hammond’s Postulate) on the mechanism of HBr addition to 1,2-dimethylcyclohexane. He prefers a concerted pathway, although that might due to the conditions he employs – in pentane, a very nonpolar solvent, polar intermediates are disfavored. - Mechanistic studies
Hydrochlorination of cyclohexene in acetic acid. Kinetic and product studies
Robert C. Fahey, Michael W. Monahan, and C. Allen McPherson
Journal of the American Chemical Society 1970 92 (9), 2810-2815
DOI: 10.1021/ja00712a034
Detailed kinetic studies of the addition of HCl to cyclohexene in acetic acid, discussing a possible third-order mechanism (rate = k[cyclohexene][HX]2). - Experimental Procedure
SPIROANNELATION OF ENOL SILANES: 2-OXO-5-METHOXYSPlRO[5.4]DECANELee, T. V.; Porter, J. R. Org. Synth. 1995, 72, 189
DOI: 10.15227/orgsyn.072.0189
The first reaction in the above procedure involves two steps – addition of HBr across the double bond and converting the aldehyde to a dimethyl acetal.
Real-Life Examples:
Org. Synth. 1938, 18, 47
DOI Link: 10.15227/orgsyn.018.0047
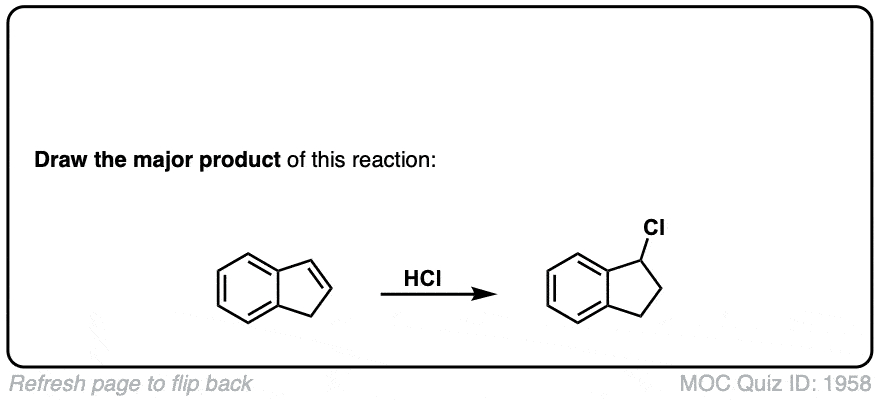
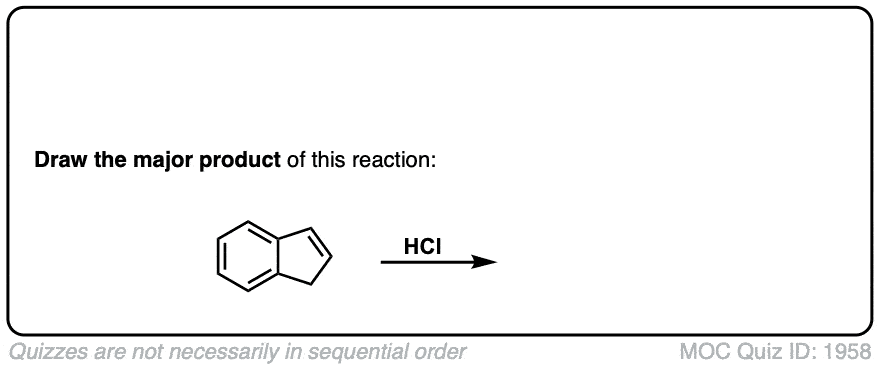 Click to Flip
Click to Flip
Org. Synth. 1940, 20, 64
DOI Link: 10.15227/orgsyn.020.0064
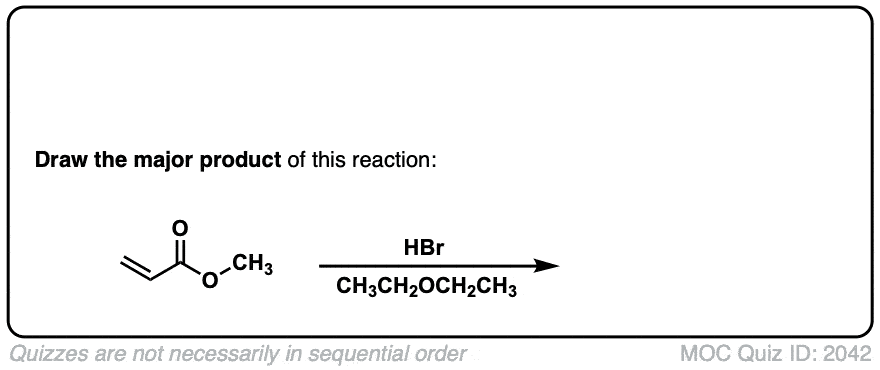 Click to Flip
Click to Flip
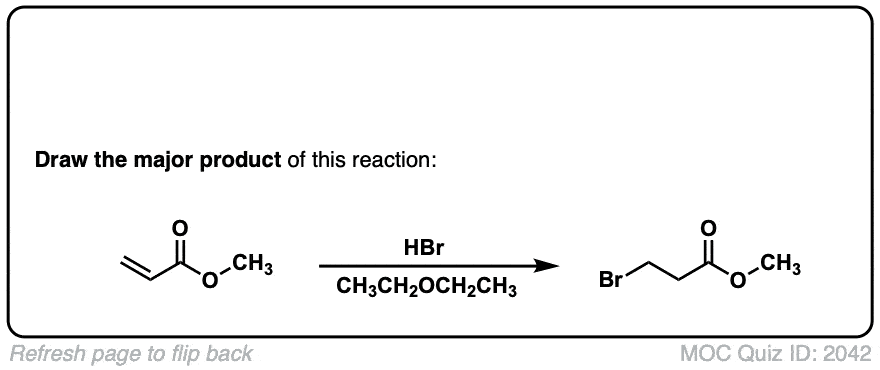
Could you please tell what happens when conc. HBr is added to a secondary alcohol
Generally, the OH is protonated to give OH2(+) and then the C-O bond breaks while a new C-Br bond forms. The net result is formation of an alkyl halide.
With secondary alkyl halides, there may be formation of an intermediate carbocation and rearrangement if a more stable carbocation can be formed.
I am looking for empirical kinetic data regarding addition rates of HX to differently substitutes alkenes (secondary, tertiary etc.).
I assume that the stability of the transition state- influenced by the stability of the differently substituted carbocation intermediate, Hammond postulate- prevails (over the opposing effect of the stability of differently substituted alkenes reactants).
A college of mine believes otherwise and neither of us could find reliable kinetic experiment to support either view.
James, could you help?
Thanks!
What happens if we add HBr in presence of CCl4
In the absence of light (hv) / peroxides then it will perform a Markovnikov addition of HBr to the alkene. Under free-radical conditions, however, it will result in free-radical anti-Markovnikov addition of HBr across the alkene.
