Amines
The Amide Functional Group: Properties, Synthesis, and Nomenclature
Last updated: January 22nd, 2026 |
Synthesis, Nomenclature, and Properties Of The Amide Functional Group
In this post, we’ll try to provide a broad overview of amides. We’ll provide a brief overview of amide nomenclature, two important properties of amides that differ greatly from amines, and go over three key strategies for amide synthesis.
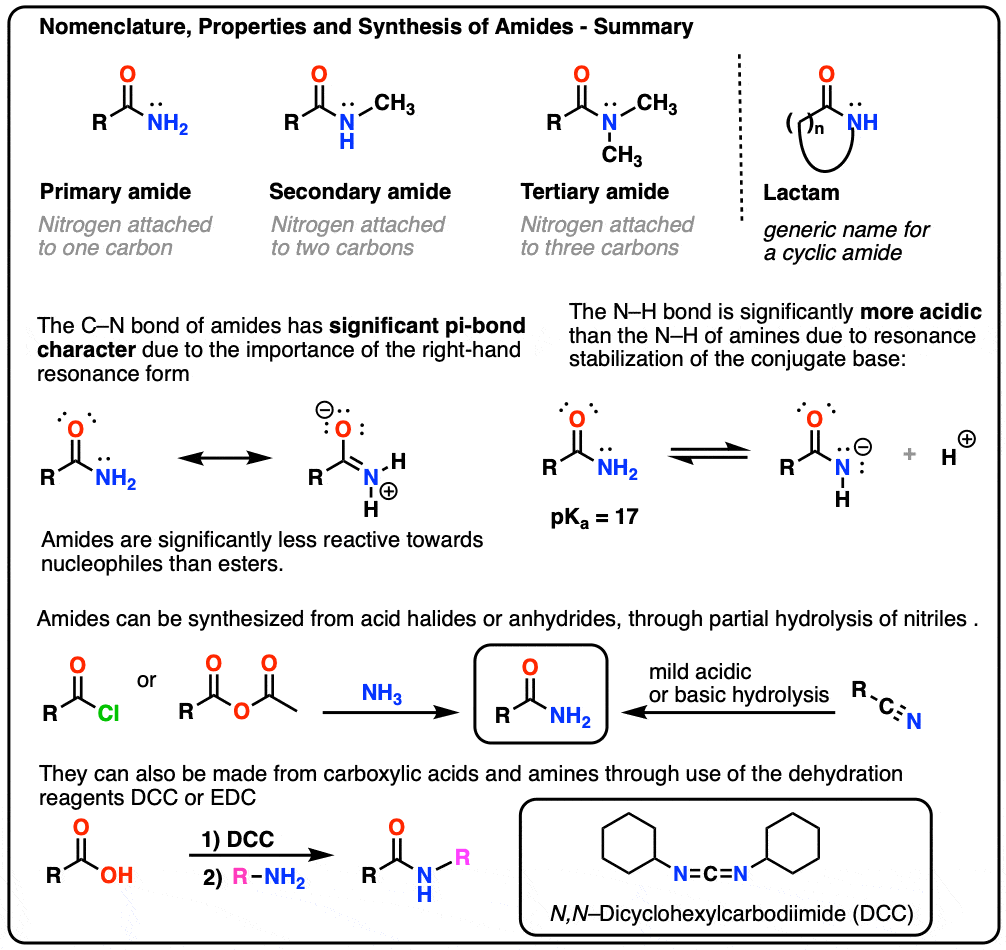
Table of Contents
- Nomenclature of The Amide Functional Group: Primary, Secondary, And Tertiary Amides
- Amides vs Amines: Less Basic, More Acidic
- Synthesis of Amides, Part 1. Nucleophilic Acyl Substitution of Acyl Halides (Or Anhydrides) With Amines
- Synthesis of Amides, Part 2: Partial Hydrolysis Of Nitriles
- Synthesis of Amides, Part 3: Use Of A Dehydrating Reagent (Such As DCC)
- Summary: Three Effective Methods For the Synthesis of Amides
- Let Us Briefly Consider A Fourth, Less Important Method: Brute Force
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Nomenclature of The Amide Functional Group: Primary, Secondary, and Tertiary Amides
“Amides” are what we call an amine that has a single attached carbonyl group. The amide functional group is to amines as esters are to alcohols.
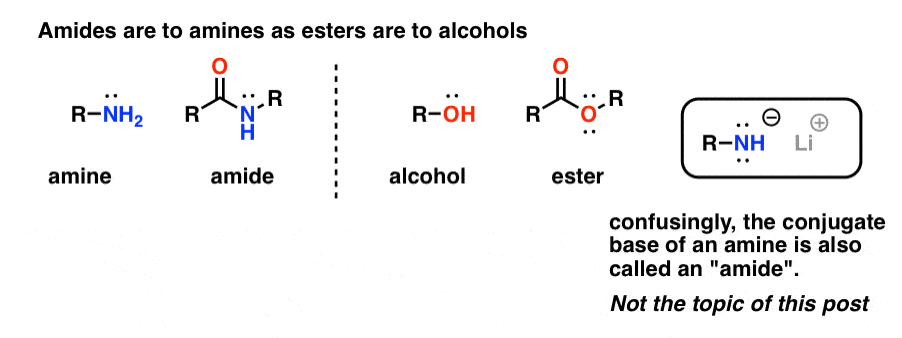
Confusingly, the word “amide” is also used to refer to the conjugate base of amines, such as sodium amide (NaNH2) and lithium di-isopropylamide (LDA). The latter are sometimes differentiated by referring to them as “amide bases”. Others will use slightly different pronunciation to differentiate the two (ayyy-myde and aaah-midd). As with any other homonym, the key is context.
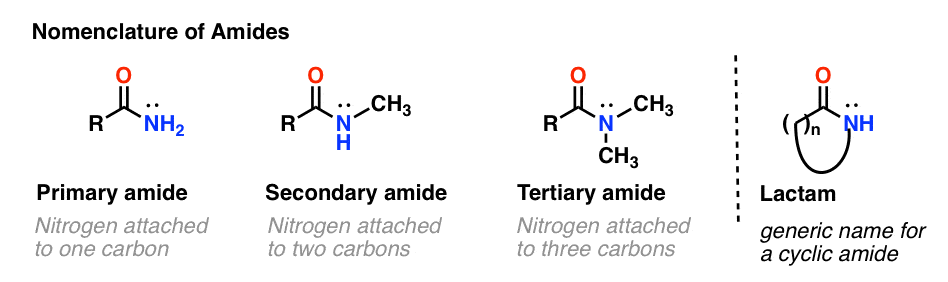
As with amines, the nomenclature used for an amide depends on the number of carbons attached to the nitrogen.
