Amines
Protecting Groups for Amines: Carbamates
Last updated: May 29th, 2026 |
Protecting Groups For Amines
- Amine protecting groups are essential for the synthesis of peptides.
- Carbamates are useful protecting groups for amines. They can be installed and removed under relatively mild conditions.
- One of the most common carbamate protecting groups is the t-butyloxycarbonyl (Boc) protecting group. It can be removed with strong acid (trifluoroacetic acid) or heat.
- The carboxybenzyl (CBz) group has a benzyl group and can be removed using catalytic hydrogenation (Pd-C, H2)
- The fluorenylmethoxy (FMoc) group can be removed with an amine base (e.g. R2NH)
- If multiple protecting groups are present on the same molecule, it’s useful to choose them such that they can each be removed under different sets of conditions (e.g. acidic, basic, hydrogenation). (We sometimes use the word “orthogonal” to describe this property).
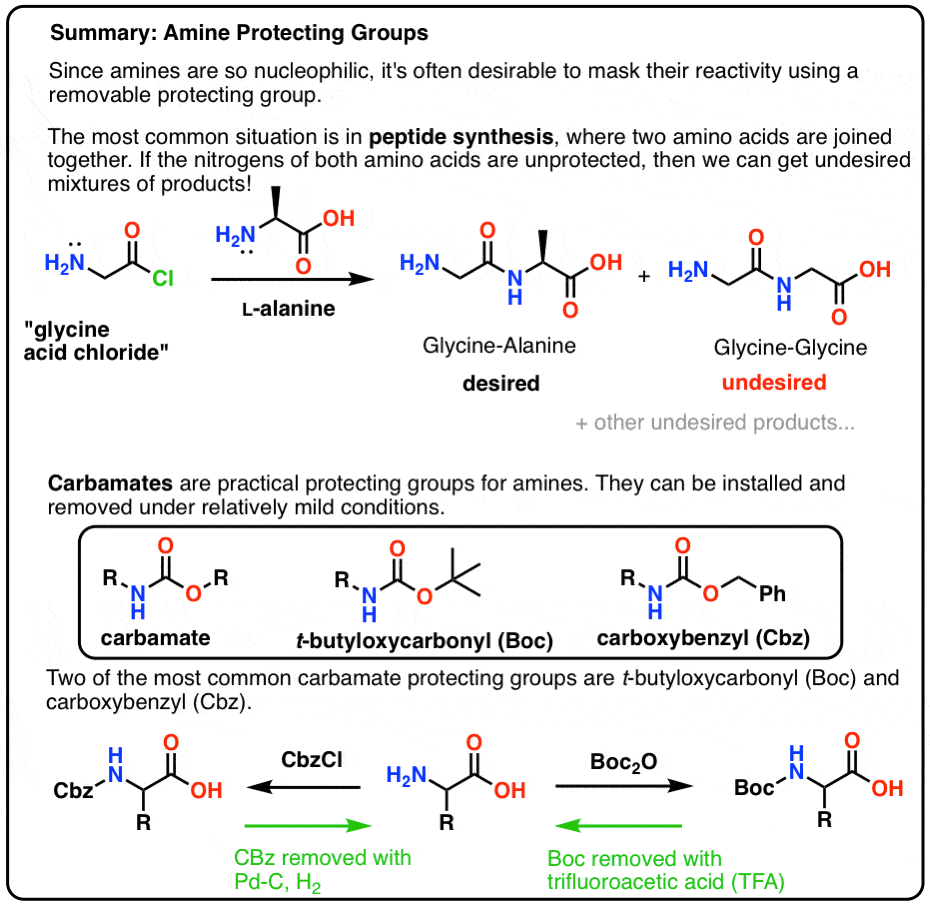
Table of Contents
- An Introduction To Simple Peptide Synthesis
- Let’s Mix Up A Batch of Gly-Ala
- A Very Bad Initial Plan
- “Hark! What Crap Is This?”
- A Protective Hat For NH2
- Mate! Use A Carbamate, Mate!
- Boc and CBz Are The Bees Knees
- Installation and Removal of the “Boc” Protecting Group
- Installation and Removal of The CBz (or “Z”) Carbamate Protecting Group
- A Simple Peptide Synthesis Using Carbamate Protecting Groups
- It Keeps Going…
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. An Introduction To Simple Peptide Synthesis
In a recent post [Amides: Synthesis, Properties, and Nomenclature] we went through 3 common ways of making amides:
- Adding amines to acyl halides / anhydrides
- Partial hydrolysis of nitriles
- Coupling of carboxylic acids with amines using a dehydrating agent like DCC (N,N’-dicyclohexylcarbodiimide).
What was missing from that post was any mention of synthesizing the great-granddaddy of most useful amide linkages known to mankind, and by that I mean peptides.
A peptide bond is the name we give the amide bond that joins together two amino acids.
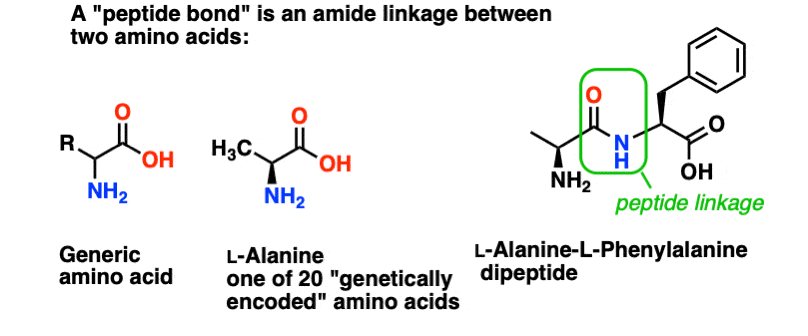
And amino acids are important because… wait. You already know why amino acids are important, right? If you don’t know this right now, come back after reading this.
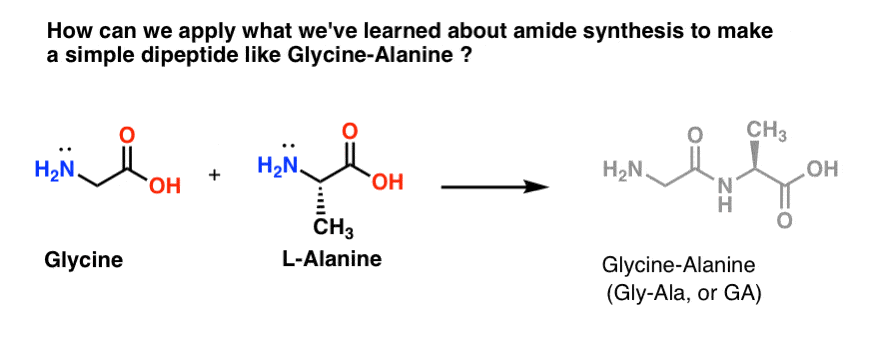
2. Let’s Mix Up A Batch of Gly-Ala
Let’s try to apply some of our newfound amide synthesis skillz to try to build a really simple dipeptide, glycine-alanine. If we can build a simple dipeptide now, we can use learn what we’ve learned to show how even more complex peptides are made in a later post. [see post: Introduction to peptide synthesis] Did you know that Nobel Prize winner Bruce Merrifield synthesized insulin by joining one amino acid together at a time? Yes, really.