Sir,if hbr in ccl4 is used,will it change the addition reaction mechanism??What role will ccl4 actually play here??
Just a non-polar organic solvent. It wouldn’t change the reaction mechanism. Although you’d want to make sure you don’t also see “light” or “peroxides” because that would indicate that you’re performing a free-radical addition of HBr across the alkene, which *does* involve a different mechanism.
How anti aromatic compounds are paramagnetic?
Look at the molecular orbital diagram. https://www.masterorganicchemistry.com/2017/03/27/antiaromaticity/
Why only hydrogenbromide follow radical pathway in addition to alkenes not other hydrogenhalides??
For the radical pathway, the free-radical pathway for addition is only favorable for HBr. Radical addition of HCl is onlky rarely observed. Here is an example: https://pubs.acs.org/doi/abs/10.1021/ja00879a034 .
The reason is that the HCl bond is too strong (103 kcal/mol) to be easily broken by the alkyl radical that forms after addition of HCl – a typical secondary C-H bond is about 96 kcal/mol, so the reaction is thermodynamically uphill.
For HI, the problem is that the C-C pi bond (60 kcal/mol) is weaker than the C-I bond (50 kcal/mol) so there is no driving force for the addition reaction to occur.
HF suffers from the same problem as HCl, but moreso – the HF bond is extremely strong. Even if you do make the F• radical, it’s nearly impossible to control.
So to summarize HBr is kind of in the “goldilocks” region for this process. The C-Br bond is stronger (66 kcal/mol) than the C-C pi bond (60 kcal/mol), facilitating addition, and the C-H bond (96 kcal/mol for a secondary carbon) is stronger than the H-Br bond (87 kcal/mol) meaning C-H abstraction will be thermodynamically downhill.
Bond strengths from here: https://labs.chem.ucsb.edu/zakarian/armen/11—bonddissociationenergy.pdf
This is in regards to the “additional example (advanced)”: I am not sure that it is reasonable to use sterics on the a carbocation to predict which diastereomer is favored. Even if you are under kinetic control as your answer would require, depending on the side of the ring it attacks there are considerations beyond sterics, such as torsional strain developing at the transition state that can trump “same side of the ring” sterics.
