Amines
Introduction to Peptide Synthesis
Last updated: May 28th, 2026 |
Peptide bonds: Forming peptides from amino acids with the use of protecting groups
Today we’ll go deeper on how to synthesize the most important amides of all – peptides – with an important contribution from protecting group chemistry.
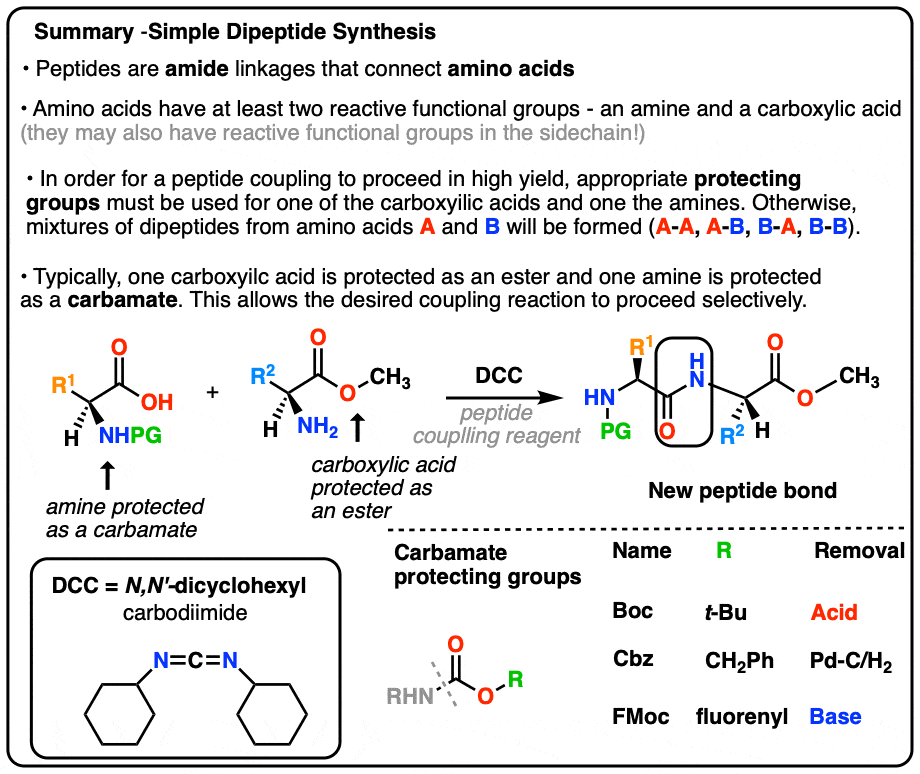
Table of Contents
- What Are Peptide Bonds?