Of the three methods we listed for making amides, only two are potentially useful for forming peptide bonds: 1) the acid chloride method, or 2) synthesis via a coupling agent like DCC. [Hydrolysis of nitriles only provides primary amides, R-CONH2 , so it is not an option for peptide synthesis].
3. A Very Bad Initial Plan
Our first stab at peptide synthesis will involve barrelling forward with a very naïve plan of attack and hope it all works out in the end. Hang on folks, as it’s going to get messy.
We’ll start by proposing the acid chloride method [or the Schotten-Baumann reaction , if you like] for the synthesis of Gly-Ala.
Looking backwards from the Gly-Ala dipeptide, our plan would have us synthesize the peptide bond through the reaction of alanine with the acid chloride derived from glycine:
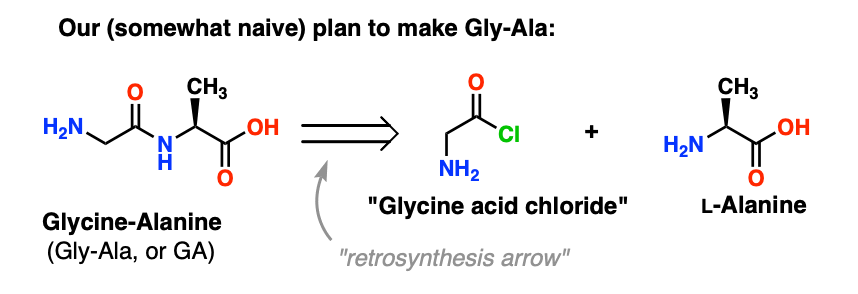
There’s a slight problem with this plan. It may be a bit hard to spot at first.
Let’s assume we’ve made the acid chloride of glycine* [Note 1] , and we have “glycine acid chloride” and alanine together in the same flask, along with some excess base to speed things along. [Note 2 – omitted a minor detail here re: the zwitterionic nature of amino acids]
We’ll draw the reaction in the forward direction:
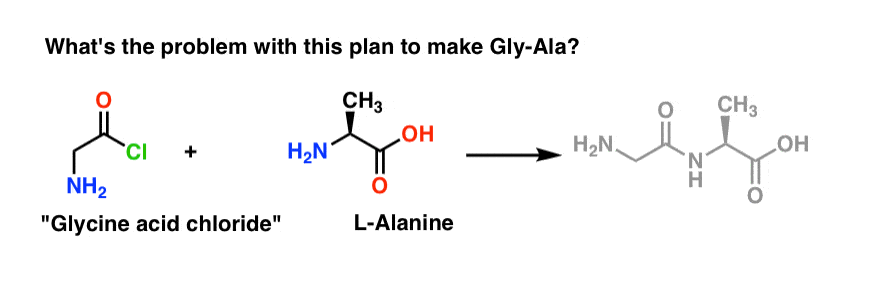
What could possibly go wrong?
4. “Hark! What Crap Is This? “
Our plan is to make a solution of the acid chloride of glycine (1 molar equivalent), and then have it patiently wait around in the flask until we added a molar equivalent of alanine, whereupon it would react with the nucleophilic NH2 group of alanine.
The problem with our plan is that we aren’t dealing with a single molecule of “glycine acid chloride” – we’re dealing with something around a mole (6.02 x 1023 molecules) of it. And the acid chloride of glycine already has a nucleophilic NH2 group!
“Glycine acid chloride” as drawn, isn’t a stable molecule, because it can react with itself.
This means that a solution of glycine acid chloride left to its own devices would form a polymer of glycine, with the structure Gly-Gly-Gly-Gly…
Even a solution of glycine acid chloride in the presence of alanine would not only form the desired Gly-Ala, but also Gly-Gly (with an attached acyl halide) which can go on to perform more mischief with another nucleophile, whether it be Gly or Ala:
(and no, there isn’t much to distinguish the NH2 of “glycine acid chloride” from the NH2 of alanine. They’re about equally reactive.)
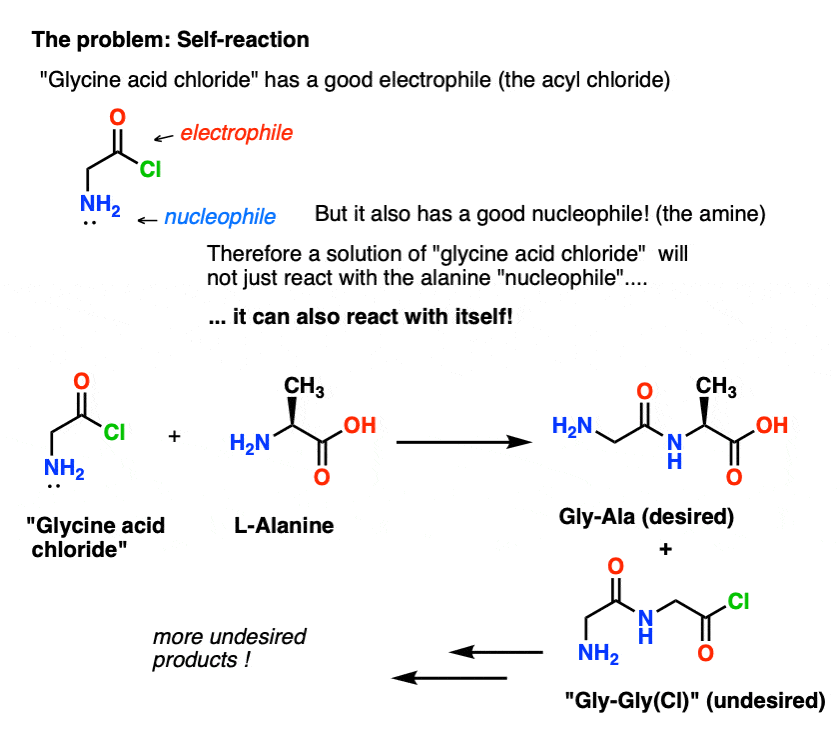
The lesson here is that when you have a solution of a molecule containing both a nucleophile and an electrophile, it can self-react. There’s a name for this process that might sound familiar: polymerization.