A primary (1°) amide has nitrogen attached to a single carbon; a secondary (2°) amide has the nitrogen attached to two carbons; a tertiary (3°) amide has the nitrogen attached to three carbons. A cyclic amide is called a lactam.
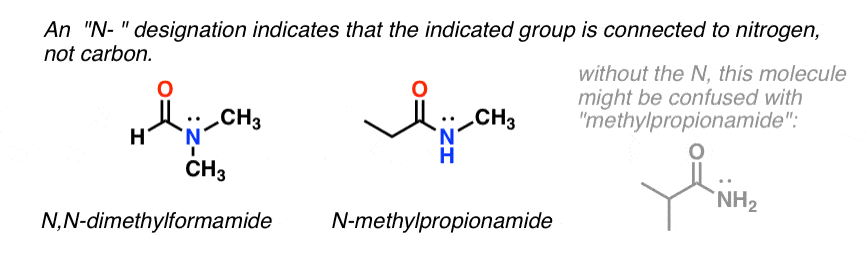
When the amide nitrogen has substituents other than hydrogen, we specify them using the prefix N- to avoid confusion. For example, N-methylpropionamide specifies the attachment of a methyl groups on the nitrogen; without the N– prefix, one might assume that the methyl group was attached to carbon, which would be an entirely different molecule.
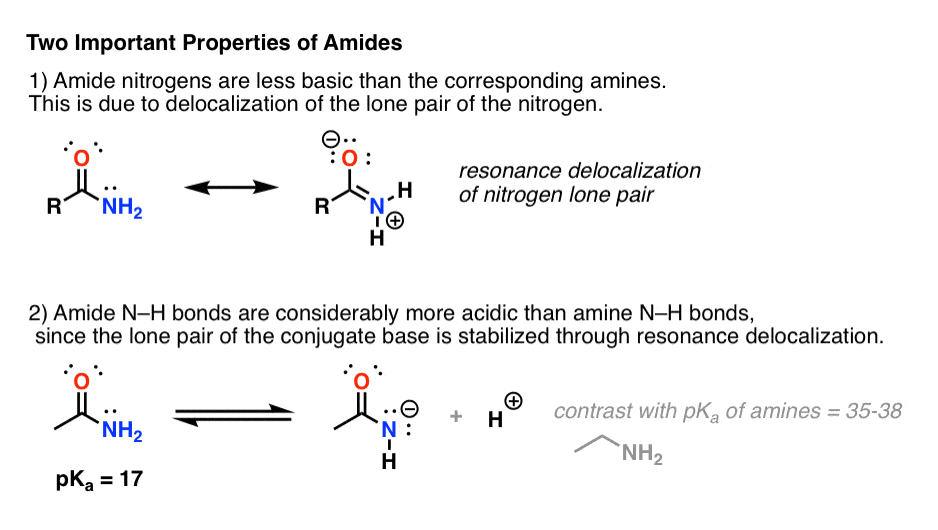
2. Amides vs Amines: Less Basic, More Acidic
Attaching a carbonyl group to an amine has two drastic effects on the properties of the nitrogen.
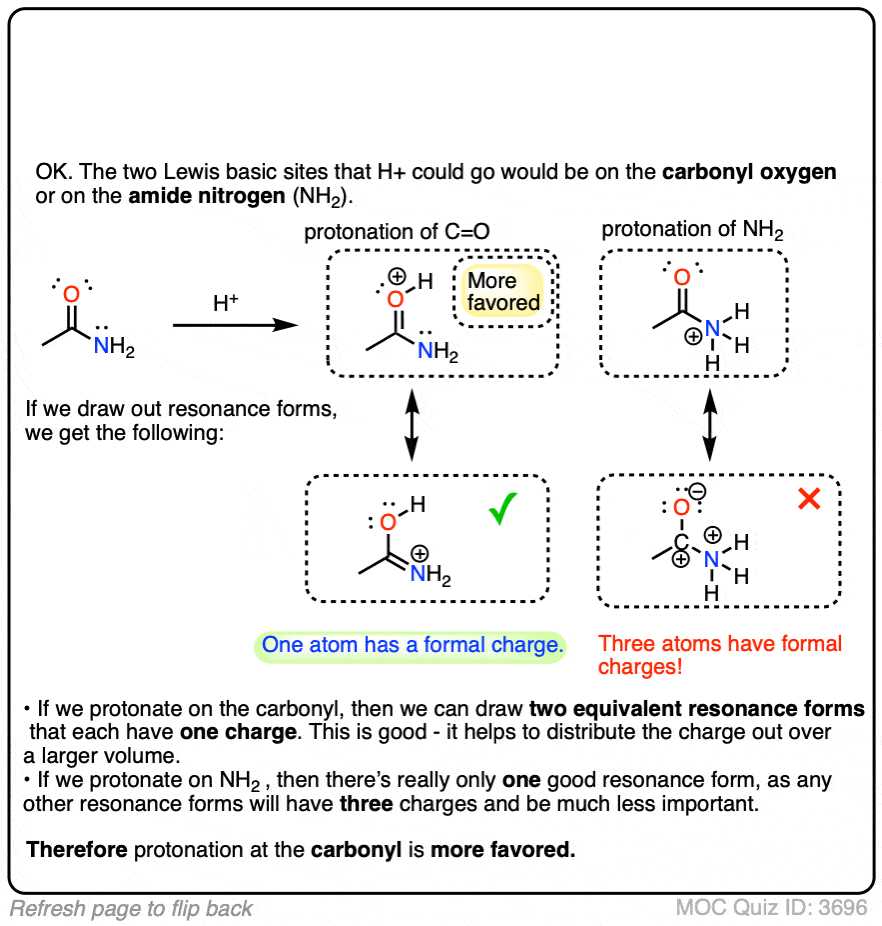
- First, amide nitrogens are considerably less basic than amine nitrogens. That’s mainly the result of the delocalization of the nitrogen lone pair into the pi bond of the carbonyl. In fact, the most basic position of an amide is not the nitrogen but the oxygen (!). [Note 1]
- Second, the N–H bonds of amides are much more acidic than the N–H bonds of amines. Why? Delocalization again. The attached carbonyl group allows the lone pair of the conjugate base to be delocalized through resonance. The pKa of acetamide (17) is about 20 orders of magnitude more acidic than ammonia (38).