- The “Proteinogenic” Amino Acids
- Synthesis of a Dipeptide Without Protecting Groups
- Synthesis of a Dipeptide Using A Protecting Group Strategy
- Synthesis of Longer Peptides – Tripeptides and Tetrapeptides
- Bonus Topic: Solid-Phase Peptide Synthesis
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
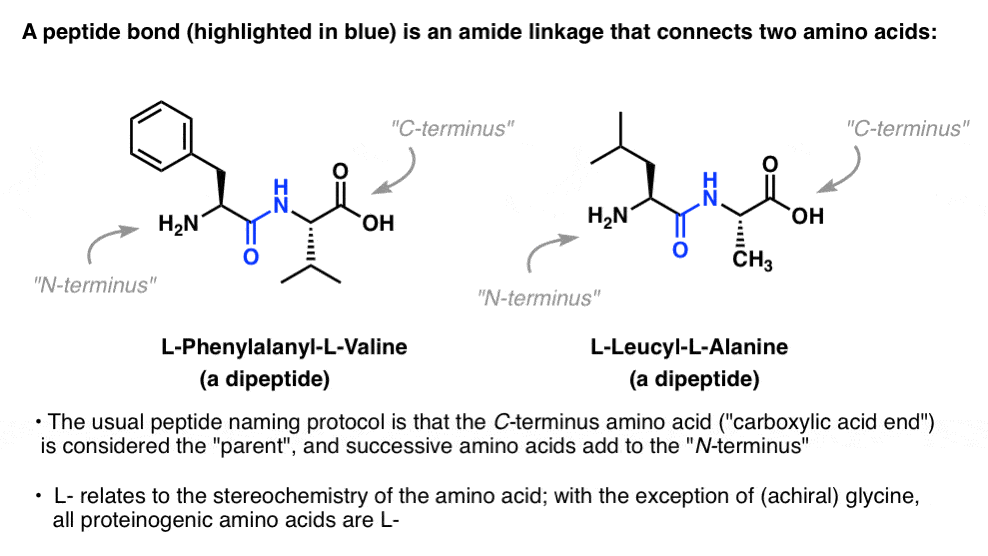
1. What Are Peptide Bonds?
A “peptide bond” is an amide linkage (see Amides: Properties. Synthesis, and Nomenclature) that connects two amino acids, as in the “dipeptides” L-phenylalanyl-L-valine (below left) and L-leucyl-L-alanine (below right):
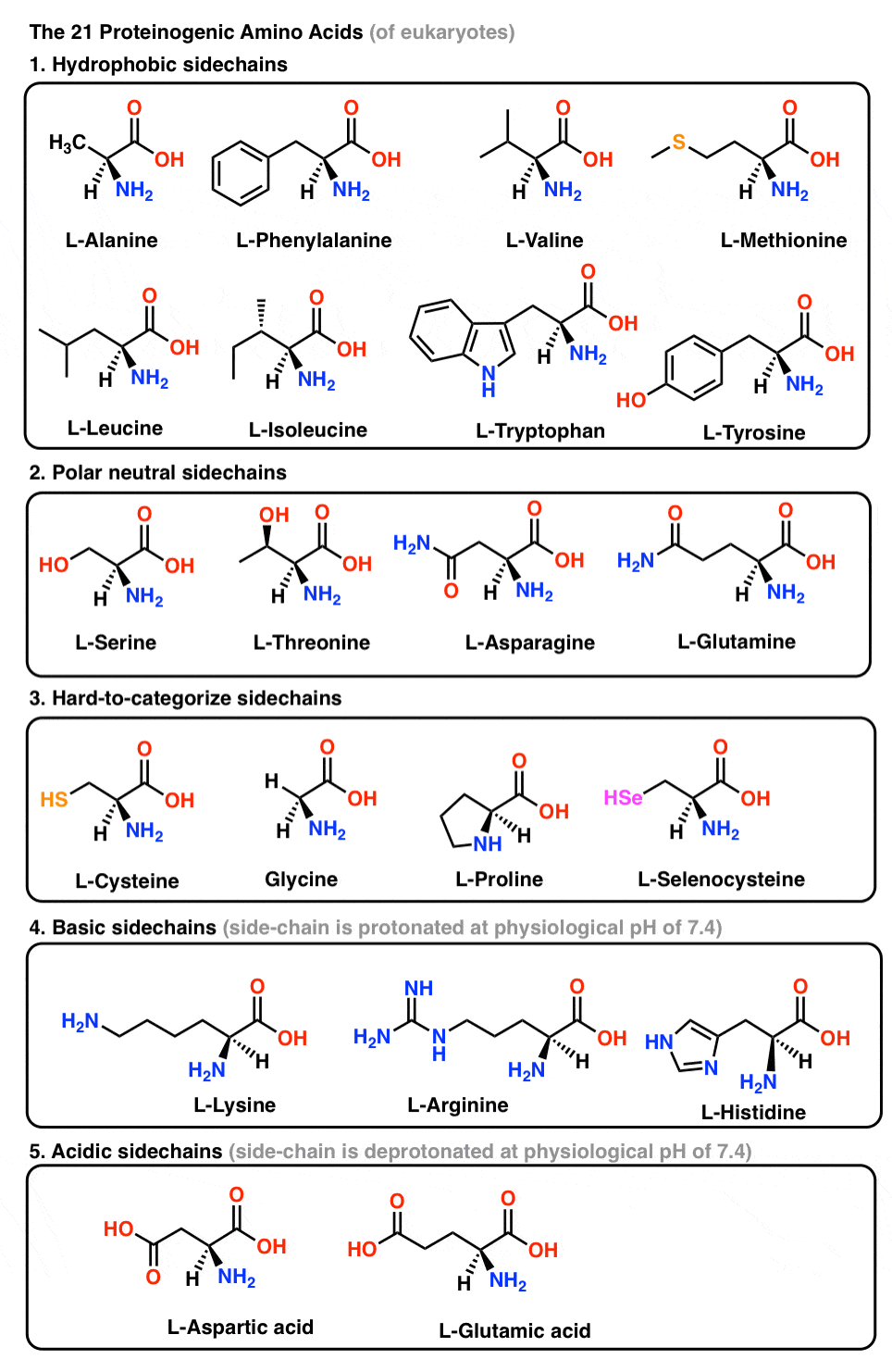
2. The “Proteinogenic” Amino Acids
Proteinogenic amino acids are the building blocks of proteins. In addition to the 20 amino acids directly encoded by the genome, two other amino acids are coded into proteins under special circumstances: selenocysteine (present in eukaryotes, including humans) and pyrrolysine (found only in methane-producing bacteria).
With the exception of (achiral) glycine, all proteinogenic amino acids are L-amino acids, where the “L-” prefix relates the stereochemistry of the amino acid relative to that of L-glyceraldehyde [See post: D and L Sugars] .
Of the chiral amino acids, all are S, with the exception of cysteine and selenocysteine [Note 1] (because sulfur and selenium have a higher priority under the CIP system. )
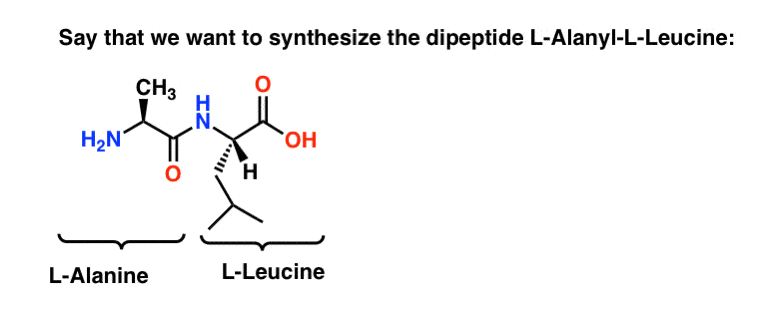
3. Synthesis of A Simple Dipeptide Without Protecting Groups (is not advisable!)
Let’s build a simple dipeptide between two of these amino acids. For simplicity’s sake, we’ll pick two from the “hydrophobic sidechain” group, alanine (Ala) and leucine (Leu), since their sidechains don’t need additional protecting groups.
What do we need to do to make L-Ala-L-Leu ?
Surveying the methods previously covered to make amides, it might seem simple.
Why not take 1 equivalent each of L-alanine and L-phenylalanine, add a coupling agent like N,N-dicyclohexylcarbodiimide (DCC) and patiently wait for our product to appear?
What could possibly go wrong?