So how do we stop this from happening?
5. A Protective Hat For NH2
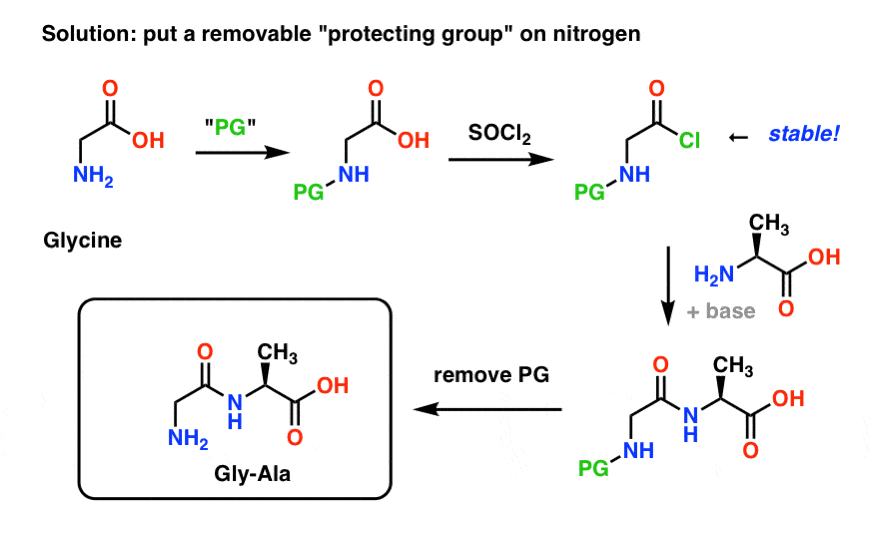
The best way is to “cap” the nitrogen somehow with a protecting group (PG) that makes the NH2 group non-nucleophilic. It should also have the following properties:
- easily and selectively installed
- inert to the desired reaction conditions (e.g. SOCl2 to make the acid chloride from the carboxylic acid)
- easily and selectively removed without affecting the final product
We’ve seen protecting group strategies before, particularly with alcohols and the use of Grignard reagents.
Here’s what a protecting group strategy for our synthesis of “Gly-Gly” might look like. We install a protecting group ( “PG”) on glycine, then make the acid chloride. The PG should be chosen such as to render the nitrogen non-nucleophilic (i.e. it won’t react with the acid chloride).
We can then form our peptide bond with unprotected alanine and then remove the PG under mild conditions.
One protecting group strategy for nitrogen we’ve explored already is the Gabriel synthesis, which uses phthalimide (you can think of a phthalimide as a protected nitrogen). This has actually been used to synthesize Gly-Gly (“glycylglycine”) [Note 2] ! One of the problems, however, is that relatively harsh conditions (ample heat) are required to both install and remove the phthalimide group, and this is not a very healthy environment for the survival of sensitive, chiral amino acids, which can easily racemize.
Another potential choice is to protect the nitrogen as an amide, but cleavage amides can require harsh conditions too. Furthermore, since we are trying to forge an amide bond (peptide) here anyway, we might have selectivity problems with its removal – destroying the village in order to save it. [Note 3]
6. Mate! Use A Carbamate, Mate!
The most popular choice of protecting group for amine nitrogen is the carbamate functional group. A carbamate looks like the bastard child of an ester and an amide, with N and O flanking a carbonyl.
The nitrogen of a carbamate is relatively non-nucleophilic, and furthermore, carbamates are:
- easily installed on nitrogen
- inert to a wide variety of reaction conditions
- easily removed without affecting existing amide groups
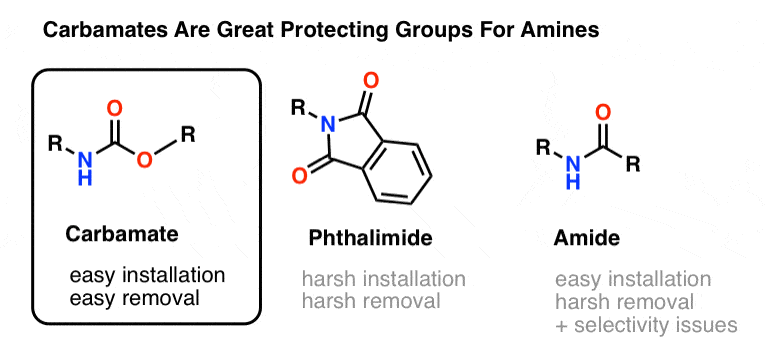
This makes them perfect for our purposes.
7. Boc and CBz Are The Bees Knees
Two popular carbamate protecting groups are Boc (t-Butyloxycarbonyl) and CBz (carboxybenzyl).
For our purposes, these two protecting groups can be thought of as more or less equivalent, as either can be used effectively for peptide synthesis.
The key difference is really in how they are removed (i.e. the “deprotection” step). Choosing between one or the other becomes crucial once you have a complex molecule with multiple protecting groups; that falls under the category of “advanced synthetic strategy”, which is more a subject for Org 3. [Note 4]
8. Installation and Removal of the “Boc” Protecting Group
The Boc group is usually installed with “Boc2O” (sometimes referred to as “Boc anhydride”), and is removed with acid. The usual choice is “neat” (i.e. undiluted) trifluoroacetic acid (TFA), which pops the Boc groups off very cleanly, liberating CO2 and t-butyl alcohol.