Lastly, bare alkenes and HX’s often follow a rate equation that is second order with respect to the HX. Therefore, the nucleophile is often delivered to the opposite face than the H+, as the developing carbocation is captured immediately as it is forming.
Final thought: as an organic chemistry professor trying to get students to appreciate “selectivity vs reactivity” and that carbocations are highly reactive intermediates, I think it’s better (and more true) to note that there is a mixture of diastereomers formed that won’t be 50/50 (because they’re of different energy).
Hello sir, useful information.
Can you please tell me, if there is option between a OH group and double bond, HBr will add to double bond or protonate OH group?
Proton transfers to OH are generally faster than is addition to a double bond. However, it’s an equilibrium situation. If your molecule contains both an alcohol and an alkene, I’d double-check to make sure that you can’t have intramolecular formation of a 5 or 6 membered ring from 1) addition of HBr across the double bond, and 2) attack of the OH on the resulting carbocation.
The other option is that the protonated OH leaves as OH2, resulting in a new carbocation, which is then attacked by a double bond as nucleophile. It really depends on your molecule.
why H2O2 is used as a catalyst in this reaction??
This is the version of HBr addition under polar conditions; H2O2 (or other peroxides) are used as catalysts in the free-radical addition of HBr to alkenes, which is covered here: https://www.masterorganicchemistry.com/reaction-guide/free-radical-addition-of-hbr-to-alkenes/
If there was an excess of HCl added to a terminal alkene, would an Sn1 reaction occur after the first Cl addition?
If Cl- is the leaving group, then what would be the nucleophile?
Let’s say you have a 3-methylcyclopentene reacted to HBr. How would it produce 1-bromo-1-methylcyclopentane? (It means the bromide ion became geminal to the methyl group)
That’s a rearrangement reaction. Look into “hydride shifts” and why they occur.
can benzene reactions with hydrogen bromide?
Yes, but you wouldn’t really see the result, since it will protonate the ring (forming C-H) and then break C-H to re-form the aromatic compound. So it looks as if no reaction is happening.
However if you use D-Br what will happen is that there will be slow exchange of H for D at the aromatic ring.
Is the production of diastereomers with the addition of HBr also true for the addition of HCl?
Yes, they’ll work exactly the same way on that substrate.
Hi I was wondering its not mentioned on here but with the stereochemistry of the product formed. Which step in the mechanism determines the stereochemistry of the product? I can’t seem to figure it out :S Does it have something to do with which carbocation intermediate is formed?
There are two steps in the mechanism which will determine the stereochemistry.
In the first step, the alkene can attack H+ from either of its two faces. If this results in a chiral center, then this step will determine if this carbon is “R” or “S”.
In the second step, the flat carbocation can be attacked from either face by the Br- nucleophile. If this results in a chiral center, then this step will determine if *this* carbon is “R” or “S”.
So if two new chiral centers are formed, it’s possible to have 4 new products! (Thankfully this won’t always be the case).
Will solvent change the product? Eg.- If water is used as a solvent will OH- attach instead of Br- as it is better nucleophile?
No as water in very weak and neutral nucleophile so its possibility is vry less
Yes. HBr in water, alkene gives alcohols and in alcohol, it gives ethers.
This is not 100% related to this video, but on the topic of alkenes in general. Why is 3-methyl-2-butene more reactive than 2-methyl-1-butene?
Think about the stability of the intermediate carbocation.
Why is there increase in energy in transition state at which Br- adds to carbocation ?
Good question! The transition state involves partial bonds and non-ideal bonding geometries, which is less stable than the carbocation itself.
Is there any variation in reaction when HBr in prescence of sunlight is used?
In the presence of ultraviolet light, the reaction undergoes radical chain addition. Covered in this post:
https://www.masterorganicchemistry.com/2013/04/12/a-fourth-alkene-addition-pattern-free-radical-addition/
This really is as good as anything out there- and I think the way it is neatly packaged makes it better than the rest. I am new as of yesterday to being a member- so if this is a frequently asked new kid question I apologize- but is there a clean way to print these? I filled out the mechanism blank sheets in Organic Chem as a Second Language and I wanted to print these out so that I can study from both my copy of notes and the reactions from this guide to make sure I have it all down- but printing it from a source like this takes a lot of pages since it prints out everything not just the text and pics about the mechanism (like this one short reaction printed is 9 pages). If not- that is okay- thanks for all of your hard work.