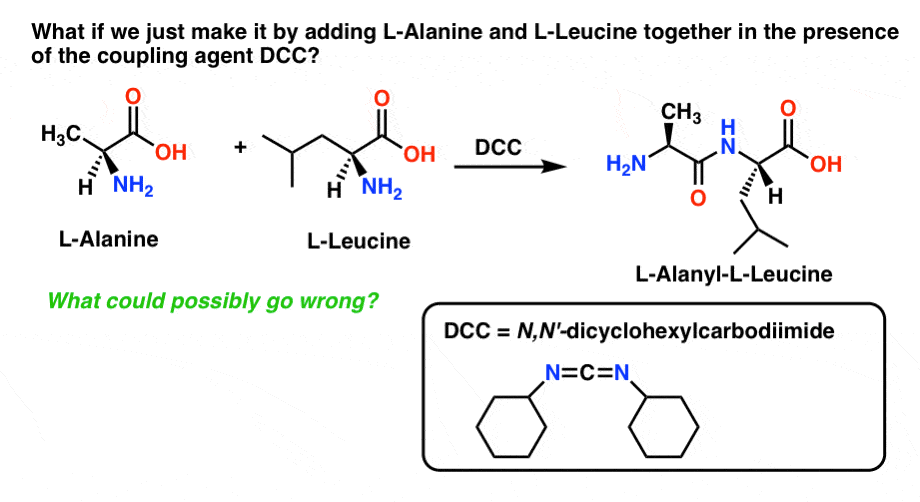
Well, this will give us some of our desired product. But it won’t do so efficiently!
That’s because each amino acid has two reactive termini – an amine and a carboxylic acid – and they can bond together in multiple ways.
Just like the letters A and L can combine to make the words AL and LA, in addition to Ala-Leu (our desired product) we will also get Leu-Ala.
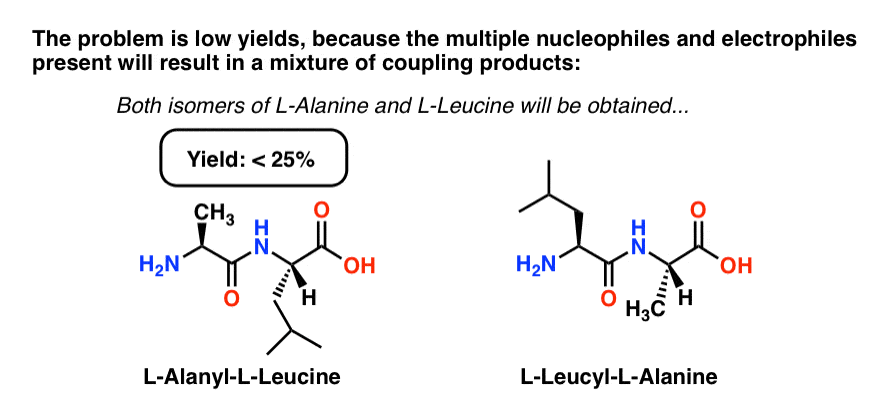
Furthermore, since we’re not adding single molecules together but molar quantities (even a millionth of a mole (a “micromole”) has 1017 molecules in it) we also have the possibility of forming the “homo-dipeptides” AA (Ala-Ala) and LL (Leu-Leu).
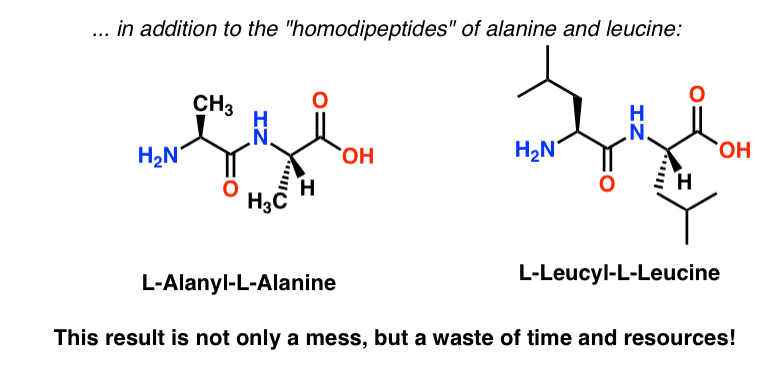
And that’s just a start. No matter how you slice it you’re looking at a low yield (<25%) of the desired material.
That’s inefficient, wasteful, and expensive! Isn’t there a better way?
4. Synthesis Of A Dipeptide Using A Protecting Group Strategy
Yes. Rather than using the native amino acids and just praying for a good yield, we can use protected versions of L-Alanine and L-Leucine.
If we protect the carboxylic acid of Leucine as an ester (e.g. a methyl ester) and protect the amine of L-Alanine as a carbamate (See: Carbamates as protecting groups) then we set up a situation where we have a single nucleophile and a single electrophile.
This results in a high yield (>95%) of a single product!