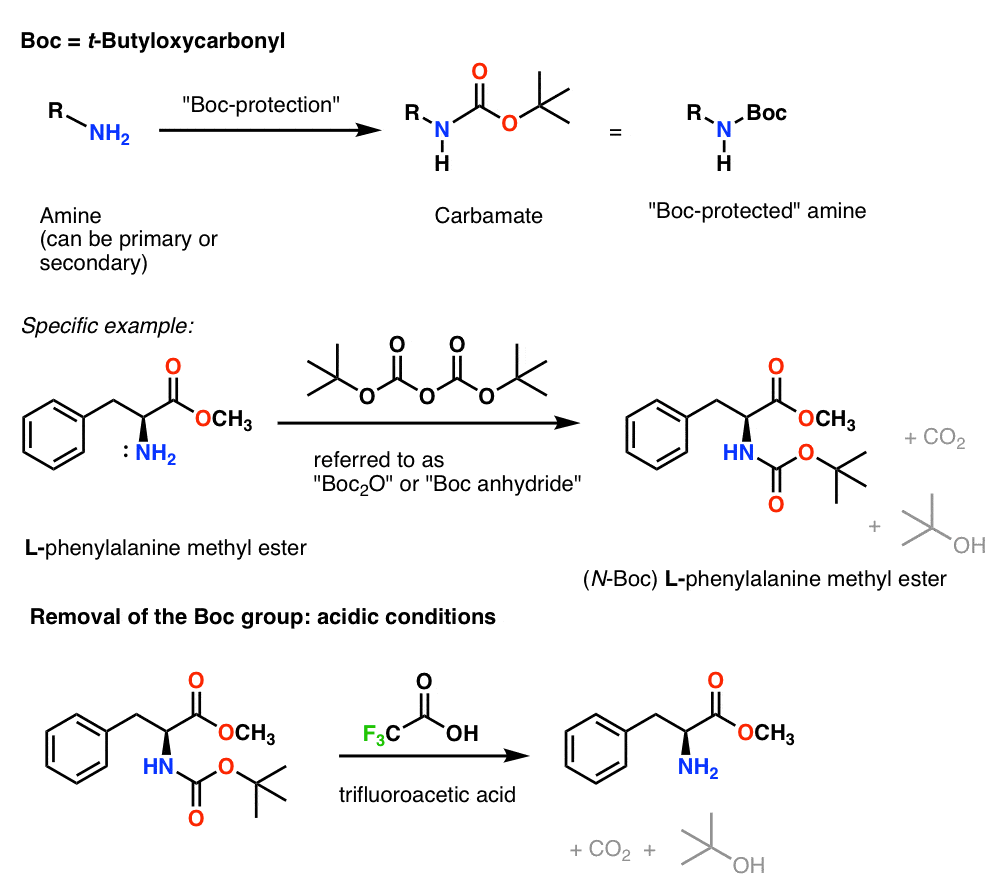
9. Installation and Removal of The CBz (or “Z”) Carbamate Protecting Group
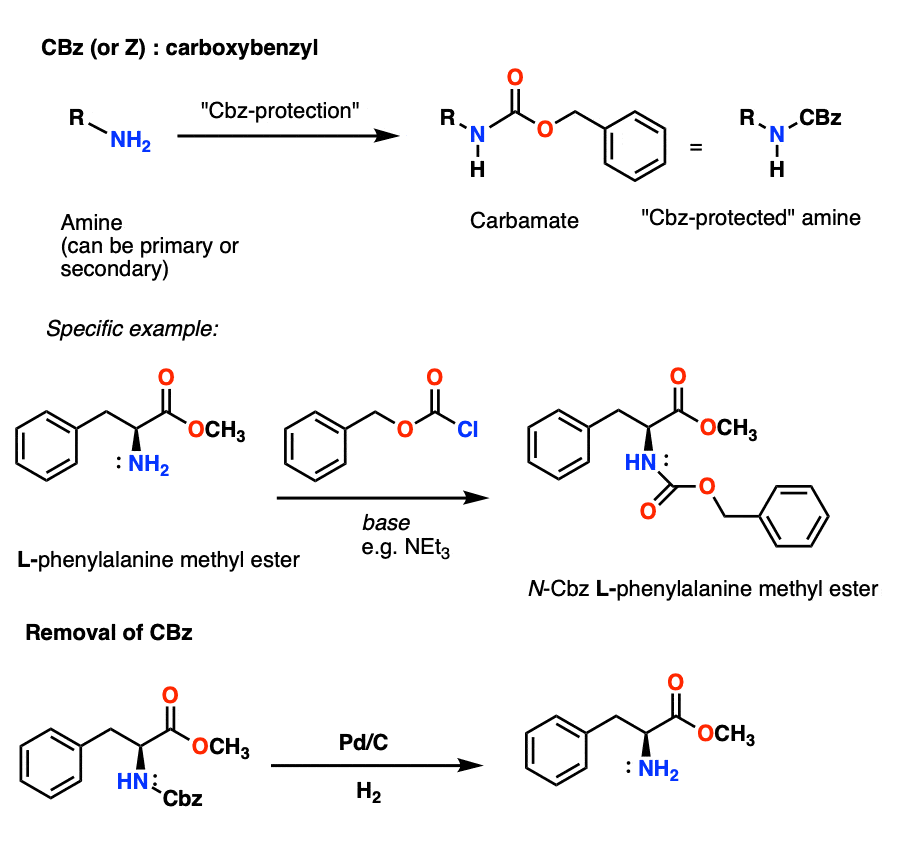
The Cbz group (sometimes further abbreviated as “Z”) can be installed with CbzCl and mild base, and is usually removed via catalytic hydrogenation (Pd-C/H2). This is extremely mild and has the advantage of occurring at neutral pH, leaving acid- or base-sensitive functional groups alone.
10. A Simple Peptide Synthesis Using Carbamate Protecting Groups
Let’s go back to peptide synthesis and apply this protecting group strategy to make Gly-Ala.
We start with an amino acid like L-alanine. Treating alanine with Boc2O, we obtain N-Boc protected L-alanine. The next step is to form an acid chloride by using SOCl2. Once formed, we then add our amine (e.g. L-valine) in the presence of excess base, forming our key amide bond. The final step to give the dipeptide is to deprotect the Boc-protected amine with trifluoroacetic acid (TFA), and voila! we have our dipeptide.
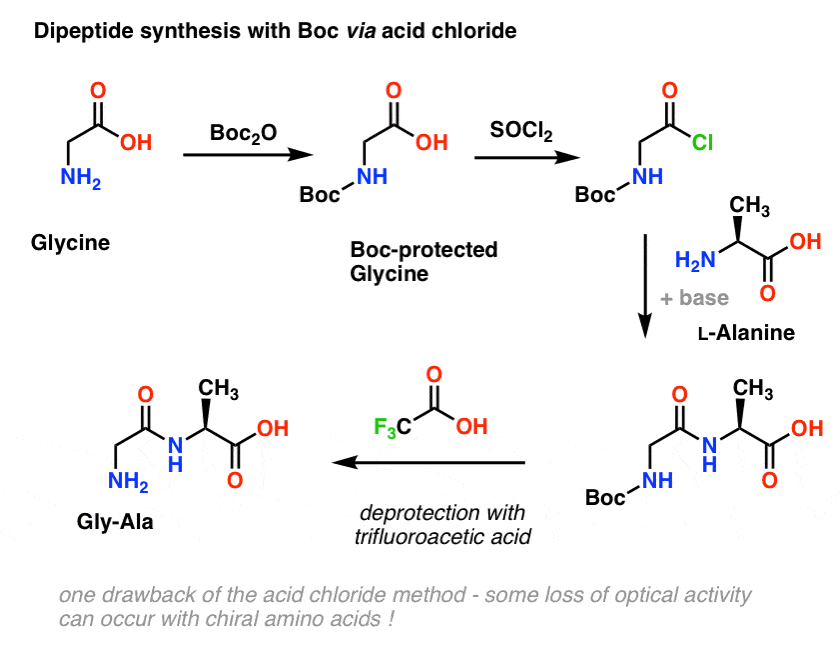
Although this method might good on paper, one problem of using acid chlorides in practice is that chiral amino acids often lose their optical purity through this method, a process sometimes referred to as “racemization”, but more correctly called”epimerization” (technically more correct, because a hydrogen on a chiral center is inverted)
Since the chirality of amino acids is essential for their biological function, a slightly milder protocol is generally used that employs DCC or a similar coupling reagent.