A third, more subtle property of amides is that they usually have restricted rotation about the C–N bond. The resonance form where there is a C-N bond makes such a significant contribution to the resonance hybrid that one can think of the C–N bond as having “partial double-bond character”. [In this post on Conjugation and Resonance, we saw that “bridgehead amides”, which lack the necessary orbital overlap for this resonance to occur, are orders of magnitude more unstable than “conventional” amides.]
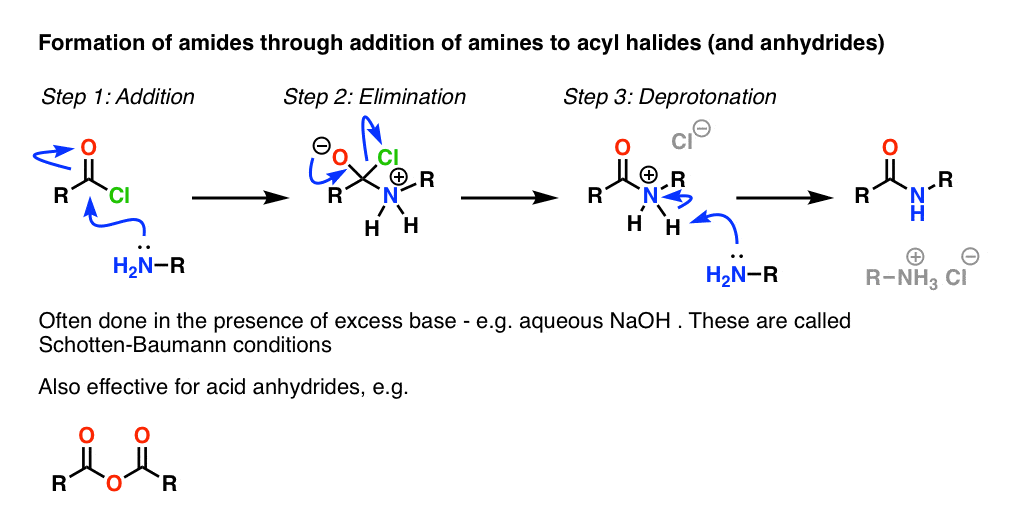
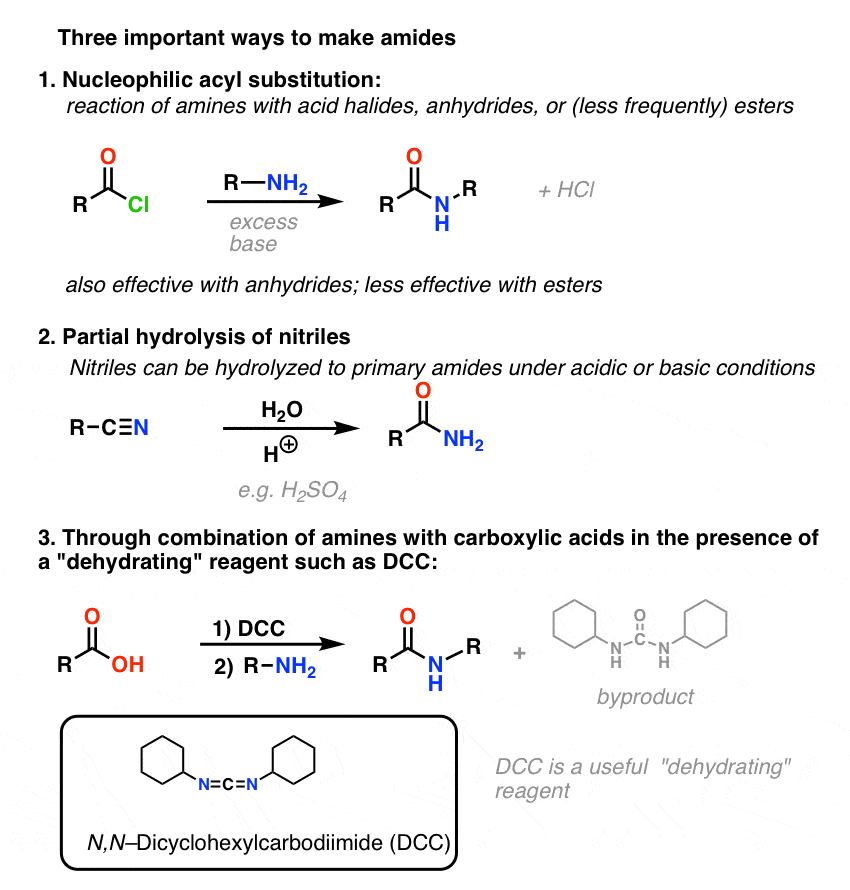
3. Synthesis of Amides, Part 1. Nucleophilic Acyl Substitution of Acyl Halides (or Anhydrides) With Amines
Acyl groups attached to a good leaving group such as acid chlorides or acid anhydrides can easily undergo nucleophilic acyl substitution with amine nucleophiles.
If only the carboxylic acid is available to start with, using a reagent like thionyl chloride (SOCl2) to convert a carboxylic acid to an acid chloride is a good first step in transforming a carboxylic acid to an amide. (PCl3, PCl5, oxalyl chloride and a host of other reagents can also work). Alternatively, treating a carboxylic acid with an acyl halide will deliver an anhydride, which can also be effective.
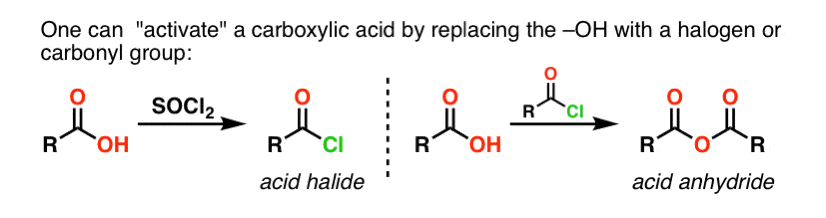
Halides (e.g. Cl– ) and carboxylates (RCO2– ) are much weaker bases, and thus much better leaving groups than HO–. So by adding an amine to an acyl halide or acid anhydride, nucleophilic acyl substitution can occur under much milder conditions, resulting in our desired amide.
(One can obtain amides through the reaction of esters with amines, but given that alkoxides [RO– ] are poorer leaving groups than halides or carboxylates, this method requires more forcing conditions. )
One thing to note with acid halides is that the process generates a single equivalent of HCl as a byproduct. In the absence of any additional base, the maximum yield of the procedure would be 50%, since the HCl would protonate any amine and render it a non-nucleophilic ammonium salt.