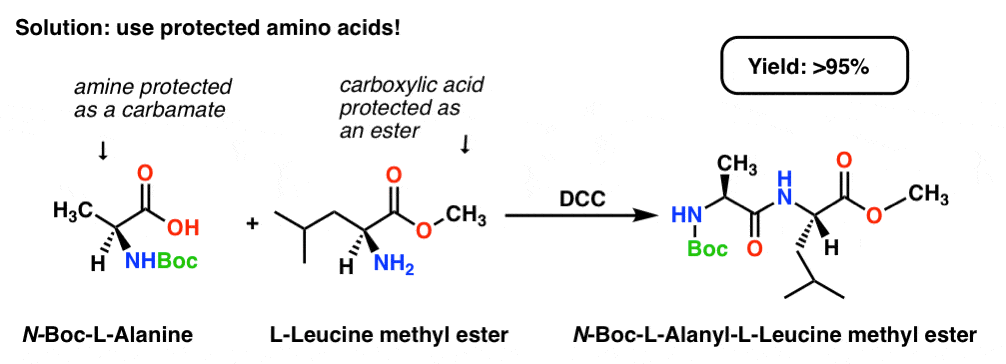
[Note: the Boc group is a popular carbamate protecting group for amines; “Boc” stands for t-butyloxycarbonyl]
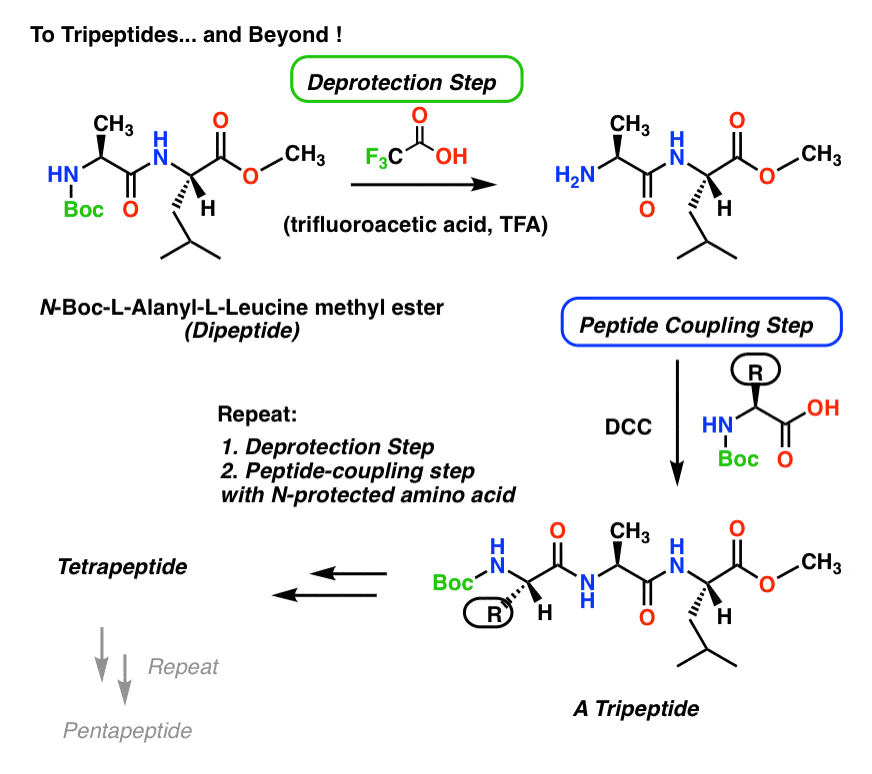
5. Synthesis of Longer Peptides – Tripeptides and Tetrapeptides
The good news is that we don’t have to stop at the dipeptide. If we choose protecting groups that can be removed selectively (and the carbamate / ester pair qualifies) then we can then deprotect the carbamate, and add a third amino acid.
The choice of carbamate protecting group here was t-butoxycarbonyl (Boc) which is removed with strong acid (trifluoroacetic acid, abbreviated as TFA).
Treatment with TFA removes the Boc group but leaves the methyl ester alone.
So if we treat the dipeptide with TFA, we liberate the amine nitrogen, and can react with another Boc-protected amino acid in the presence of DCC to get a tripeptide.
If we’re keen, we can even extend the same method to build a tetrapeptide, a pentapeptide… or beyond!
It’s not unreasonable to consider this method for longer peptides.
For instance, take something like bradykinin, a 9-peptide chain that causes dilation of blood vessels leading to a rapid drop in blood pressure. (Your body releases bradykinin in response to snake bites, which is how it was originally discovered.)
It might be interesting to synthesize variants of bradykinin where some of the amino acids are swapped out for other ones. In order to do that, we’d need to be able to synthesize it.
So how effective could it be?
If each peptide coupling step has a yield of about 95%, then our overall yield for making bradykinin would be (0.95)9 , or 63%. That’s actually pretty good! A lot of chemists would be happy to get a yield of 63% for a single reaction, let me tell you.
If the yields are high enough, one can even imagine building something crazy like insulin (51 peptide residues). That’s 7% yield for 51 steps.
Is this possible?
Yes… but it requires a clever modification that won its inventor, Bruce Merrifield, the 1984 Nobel Prize in Chemistry.
6. Bonus Topic: Solid-Phase Peptide Synthesis
What follows below is more supplemental than anything else, but given the importance of the topic, both interesting and useful.
In 1963 a chemist at Rockefeller University named Bruce Merrifield published a paper that would revolutionize how peptides were synthesized, and eventually make the synthesis of long peptides routine.
It was entitled: “Solid-Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide“.
Here’s the key idea.
Recall that in our original scheme (above) we protected the carboxylic acid as a methyl ester, which stays the same throughout the whole peptide synthesis.
Merrifield’s idea was: what if we find a way to attach the carboxylic acid to a functional group that is itself linked to a polymer bead? Not only would this also protect the carboxylic acid, it would drastically improve the ease of separations.
Why? Because instead of having to purify the final product by crystallization or column chromatography, you purify by filtering off the polymer beads (each 200-500 μm) and washing them to remove excess reagents.
The polymer beads themselves are pretty small. A typical size is 200 micrometers. Each bead can load about 4 nanomoles of amino acid.
[Brandon Finlay from ChemTips shows a picture here]
The video posted below is not mine, but it gives you an idea of the process.
At about 0:34 you can see how small the beads are.
The starting point for the Merrifield process is crosslinked polystyrene. which behaves like one big interlinked molecule. Polystyrene is then attached to a linker, which usually terminates with an NH2 group. This itself is usually protected; in order to activate the linker, you need to remove the protecting group cap.