Here, we treat Boc-protected glycine with DCC to activate the carboxylic acid. Then we add our amino acid nucleophile (L-alanine) which forms the dipeptide. If we want to isolate the Gly-Ala dipeptide at this point, we can then remove the Boc group with TFA.
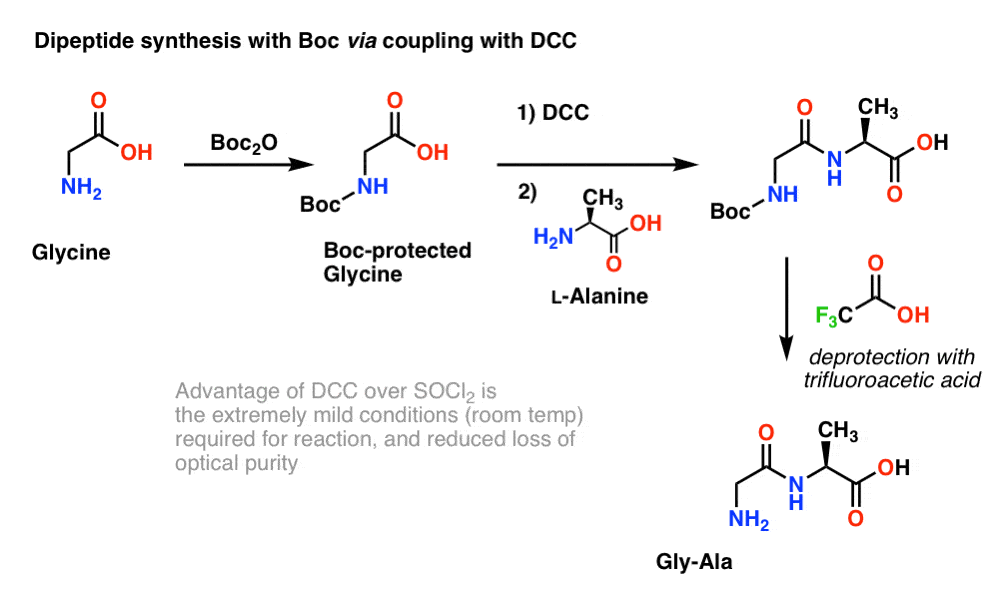
(Note 5 about this scheme)
11. It Keeps Going…
Note that if we wanted to make a tri-peptide, we can just keep performing cycles of adding DCC (to activate the carboxylic acid) followed by addition of new amino acids, building up the peptide one unit at a time!
There’s a particularly effective method for building longer peptides pioneered by Bruce Merrifield (and applied in the synthesis of insulin, among others) called solid-phase peptide synthesis, which we’ll cover the next time we’re on this topic.
Notes
Note 1. Similar to the old joke about the economist who proposes a plan for getting off a desert island:
“Assume we have a boat”.
Glycine (like all amino acids) itself is a zwitterion. Treatment of glycine with SOCl2 should yield the acid chloride with a protonated amine. This should be relatively stable in solution so long as no base is added.
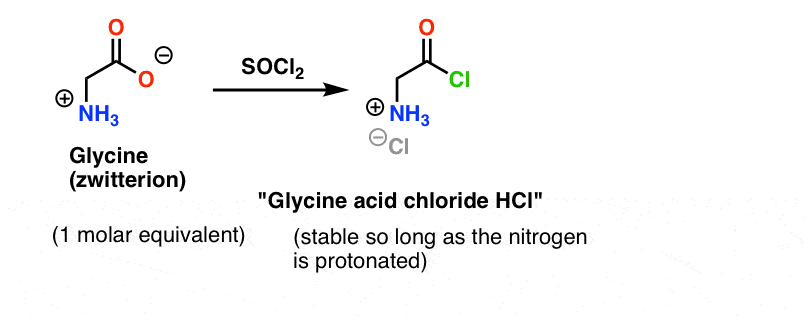
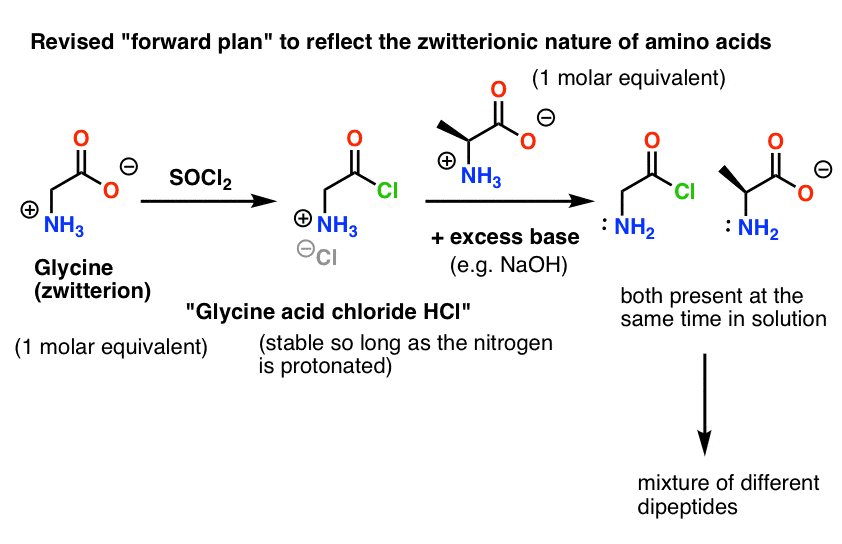
Here’s the problem. Since our amino acid nucleophile (alanine) is also zwitterionic, no reaction can occur until excess base is added to liberate a lone pair on the alanine nitrogen. After addition of base, we have “glycine acid chloride” and alanine together in solution. There is no appreciable difference in nucleophilicity between the nitrogens of these two species, and each of them will compete to react with the acid chloride nucleophile, leading to a mixture of Gly-Ala and Gly-GlyCl, and the Gly-GlyCl can then react further with the various nucleophiles preset in solution to give tri-, tetra- and higher peptides.
Note 2. John Sheehan, whom we met earlier as the inventor of DCC en route to the first to synthesize penicillin, also made phthalyl-protected Gly-Gly through a Gabriel synthesis:
Reference here (JACS, 1949, 71, 1856)
Note 3. Another problem with using amide protecting groups is azlactone formation, which can lead to epimerization of chiral amino acids. Also see this problem set.