One way to ensure the reaction proceeds to completion is to add a second equivalent of amine. There are other practical ways of resolving this issue, which I’ve consigned to Note 2 .
If you need a refresher on the mechanism of nucleophilic acyl substitution hover here for a pop-up image or open image link here:
[click here and an image of the mechanism will pop up].
{kind=link}
4. Synthesis of Amides, Part 2: Partial Hydrolysis of Nitriles
One way to think of nitriles is that they are masked carboxylic acids. If treated with aqueous acid and a lot of heat – sledgehammer conditions – they can be hydrolyzed to carboxylic acids.
One of the intermediates in this process is a primary amide.
So if we use a slightly kinder, gentler sledgehammer technique, sometimes it’s possible to salvage the amide out of our reaction mixture before it’s hydrolyzed to the carboxylic acid.
The image below shows how one could synthesize an amide from an alkyl halide precursor, via the good-ol’ SN2 reaction:
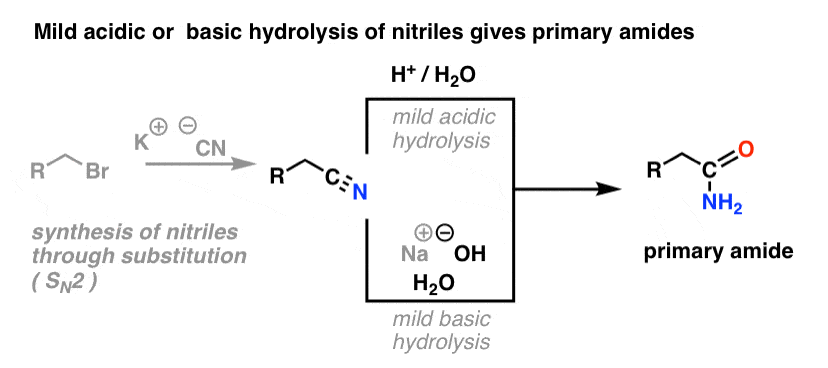
What’s meant by “mild” ? One set of conditions for the hydrolysis of phenylacetonitrile (PhCH2CN) to phenylacetamide (PhCH2CONH2 ) gives the reaction conditions as “HCl, H2O, 40-50°C, 1h “. [for a lovingly detailed experimental procedure, go here]
5. Synthesis Of Amides, Part 3: Use Of A Dehydrating Reagent (Such As DCC)
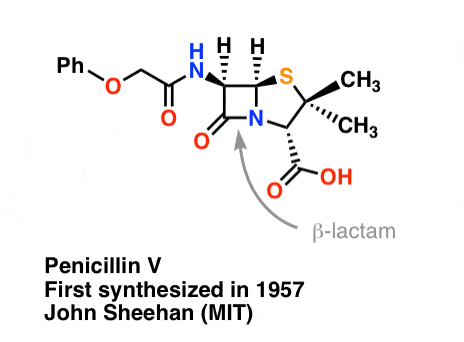
The synthesis of penicillin V in 1957 by John Sheehan’s group at MIT stands as one of the heroic achievements of postwar-era organic chemistry. The key problem was construction of a cyclic amide (the β-lactam ring) which is extremely unstable under acidic conditions. This was of no small importance, as the β-lactam is also key to penicillin’s mechanism of action: interfering with synthesis of the bacterial cell wall. Attempts to make this cyclic amide by converting a carboxylic acid to an acyl halide with SOCl2, PCl3, PCl5, and a host of other methods all failed. [Note 3]
In response, Sheehan’s group cleverly invented a very mild dehydrating reagent: N, N’-dicyclohexylcarbodiimide (DCC) which allowed for the formation of amides under very mild conditions at neutral pH.
Today, DCC (and its more practical (Note 4) cousin, EDC ) are extensively used for the synthesis of sensitive amides – particularly peptides – under very mild conditions.
Under the reaction conditions, the carboxylate oxygen attaches to the electrophilic carbon of DCC, making what we call an “active ester” – in other words, an ester that actually has a decent leaving group (unlike most esters, which don’t). The active ester is then attacked by the amine in a classic nucleophilic acyl substitution, which leads to the formation of the amide.
Looking at the byproduct, note that there are two hydrogens (each attached to nitrogen) and an oxygen (attached to the central carbon). That’s where the H2O has gone!
Because this post is likely long enough as it is, hover here for a pop-up image or open image link here:
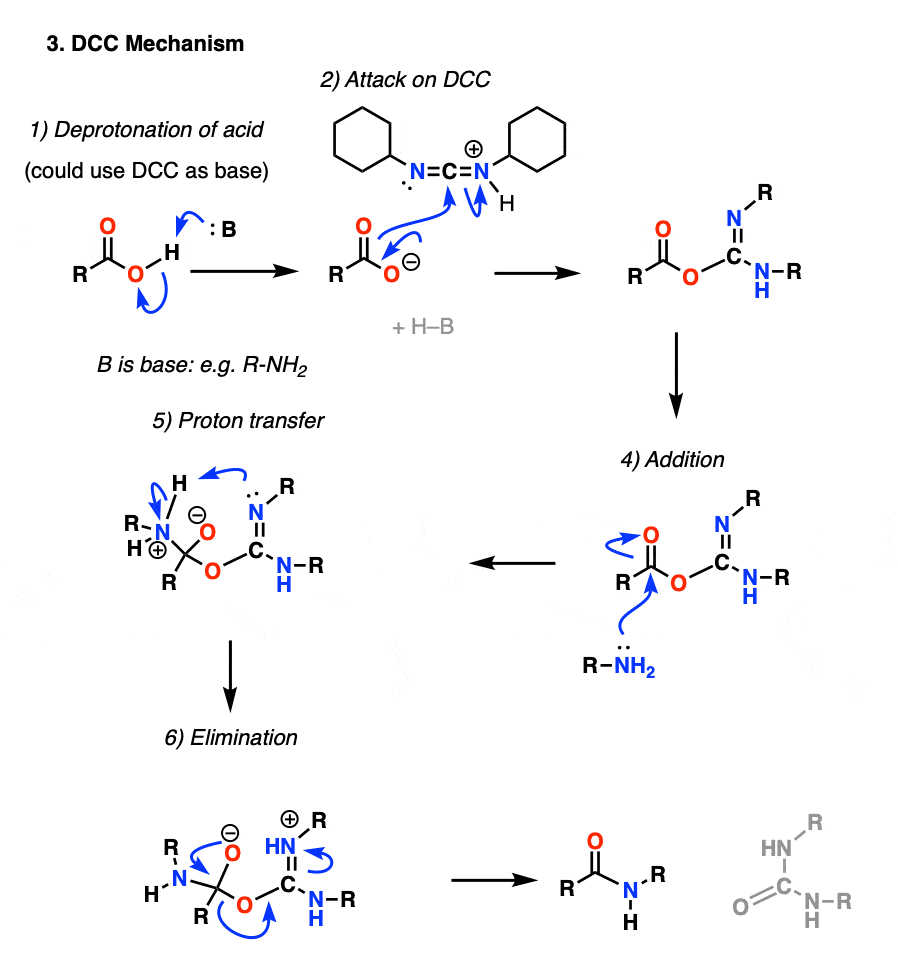{kind=link}
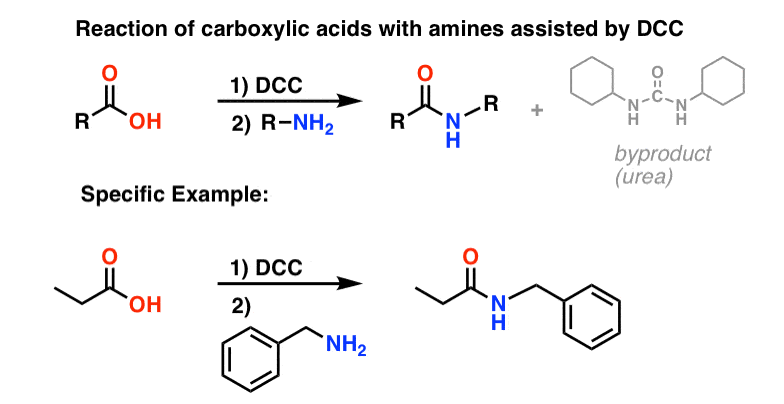
Since this reaction occurs under neutral conditions, it is extremely useful in the synthesis of peptides, which can undergo racemization (epimerization, actually) under both basic and acidic conditions.