The polymer bead needs to swell in a solvent in order for functional groups on the solid support to undergo reactions efficiently.
The essential procedure is: swell –> add reagents –> wait –> filter –> wash, and repeat. Beads stay in the reaction vessel the whole time. There’s also usually some kind of capping step to make sure any unreacted amines don’t participate in the next reaction.
It’s possible to make peptides up to about 50 units this way. In highly automated systems one can be even more ambitious.
Merrifield started knocking off peptides in the 1960s. Bradykinin was made in 8 days and 68% overall yield. one example. Insulin was made two years later. The crowning achievement of this initial period was probably ribonuclease A, which has 150 amino acid residues.
The original Merrifield process has been significantly modified and improved. Originally, removal of the linker required harsh conditions (strong acid). Today, procedures usually employ FMOC protecting groups instead of BOC, which allow for deprotection with mild amine base (piperidine). A galaxy of new resins, linkers, and coupling procedures have been subsequently developed. The Wikipedia article on solid-phase peptide synthesis is an OK place to start.
Notes
Note 1. Cysteine (and selenocysteine) are L, but their stereocenters are (R), because sulfur and selenium have higher priority within the Cahn-Ingold-Prelog system.
Quiz Yourself!
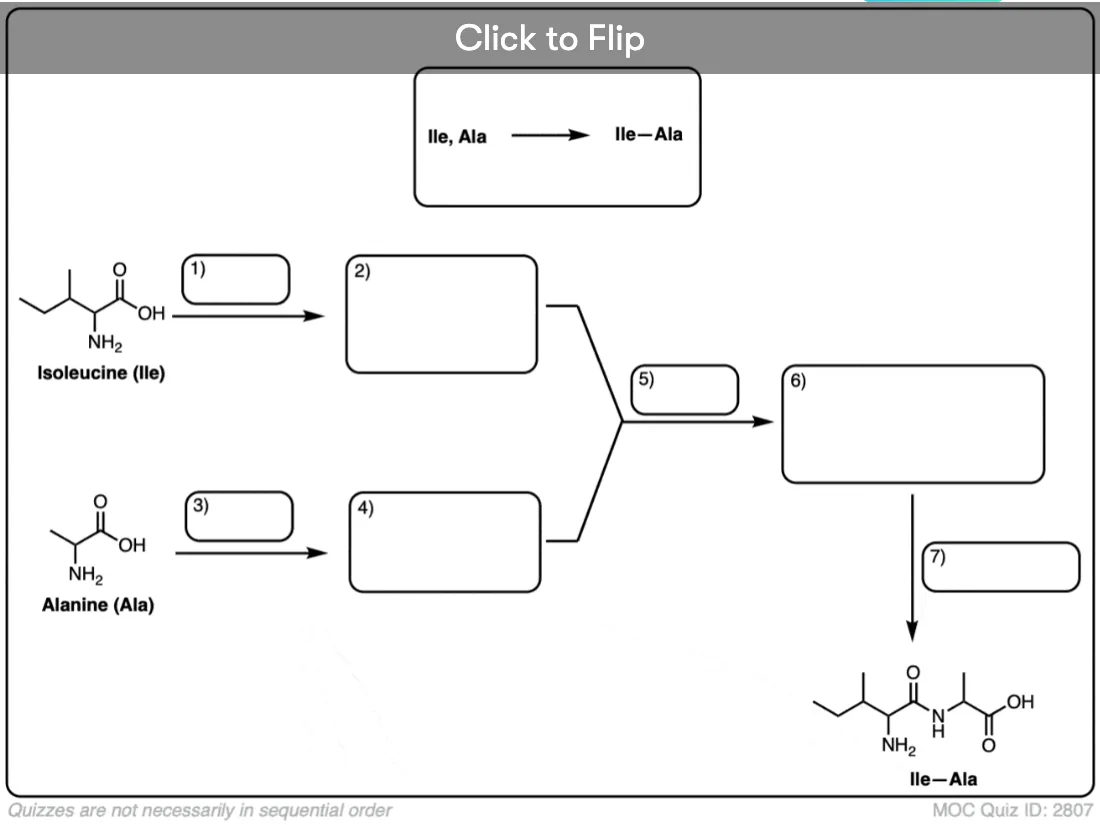
Become a MOC member to see the clickable quiz with answers on the back.