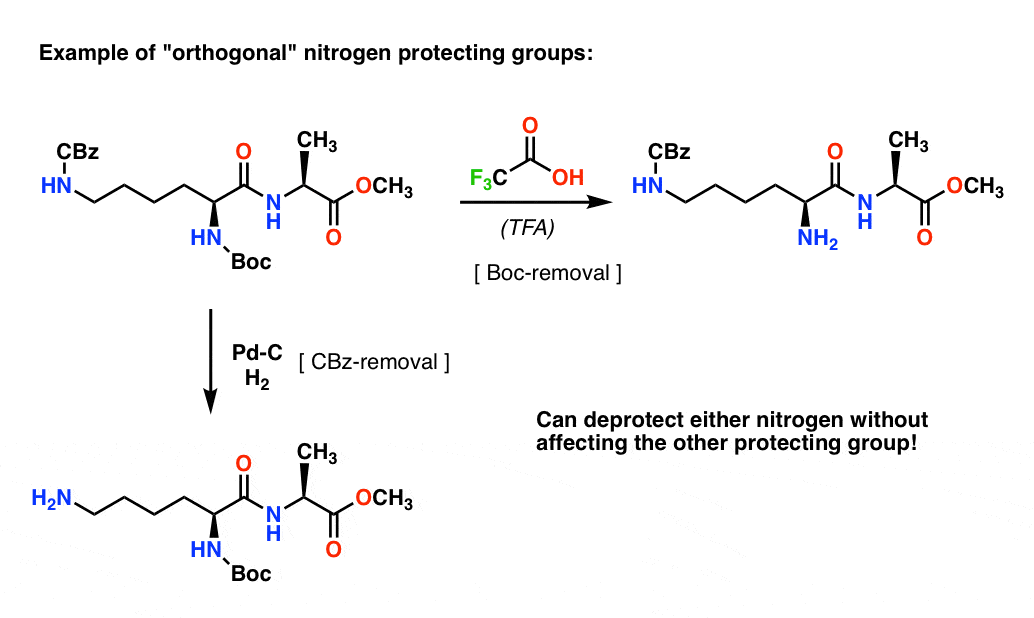
Note 4. “Orthogonal Protecting Groups”. In synthetic planning it’s often crucially important to have protecting groups that are removable under distinctly different conditions. This property is often referred to as “orthogonality”
For example in the following dipeptide we have two different protecting groups on nitrogen – one Boc and one CBz. By selecting “orthogonal” protecting groups, each nitrogen is addressable – we can choose which protecting group to remove, and our synthesis can proceed from there. This avoids getting into a situation where we have two unprotected amines and have to rely on one being more reactive than the other. These approaches very rarely work!
Note 5. One note – for simplicity here alanine is depicted with a free carboxylic acid, but a slightly better approach would be to use the methyl ester of alanine to avoid any self-coupling between the free amine of alanine and the free carboxylic acid.
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
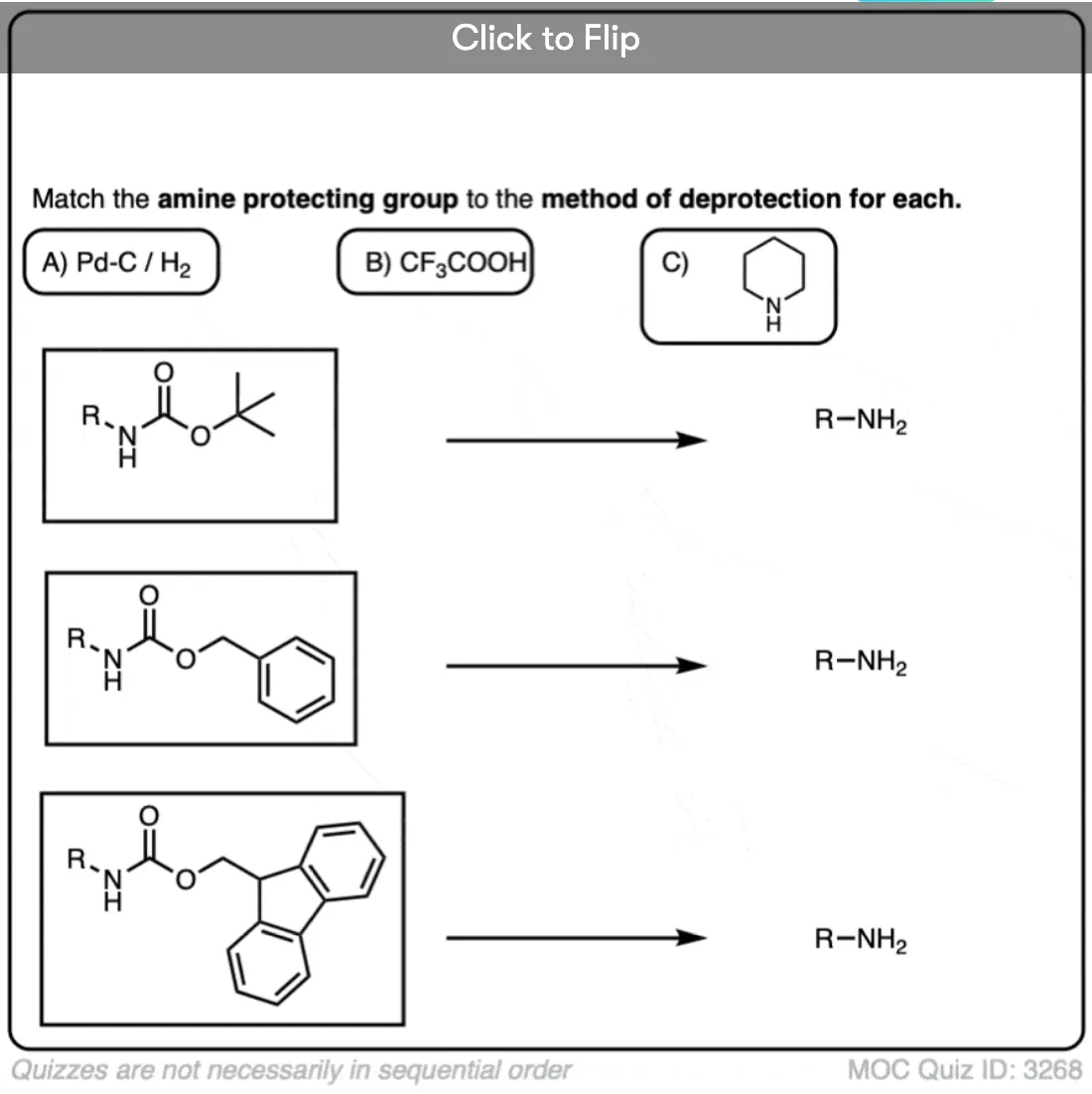
Become a MOC member to see the clickable quiz with answers on the back.