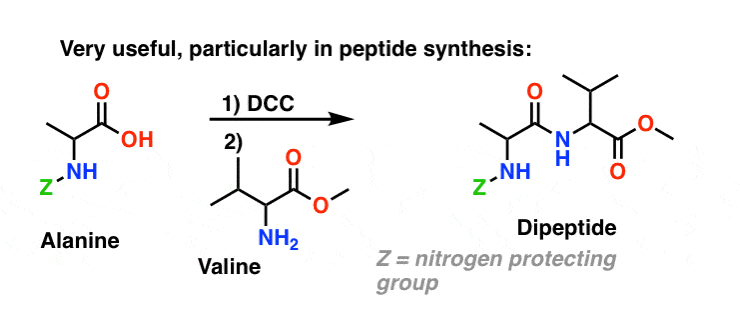
6. Summary: Three Effective Methods For The Synthesis of Amides
Let’s end by summarizing these three important (but by no means exhaustive) ways to make amides:
This concludes our post on the main points of amide nomenclature, properties, and synthesis. For a bonus method of amide synthesis, read on.
7. Let Us Briefly Consider A Fourth, Less Important Method: Brute Force
Since it’s usually covered in the textbooks, let’s conclude by considering a fourth possibility – the simplest one imaginable. What if we take a carboxylic acid and combine it with an amine, hoping that an amide will form. What happens?
Amines are bases, and carboxylic acids are, well, acids. Add the two together and you get an innocuous salt.
Sometimes one can make amides through heating the living daylights out of this salt in a sealed tube, driving off an equivalent of water. This method is called pyrolysis.
The method is nothing if not direct, and has all the subtlety of a howitzer.
The problem with pyrolysis is that the HO– group of a carboxylic acid is a terrible leaving group.
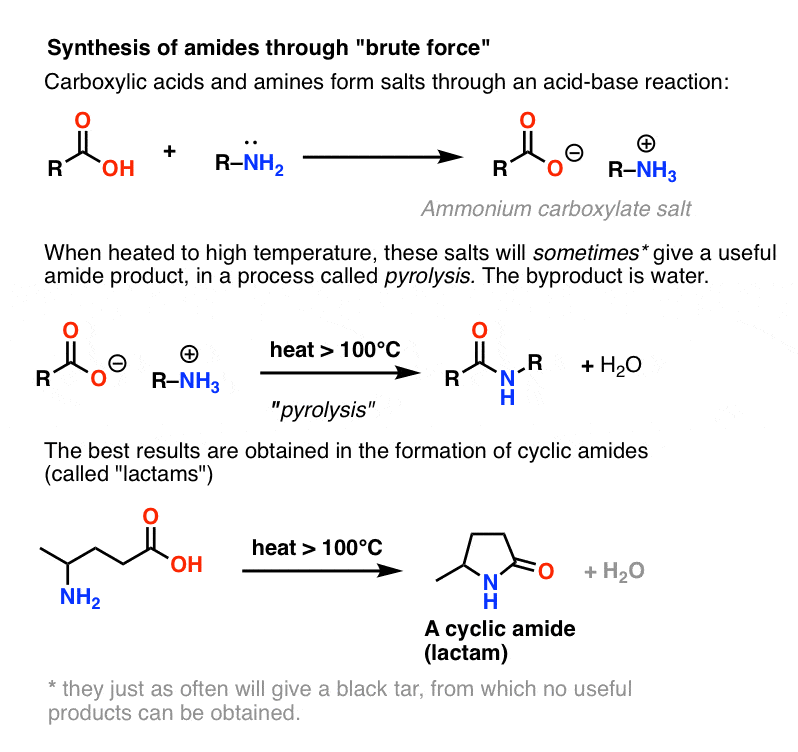
In order to form an amide from this species, the carboxylate oxygen (O– ) must somehow depart. This is not an easy thing to do, as the conjugate base of O– is the double anion O2– . In any list of leaving groups, O2– would rank somewhere between, “shitty” and “f&cking awful”.
However, if one hits this salt with the chemical equivalent of the Hammer of Thor: brute-force, high heat, a series of proton transfers from the ammonium salt can occur. eventually liberating H2O and forming the C-N bond.
This process is called pyrolysis (pyro = fire, lysis = breaking).
In certain cases, especially simple amides, and also in the formation of simple lactams, the process can be satisfactory.
In many other cases, however, it results in the formation of a black tar at the bottom of your flask from which no useful product may be obtained.
As any organic chemist can tell you, there are many diverse ways to create intractable black tars at the bottom of your flask, and this is just one method. Think of how much you still have to discover!

“Hammer of Thor” Google Image Search was chemistry-related, but surprisingly NSFW.