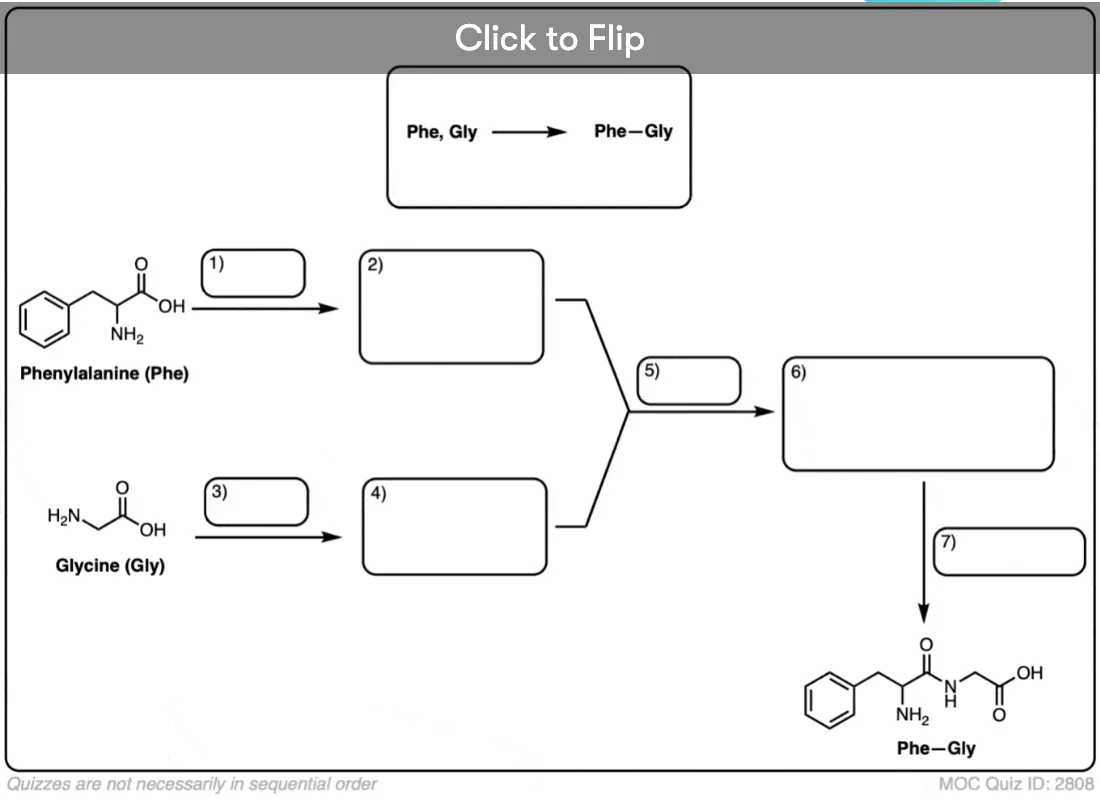
Become a MOC member to see the clickable quiz with answers on the back.
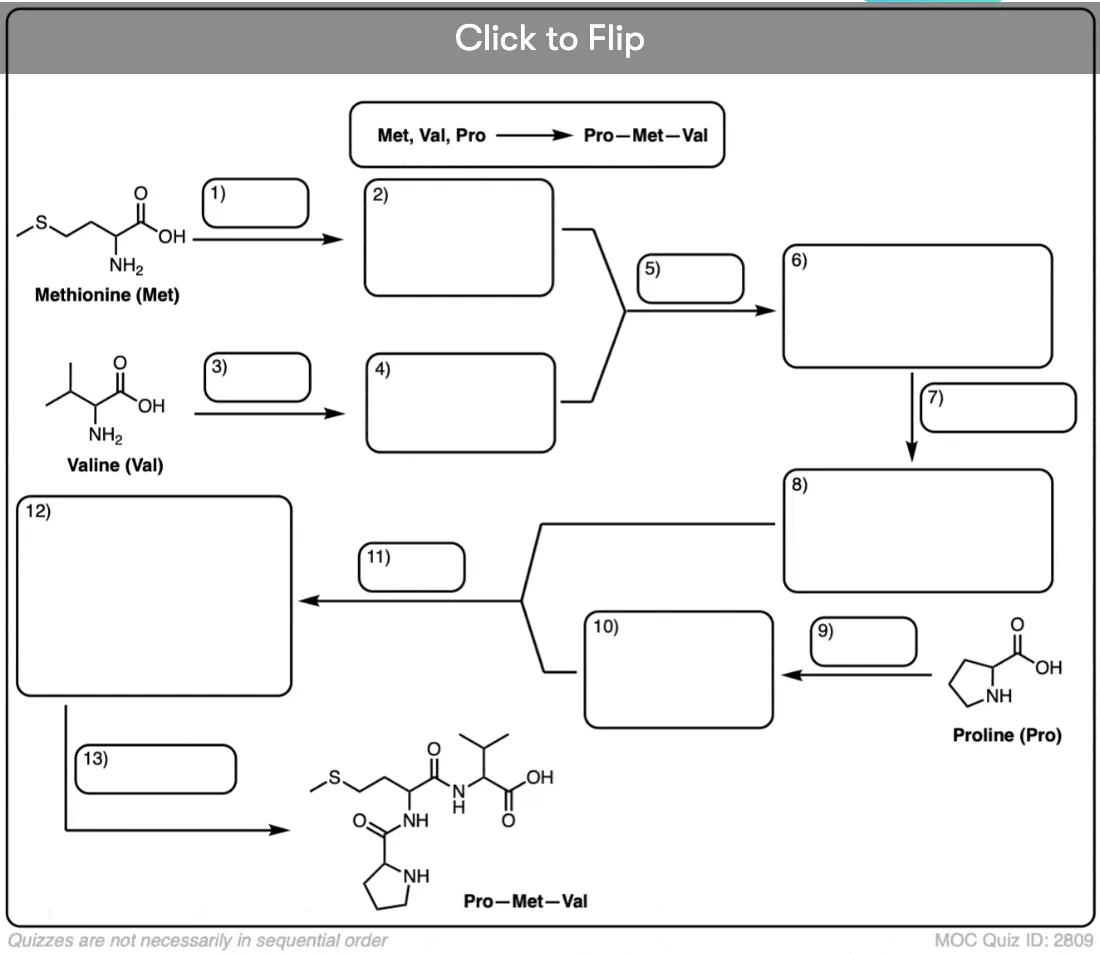
Become a MOC member to see the clickable quiz with answers on the back.