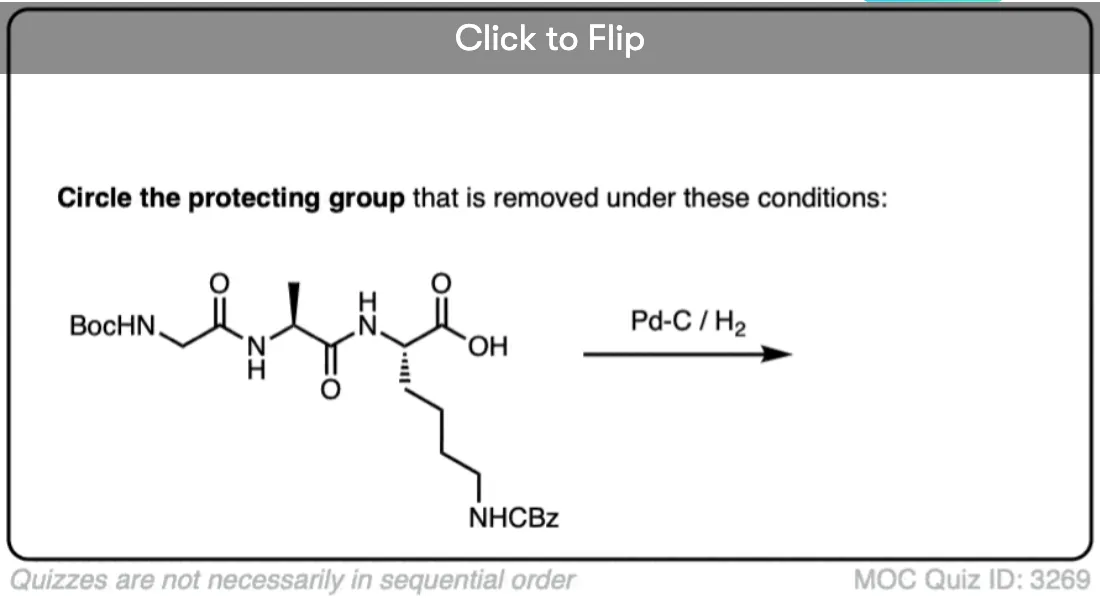
Become a MOC member to see the clickable quiz with answers on the back.
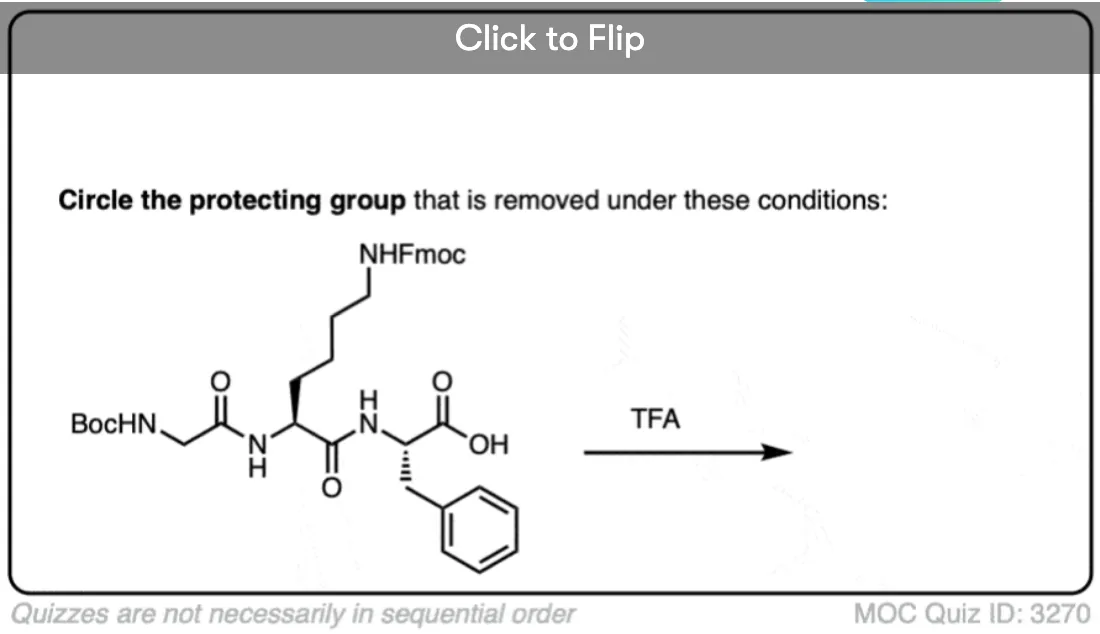
Become a MOC member to see the clickable quiz with answers on the back.
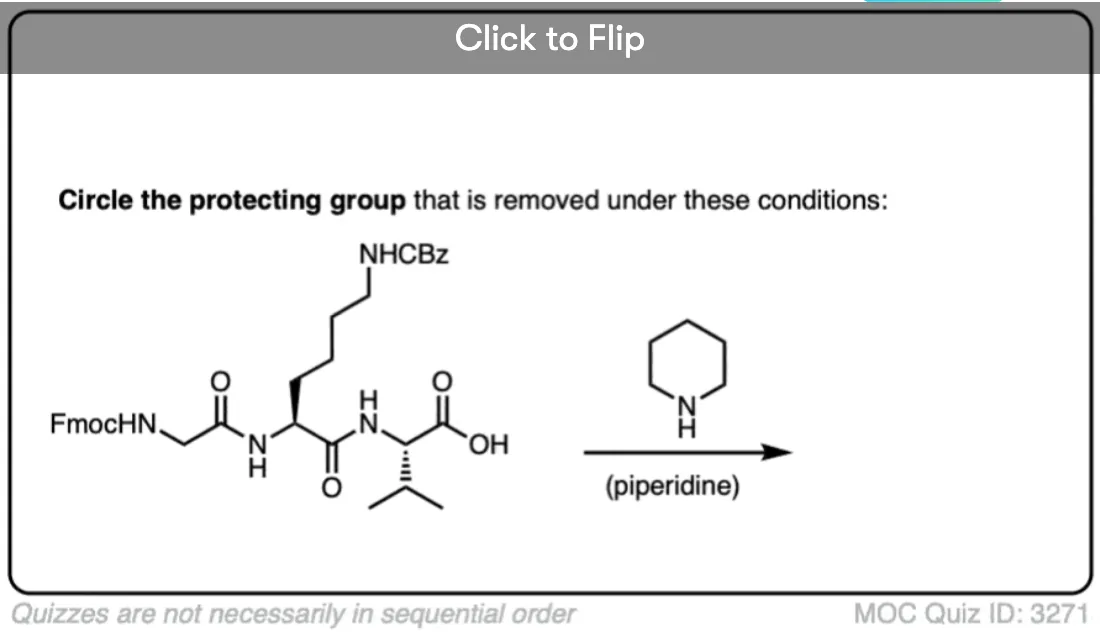
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
Carbamates are useful as protecting groups for amines, and the most commonly employed are -Boc, -Cbz, and -Fmoc.