Notes
[related articles]
A fun, related article: Amides: Humble But Useful (from Chemical & Engineering News).
Note 1. There’s also an inductive effect, whereby the electronegative oxygen (electronegativity of 3.44) tugs on the electrons of the attached carbon, which in turn tugs on the electrons of the nitrogen.
Note 2. A very common way of carrying out this reaction is to use what are called, Schotten-Baumann conditions, where one takes up the reactants in solvent like diethyl ether or dichoromethane, and adds an aqueous solution of NaOH, resulting in a biphasic mixture. Any ammonium salts that form can dissolve in the aqueous phase, whereupon they are neutralized by the excess base and return to the organic phase. Amines are generally far more nucleophilic than hydroxide ions, so hydrolysis of the acid chloride to give a carboxylic acid is generally not a problem.
“At the time of my successful synthesis of penicillin V in 1957, I compared the problem of trying to synthesize penicillin by classical methods to that of attempting to repair the mainspring of a fine watch with a blacksmith’s anvil, hammer, and tongs” – John C. Sheehan
Note 4. The problem with using DCC is that the byproduct, DCU, is a tremendous pain in the ass to get rid of. Most byproducts are easily removed using column chromatography. Not DCU. Paying little heed to solvent polarity , DCU emerges from a column slowly, in drips and drabs, contaminating every fraction as it goes. EDC [1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide] is a variant of DCC that has a tertiary amine unit; thus, a simple acid wash during workup will remove all the urea, saving a lot of time and headache.
The following image shows the last step of Sheehan’s synthesis using DCC.
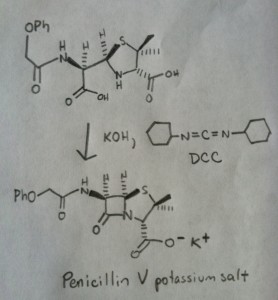
Image: Carmen Drahl/Chemical & Engineering News
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
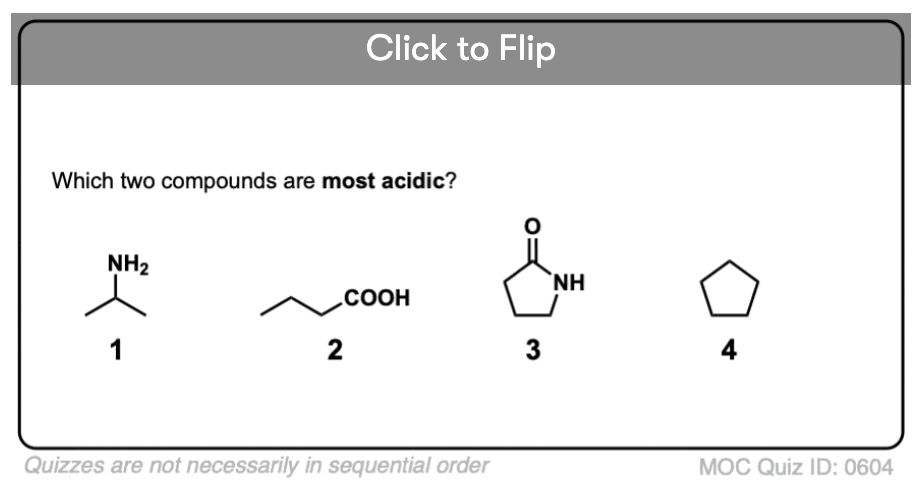
Become a MOC member to see the clickable quiz with answers on the back.
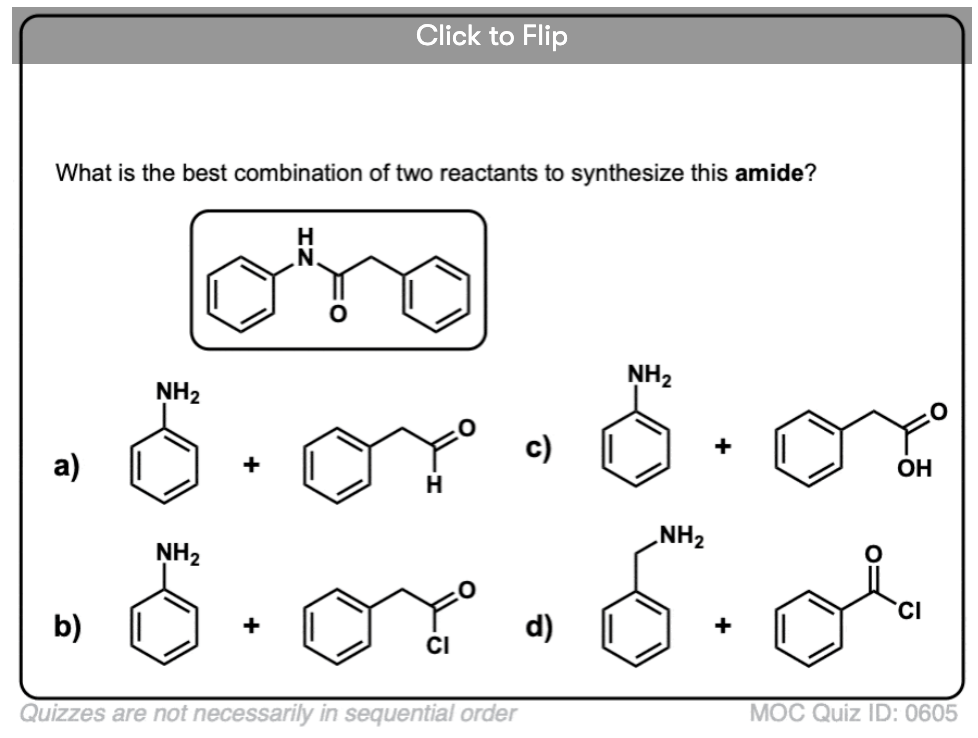
Become a MOC member to see the clickable quiz with answers on the back.