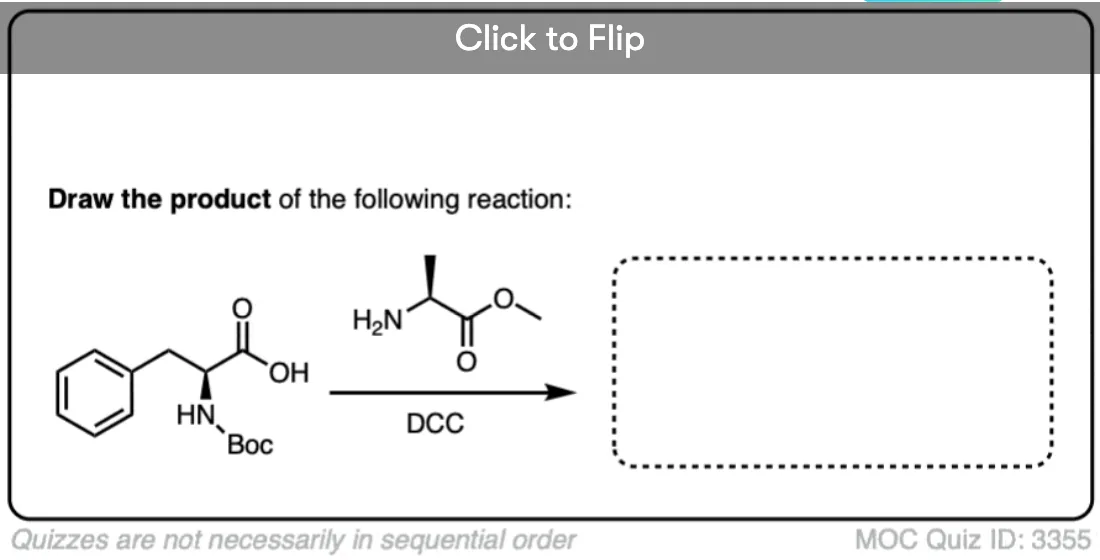
Become a MOC member to see the clickable quiz with answers on the back.
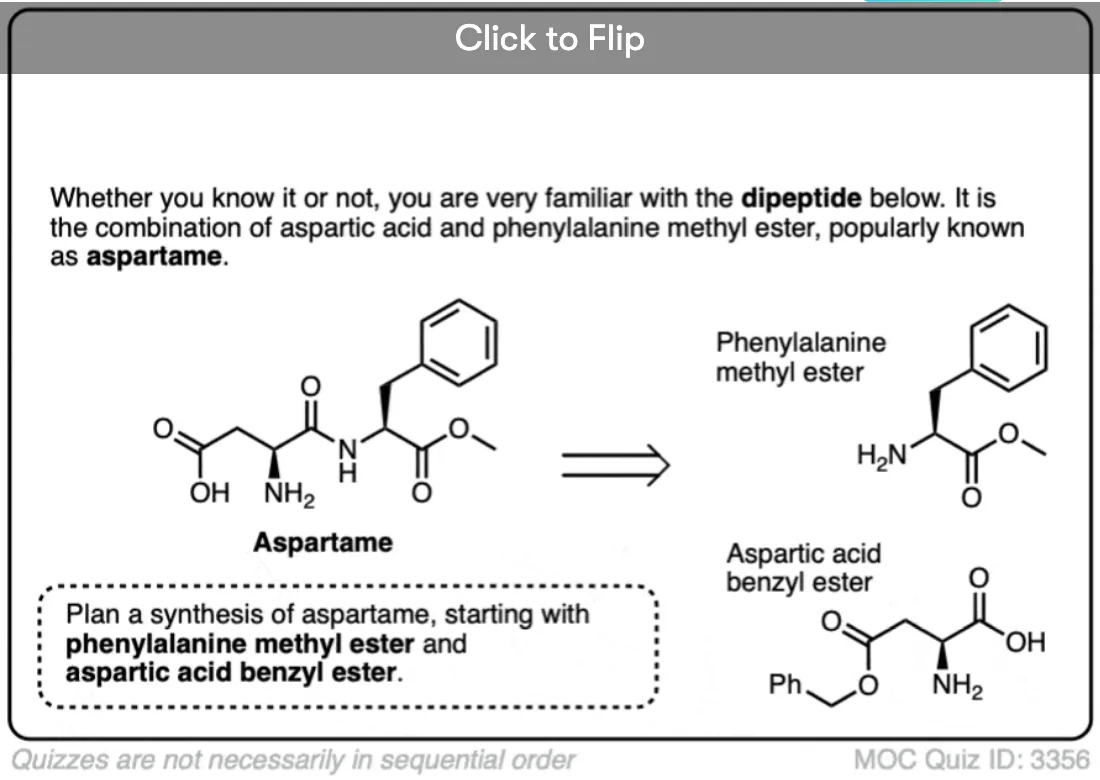
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
This is a major topic, as the synthesis of peptides is a global billion-dollar industry.