- Über ein allgemeines Verfahren der Peptid‐Synthese
Max Bergmann and Leonidas Zervas
Ber. 1932, 65 (7), 1192-1201
DOI: 10.1002/cber.19320650722
The -Cbz (carboxybenzyl) protecting group was first used by Max Bergmann and Leonidas Zervas in 1932 for the synthesis of peptides, and is sometimes abbreviated “-Z” in honor of Zervas. - REMOVAL OF t‐BUTYL AND t‐BUTOXYCARBONYL PROTECTING GROUPS WITH TRIFLUOROACETIC ACID
Mechanisms, Biproduct Formation and Evaluation of Scavengers
Behrend F. Lundt, Nils L. Johansen, Aage Vølund, and Jan Markussen
J. Pept. Prot. Res. 1978, 12 (5), 258-268
DOI: 10.1111/j.1399-3011.1978.tb02896.x
In practice, nucleophilic scavengers (e.g. thiols) are usually added to the acid cocktail (TFA) when deprotecting, since deprotection of Boc will give electrophilic t-butyl species (e.g. t-butyl trifluoroacetate) that can react with sensitive residues (e.g. Trp or Cys). - A Method of Synthesis of Long Peptide Chains Using a Synthesis of Oxytocin as an Example
Miklos Bodanszky and Vincent du Vigneaud
Journal of the American Chemical Society 1959, 81 (21), 5688-5691
DOI: 1021/ja01530a040
In the first half of the 20th century, peptide synthesis was done using standard organic chemistry solution phase techniques. This is now known as LPPS (liquid-phase peptide synthesis). du Vigneaud received the Nobel Prize in chemistry in 1955 for his work in showing that peptide synthesis could be achieved, using the correct choice of protecting groups and synthetic strategies. - THE TEMPORARY FORMATION OF THE AZLACTONE RING IN THE RACEMIZATION OF ACYL DERIVATIVES OF AMINO ACIDS WITH ACETIC ANHYDRIDE
Vincent du Vigneaud and Curtis E. Meyer
J. Biol. Chem. 1932, 99:143-151
DOI: 10.1016/S0021-9258(18)76075-4
Another problem with using amide protecting groups is azlactone formation, which can lead to epimerization of chiral amino acids. - A New Synthetic Route to Peptides
John C. Sheehan and Victor S. Frank
Journal of the American Chemical Society 1949, 71 (5), 1856-1861
DOI: 10.1021/ja01173a095
John Sheehan, the inventor of DCC en route to the first synthesis of penicillin, also made phthalyl-protected Gly-Gly through a Gabriel synthesis. - A New Method of Forming Peptide Bonds
John C. Sheehan and George P. Hess
Journal of the American Chemical Society 1955, 77 (4), 1067-1068
DOI: 1021/ja01609a099
Original paper on the synthesis of peptide bonds/amide bonds using DCC. - 9-Fluorenylmethoxycarbonyl function, a new base-sensitive amino-protecting group
Louis A. Carpino and Grace Y. Han
Journal of the American Chemical Society 1970, 92 (19), 5748-5749
DOI: 10.1021/ja00722a043 - 9-Fluorenylmethoxycarbonyl amino-protecting group
Louis A. Carpino and Grace Y. Han
The Journal of Organic Chemistry 1972 37 (22), 3404-3409
DOI: 10.1021/jo00795a005
The discovery and development of the -Fmoc protecting group for amines adds another layer orthogonality to amine protection/deprotection strategies. The -Fmoc group is base labile, and in peptide synthesis is typically removed with 20% piperidine in DMF. Cbz is removed by hydrogenation, -Boc removed with acid, and -Fmoc with base.
Hi,
I noticed that the stereochemistry of phenylalanine is wrong in the first dipeptide.
Thanks for the web.
Rubén
Thanks so much for this module, it was super helpful! I’m wondering how these procedures would need to be modified when dealing with the synthesis of a peptide bond involving an amino acid like lysine, which has an NH2 group that could act as a nucleophile. Is there a second protecting group that would need to be added to prevent side reactions between the nucleophilic side group at the carbonyls of the incoming amino acids? Thanks!
Yes Zoe, absolutely there is. One would need to protect the lysine nitrogen separately, and ideally be able to deprotect the lysine nitrogen under conditions that leave other nitrogens untouched. Functional groups that can be removed without affecting other existing protecting groups are said to be “orthogonal”, using an analogy from geometry. This is why such a large number of protecting groups have been developed.
For example one could use a benzyl protecting group (PhCH2) on the lysine nitrogen and remove it with Pd-C / H2 without affecting any Boc or FMOC protecting groups. One could deprotect FMOC with base (piperidine) without affecting the other two, or remove BOC with strong acid (TFA).
Does that answer your question? James
In the orthogonal protecting groups, how is it possible to protect one nitrogen leaving the other unprotected which is then protected with a different group. And how do you choose whether to use first Boc or Cbz?
Thank you!
The usual approach is to protect the nitrogen(s) before you bring them together in the same molecule. If you’re in a situation where you have two chemically similar amines and have to selectively protect one, that’s a planning problem. There are a lot of variables so it depends on the particulars of your molecule, but there’s almost always a way to do it!
Whether you use Boc or CBz depends on the other functional groups in your molecule. The acid required for Boc deprotection can sometimes affect sensitive functional groups in your molecule; CBz deprotection is much milder. It would be easier to give you a more specific answer if you showed me a specific problem.
How to protect Glutamine’s side chain for easy and mild removal of the protection group under the physilogical condition without denaturing proteins?
I don’t know. I’m unaware of any schemes for deprotecting the glutamine sidechain in vivo.
Why TFA is the best for the deprotection step? What about other acids? It does require very low pH to work? Thanks
Other acids work too, such as bubbling HCl gas through the solution. However using neat (i.e. undiluted) TFA is just such a clean reaction that it’s typically the first choice. You can literally just dissolve your reactant in dicholoromethane in a stirred, open flask, get out a pipet, drop neat TFA into solution and wait for the bubbling to stop (the byproduct is isobutylene, aka 2-methyl-propene) and the reaction is done. It’s extraordinarily easy to do.