Aldehydes and Ketones
Breaking Down Carbonyl Reaction Mechanisms: Reactions of Anionic Nucleophiles (Part 2)
Last updated: December 29th, 2022 |
Continuing from Part 1, today I’ll cover the remaining 3 classes of mechanistic pathways for the reactions of anionic nucleophiles with carbonyl compounds, using the reactions on this sheet as a guide. Remember, that here we’re just talking about anionic nucleophiles here. The neutral nucleophiles (e.g. amines, alcohols, water) will be dealt with seperately.
Case 3: 1,4-addition followed by protonation:
Relevant electrophiles: α,β unsaturated carbonyls [example – row E]
Relevant (anionic) nucleophiles: Grignards, enolates, alkoxides/hydroxide, hydrides.
Key concepts to know: 1) pKa (the reactivity of an α, β unsaturated carbonyl towards nucleophiles INCREASES as the enolate becomes more stable). 2) Thermodynamic versus kinetic control (for Grignards and hydrides, which can competitively add [1,2] or [1,4]).
Examples: Michael reaction
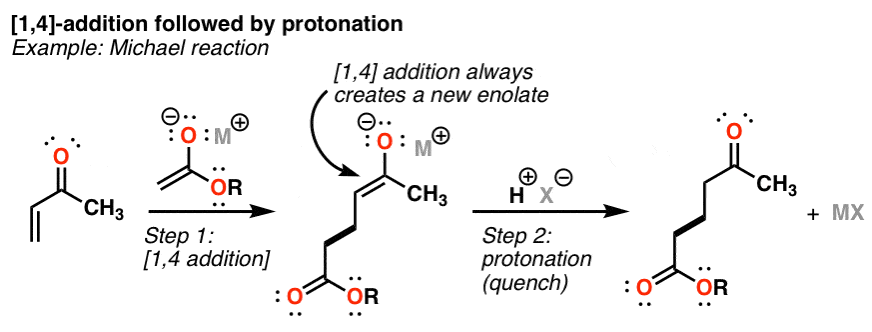
When a π bond is attached to an electron withdrawing group like a ketone or other strongly electron withdrawing species, it becomes susceptible to nucleophilic attack. The reason is that the negative charge generated by this addition reaction [which we call “1,4 addition” or “conjugate addition”] can be stabilized through resonance with the electron withdrawing group. Addition of a nucleophile at the terminus of the double bond thus forms an enolate.
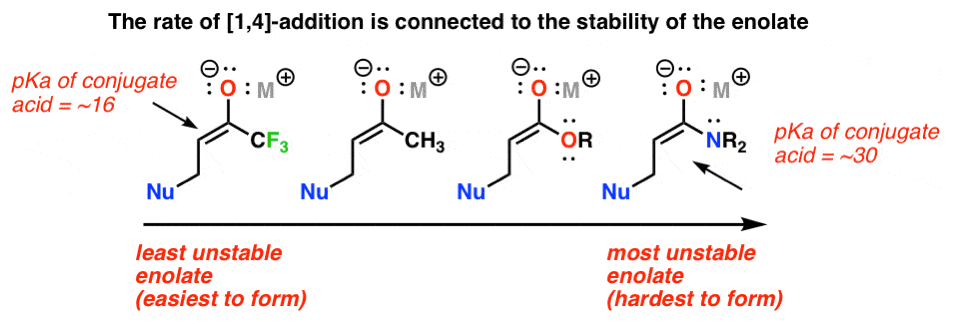
The rate at which 1.4 addition occurs is dependent on the stability of the resulting enolate. This might be tough to see at first, so let me stop to explain. If you look at your pKa table, you’ll see that your typical ester has a pKa of about 25 and that of a ketone is about 20. The ester is less acidic, meaning its enolate is less stable. This also means that all else being equal, the rate of conjugate addition at an α, β unsaturated ester will be slower (i.e. more difficult, less reactive) than that for an α, β
unsaturated ketone, because you are forming a stronger (i.e. less stable) base.
Things get complicated when the nucleophile can attack either [1,2] or [1,4]. This is the case for Grignards and certain reducing groups: attack is faster at the carbonyl (because of its higher partial positive charge) but more thermodynamically favored at the alkene (because the C-C π bond is weaker than the C-O π bond). This leads into a big topic which I don’t want to get into right now, which is thermodynamic versus kinetic control, so for now, I’ll just say that if you need to add a carbon nucleophile to the β position of an α, β unsaturated ketone, use an organocuprate (Gilman reagent) instead.
Case 4: SN2
Relevant electrophiles: alkyl halides
Relevant (anionic) nucleophiles. enolates, cyanide, alkoxides, lithium aluminum hydride.
Relevant electrophiles: alkyl halides [or alkyl groups with any other good leaving group].