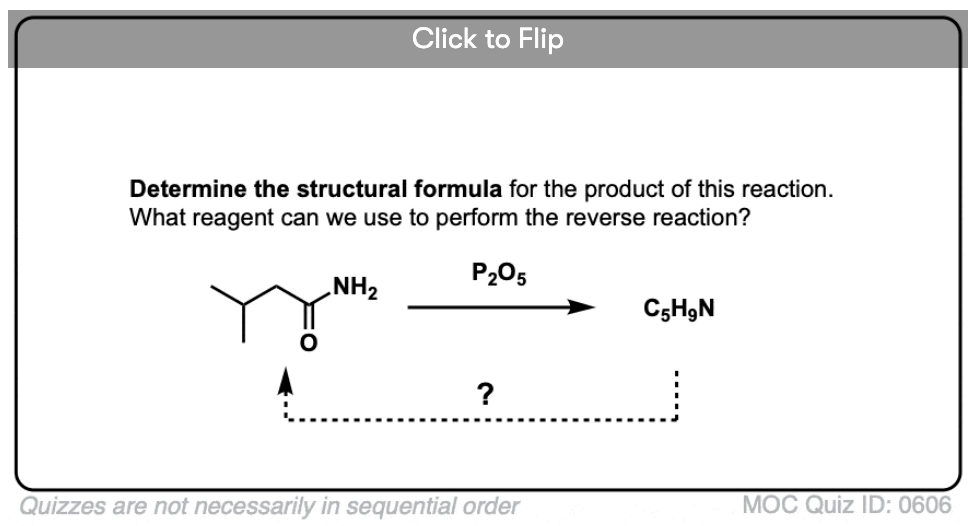
Become a MOC member to see the clickable quiz with answers on the back.
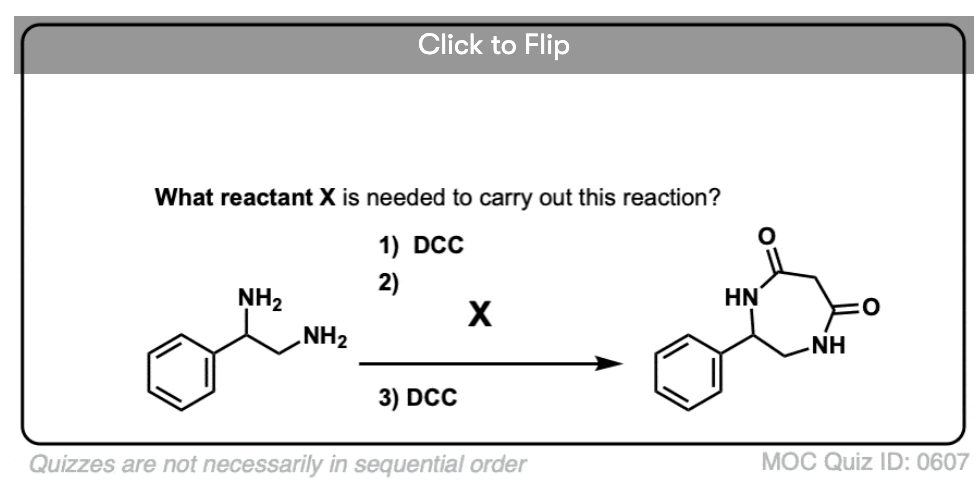
Become a MOC member to see the clickable quiz with answers on the back.
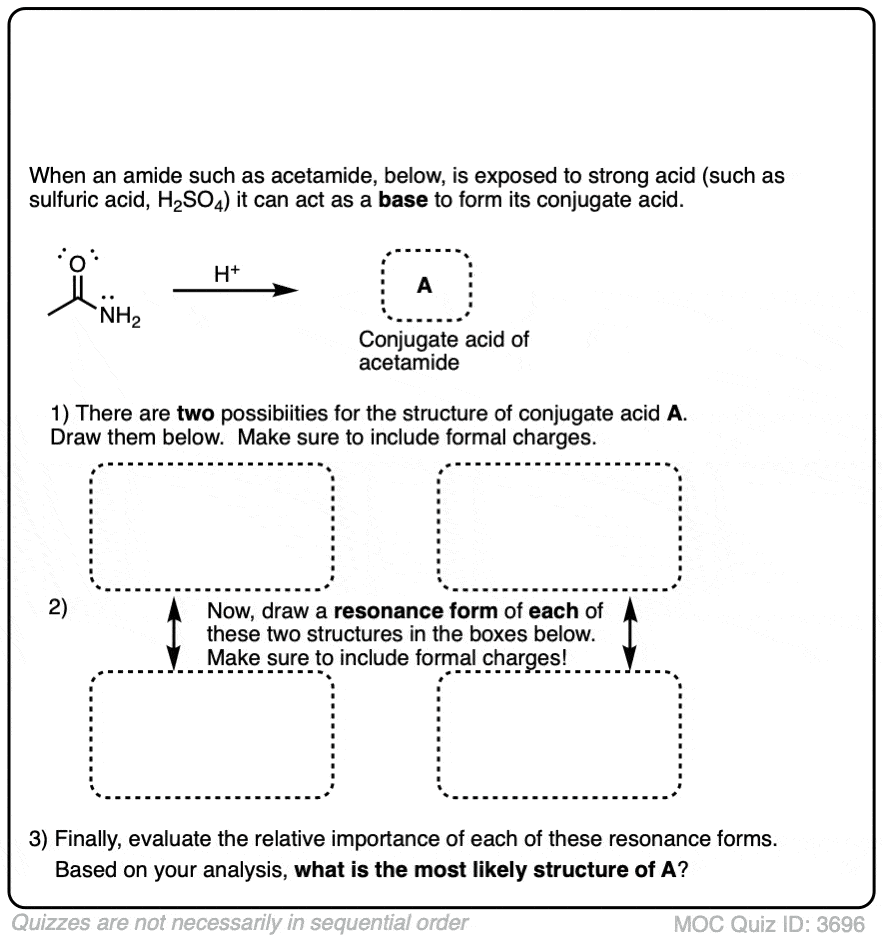 Click to Flip
Click to Flip
(Advanced) References and Further Reading
Nitrile hydrolysis:
- PHENYLACETAMIDE
Wilhelm Wenner
Org Synth. 1952, 32, 92
DOI: 10.15227/orgsyn.032.0092
The conditions used here for hydration of the nitrile to the amide are rather gentle – this uses a temperature of 40 °C for approximately 1 hr. - Halide-directed nitrile hydrolysis
James M. Photis
Tetrahedron Lett. 1980, 21 (37), 3539-3540
DOI: 10.1016/0040-4039(80)80228-0
This is a useful procedure for selective hydrolysis of nitriles to primary amides, especially in the case of aroyl cyanides (e.g. PhCOCN). - Facile and Highly Selective Conversion of Nitriles to Amides via Indirect Acid-Catalyzed Hydration Using TFA or AcOH−H2SO4
Jarugu Narasimha Moorthy and Nidhi Singhal
The Journal of Organic Chemistry 2005, 70 (5), 1926-1929
DOI: 10.1021/jo048240aSchotten-Bauman Reaction:
#4 and #5 are the original papers by Schotten and Baumann on a simple biphase amide synthesis. - Ueber die Oxydation des Piperidins
Schotten, C.