- THE SYNTHESIS OF AN OCTAPEPTIDE AMIDE WITH THE HORMONAL ACTIVITY OF OXYTOCIN
Vincent du Vigneaud, Charlotte Ressler, College John M. Swan, Carleton W. Roberts, Panayotis G. Katsoyannis, and Samuel Gordon
Journal of the American Chemical Society 1953, 75 (19), 4879-4880
DOI: 1021/ja01115a553 - The Synthesis of Oxytocin
Vincent du Vigneaud, Charlotte Ressler, John M. Swan, Carleton W. Roberts, and Panayotis G. Katsoyannis
Journal of the American Chemical Society 1954, 76 (12), 3115-3121
DOI: 1021/ja01641a004 - A Method of Synthesis of Long Peptide Chains Using a Synthesis of Oxytocin as an Example
Miklos Bodanszky and Vincent du Vigneaud
Journal of the American Chemical Society 1959, 81 (21), 5688-5691
DOI: 1021/ja01530a040
In the first half of the 20th century, peptide synthesis was done using standard organic chemistry solution phase techniques. This is now known as LPPS (liquid-phase peptide synthesis). du Vigneaud received the Nobel Prize in chemistry in 1955 for his work in showing that peptide synthesis could be achieved, using the correct choice of protecting groups and synthetic strategies.In 1963, Prof. Robert Bruce Merrifield (Rockefeller U., New York) revolutionized peptide synthesis by coming up with the SPPS (Solid-Phase Peptide Synthesis) method, making the synthesis of long peptide chains much more feasible. The C-terminus is bound to a polymer resin, and the amino acids are added one at a time following the same cycle: deprotect, wash, couple the next amino acid (with a peptide-coupling reagent such as DCC), wash, deprotect the N-terminus again, and so on. Merrifield’s method came to be called the Boc/Bzl strategy due to the protecting groups employed (Boc for the nitrogen atoms, and Bzl (benzyl) for the side chains). The catch is that final cleavage of the peptide from the resin using this method requires anhydrous HF, which is not easy to handle. - Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide
R. B. Merrifield
Journal of the American Chemical Society 1963, 85 (14), 2149-2154
DOI: 10.1021/ja00897a025
This is the paper that started it all – a single-author publication by Prof. Merrifield using the SPPS method to make a tetrapeptide. This remains one of the most highly-viewed and highly-cited papers in JACS, even today. - The Synthesis of Bovine Insulin by the Solid Phase Method
Marglin and R. B. Merrifield
Journal of the American Chemical Society 1966, 88 (21), 5051-5052
DOI: 10.1021/ja00973a068
Insulin is a billion-dollar hormone as its dysregulation is what causes diabetes. This paper shows that insulin can be made through SPPS methods. Insulin is tricky to make as it has 2 chains (the A and B chain) connected through disulfide bonds. Interestingly, Merrifield synthesized the active hormone by combining both chains with protected thiols (protected as sulfonates), which he then reduced to the thiol, and then oxidized in air in a basic medium (pH 10). This is called undirected or air oxidation since the disulfide bonds are not being formed selectively; luckily they formed correctly in this case. Insulin today is not manufactured by the SPPS method due to these complications; instead it is made through a recombinant process. - The Synthesis of Ribonuclease A
Bernd Gutte and R. B. Merrifield
Journal of Biological Chemistry 1971, 246 (6), 1922-1941
DOI: 10.1016/S0021-9258(18)62396-8
This is the crowning achievement of SPPS – the synthesis of a 124-mer peptide (or protein, at this point), RNAse A. - The Solid Phase Synthesis of Ribonuclease A by Robert Bruce Merrifield
Nicole Kresge, Robert D. Simoni and Robert L. Hill
Journal of Biological Chemistry 2006, 281 (26), e21
DOI: 10.1016/S0021-9258(20)55702-5
A short biographical account of Prof. Merrifield’s life. This mentions that he developed the first prototype of an automated peptide synthesizer working in the basement of his house in 1965! - Solid Phase Synthesis
Nobel Lecture, December 8, 1984 by Bruce Merrifield
Merrifield’s Nobel Lecture upon receiving the Nobel Prize in Chemistry in 1984. This describes his life, how he conceived of and developed the SPPS process, and all the breakthroughs it has enabled. - 9-Fluorenylmethoxycarbonyl function, a new base-sensitive amino-protecting group
Louis A. Carpino and Grace Y. Han
Journal of the American Chemical Society 1970, 92 (19), 5748-5749
DOI: 10.1021/ja00722a043 - 9-Fluorenylmethoxycarbonyl amino-protecting group
Louis A. Carpino and Grace Y. Han
The Journal of Organic Chemistry 1972 37 (22), 3404-3409
DOI: 10.1021/jo00795a005
The discovery and development of the Fmoc (9-fluorenylmethoxycarbonyl) group as a protecting group for amines has also improved the practice of peptide synthesis and SPPS. - A mild procedure for solid phase peptide synthesis: use of fluorenylmethoxycarbonylamino-acids
Atherton, Hazel Fox, Diana Harkiss, C. J. Logan, R. C. Sheppard and B. J. Williams
J. Chem. Soc. Chem. Comm. 1978, 537-539
DOI: 10.1039/C39780000537
This is the first paper describing what is now known as the Fmoc/tBu SPPS process, which has largely supplanted the original Boc/Bzl process developed by Prof. Merrifield. The advantages with Fmoc-SPPS are multifold, but the main ones are simplicity and orthogonality. Amine deprotection is done with a base (20% piperidine in DMF is sufficient to deprotect an Fmoc-amine), and final cleavage of the peptide from the resin can be done with TFA (trifluoroacetic acid), which is much easier to handle than HF. - Advances in Fmoc solid-phase peptide synthesis
Raymond Behrendt, Peter White, and John Offer
Peptide Sci. 2016, 22, 4-27
DOI: 10.1002/psc.2836
A modern review on Fmoc-SPPS that describes how far we have come and some of the challenges that remain. For instance, aggregation of peptide chains on the resin is a major issue in Fmoc-SPPS when synthesizing especially hydrophobic peptides, and there are ways to deal with it, such as the introduction of ‘kinking’ residues, like Pro or the use of pseudoprolines, which will revert back to the desired amino acids when the peptide is cleaved with TFA. Native Chemical Ligation is also used nowadays for the synthesis of especially long peptides, like Merrifield’s RNAse A.
Thanks for very good explanation and excellent step-by-step video!!
These tips are awesome, I was looking for something like that. Thanks!
OK. Do you guys actually do peptide synthesis or do you just drop ship?
We, or least, in my school do cover this topic extensively. We need to learn fischer esterfication of them, acylation, protecting group although it was different than the protecting group you used It had two benzene rings and a five member ring in the middle.
Good to know! Sounds like you’re talking about FMOC. https://en.wikipedia.org/wiki/Fluorenylmethyloxycarbonyl_protecting_group
I should update to include that.
Thank you very much!