Ber. 1884, 17 (2), 2544-2547
DOI: 10.1002/cber.188401702178 - Ueber eine einfache Methode der Darstellung von Benzoësäureäthern
Baumann, E.
Ber. 1886, 19 (2), 3218-3222
DOI: 10.1002/cber.188601902348 - Enantioselective Total Synthesis of (−)-Kibdelone C
John R. Butler, Chao Wang, Jianwei Bian, and Joseph M. Ready
Journal of the American Chemical Society 2011, 133 (26), 9956-9959
DOI: 1021/ja204040k
The humble Schotten-Baumann reaction is even used in demanding total syntheses – in this case, it is used to make the lactam in 4 from 5 and 6! - BENZOYL PIPERIDINE
Marvel, C. S.; Lazier, W. A.
Org. Synth. 1929, 9, 16
DOI: 10.15227/orgsyn.009.0016
This procedure from Organic Syntheses, a source of independently tested and reproducible synthetic organic laboratory procedures, is a classic Schotten-Baumann amide synthesis. - A High-Throughput Process for Valsartan
Ulrich Beutler, Matthias Boehm, Peter C. Fuenfschilling, Thomas Heinz, Jean-Paul Mutz, Ulrich Onken, Martin Mueller, and Werner Zaugg
Organic Process Research & Development 2007, 11 (5), 892-898
DOI: 1021/op700120n
Organic Process & Research Development (“OPRD”) is a great journal for process or scale-up chemistry. This paper shows how the Schotten-Baumann reaction (4 to 3) is preferred for large-scale reactions as it is simple, robust, easily carried out, and does not have large exotherms (unlike the Grignard reaction, for instance). - The nylon rope trick: Demonstration of condensation polymerization
Paul W. Morgan and Stephanie L. Kwolek
Journal of Chemical Education 1959, 36 (4), 182
DOI: 1021/ed036p182
The classic ‘nylon rope trick’ in which one pulls a string of nylon from a biphasic mixture of hexamethylenediamine and sebacoyl chloride can be considered a type of Schotten-Baumann reaction, in that it forms a polyamide! This was first developed by Stephanie Kwolek, who was a distinguished chemist at DuPont for over 40 years and was responsible for discovering Kevlar and developing the chemistry of aramids and other high-tensile strength materials.DCC: - A New Method of Forming Peptide Bonds
John C. Sheehan and George P. Hess
Journal of the American Chemical Society 1955, 77 (4), 1067-1068
DOI: 1021/ja01609a099
Original paper on the synthesis of peptide bonds/amide bonds using DCC.DeTar has published a series of papers studying the mechanism of the bond-forming reactions mediated by DCC and other carbodiimides, and here are the first two: - Reactions of Carbodiimides. I. The Mechanisms of the Reactions of Acetic Acid with Dicyclohexylcarbodiimide
DeLos F. DeTar and Richard Silverstein
Journal of the American Chemical Society 1966, 88 (5), 1013-1019
DOI: 10.1021/ja00957a027 - Reactions of Carbodiimides. II. The Reactions of Dicyclohexylcarbodiimide with Carboxylic Acids in the Presence of Amines and Phenols
DeLos F. DeTar and Richard Silverstein
Journal of the American Chemical Society 1966, 88 (5), 1020-1023
DOI:1021/ja00957a028 - The Chemistry of Carbodiimides.
G. Khorana
Chemical Reviews 1953, 53 (2), 145-166
DOI: 10.1021/cr60165a001
An old review by Prof. Har Gobind Khorana, who later received the Nobel Prize in Medicine for his work demonstrating that the nucleotides in DNA and RNA code for protein synthesis. - ESTERIFICATION OF CARBOXYLIC ACIDS WITH DICYCLOHEXYLCARBODIIMIDE/4-DIMETHYLAMINOPYRIDINE: tert-BUTYL ETHYL FUMARATE
Neises and Wolfgang Steglich
Org. Synth. 1985, 63, 183
DOI: 10.15227/orgsyn.063.0183
This is a procedure for selective esterification using DCC – this avoids transesterification that would occur under usual Fischer esterification conditions. This procedure is from Organic Syntheses, a source of reliable, independently tested synthetic organic reactions. - Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide
B. Merrifield
Journal of the American Chemical Society 1963, 85 (14), 2149-2154
DOI: 10.1021/ja00897a025
This is one of the most highly cited papers in JACS, and for good reason – it basically lays the foundation of SPPS, and what is now a billion-dollar industry. This work led to a Nobel Prize in Chemistry for the author, Prof. R. Bruce Merrifield (Rockefeller U.). The peptide couplings are done using none other than DCC. - Notes- A Convenient Synthesis of Water-Soluble Carbodiimides.
John Sheehan, Philip Cruickshank, and Gregory Boshart
The Journal of Organic Chemistry 1961, 26 (7), 2525-2528
DOI:1021/jo01351a600
The major drawback with DCC is that separating the DCU (dicyclohexylurea) thus produced can be cumbersome. Thus other reagents, such as EDC (1‐Ethyl‐3‐(3′‐dimethylaminopropyl)carbodiimide) have been developed, for which the resulting urea is water-soluble and easily removed by extraction. - Total synthesis of a monocyclic peptide lactone antibiotic, etamycin
John C. Sheehan and Stephen L. Ledis
Journal of the American Chemical Society 1973, 95 (3), 875-879
DOI:1021/ja00784a041
EDC was used for most of the peptide couplings in the synthesis of this peptide, which is one of the first cyclic peptides to be synthetically produced.
I think it meant phenylacetonitrile (PhCH₂CN) here:
One set of conditions for the hydrolysis of phenylacetamide (PhCH2CN) to PhCH2CONH2 gives the reaction conditions as “HCl, H2O, 40-50°C, 1h
yes, that was a typo – thank you very much for the spot, I renamed it to phenylacetonitrile.
Does this paragraph imply that to make an amide directly from a carboxylic acid, we would just need harsher conditions:
Halides (e.g. Cl– ) and carboxylates (RCO2– ) are much weaker bases, and thus much better leaving groups than HO–. So by adding an amine to an acyl halide or acid anhydride, nucleophilic acyl substitution can occur under much milder conditions, resulting in our desired amide.
Sir how can I download the pdf files?
What pdf files?
Hi James!
I’m an Italian young chemistry student.
I’m studying for the International Chemistry Olympiad, and your site is very very useful!!
In note 2, you said that amines are generally more nucleophilic than OH-.
Can you explain me why?
Thank you very much.