Key concepts to understand: 1) Leaving group ability (related to pKa) – the better the leaving group, the more reactive it will be in an SN2. 2) Steric hindrance (primary alkyl halides are more reactive than secondary). 3) Stereochemistry – SN2 occurs with inversion.
Examples: Enolate alkylation
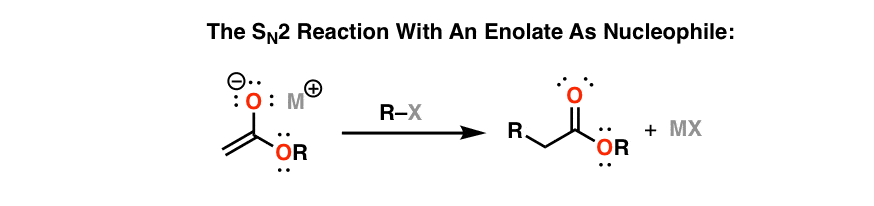
The SN2 is a standard part of every organic chemists’ toolkit, and nothing here has changed since you took Org 1. The SN2 is favored by using a primary alkyl group with a good leaving group attached (alkyl halides, alkyl mesylates, alkyl tosylates). The only twist here is that you’ve been introduced to some new classes of nucleophiles – enolates, in particular. Enloates are great nucleophiles and they react readily with alkyl halides and other species in the SN2 reaction.
Case 5: What doesn’t work
There are several places on the chart where the cell reads “NR” or “borderline”. This means that the nucleophile and electrophile do not react in a way that leads to a productive bond formation. Instead, what occurs is one of several side reactions (or lack of reaction, for that matter)
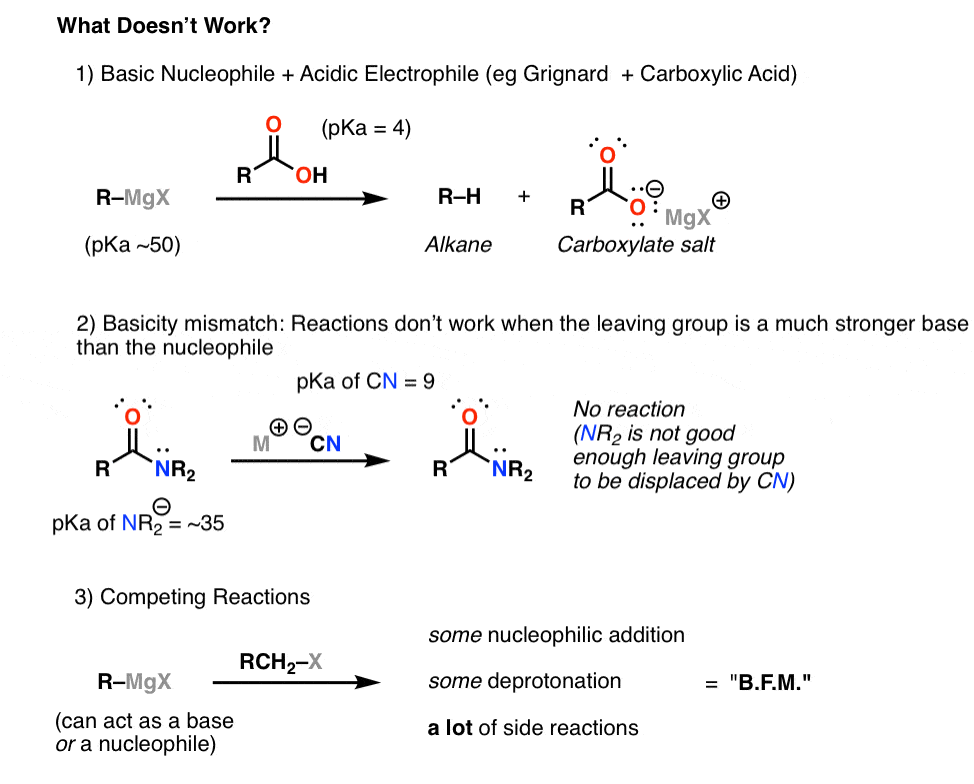
1) Basic nucleophiles/acidic electrophiles. As you might expect, the carbonyl carbon of carboxylic acids is a poor electrophile under basic conditions, since the acidic -COOH group protonates basic nucleophiles like Griganards and enolates, turning it into a carboxylate (negatively charged carboxylic acid). The negatively charged oxygen donates strongly into the carbonyl carbon, making it extremely unreactive towards nucleophiles. Another prominent case is that of primary amides, which are fairly acidic (pKa ~17) and will protonate Grignards and enolates. This is yet another prominent example where knowing your pKas comes in really handy.
2) Basicity mismatch. Some reactions fail because it is simply too difficult to displace a strong base with a weaker base. An example on this chart would be the attempted reaction of cyanide ion (pKa = 4) with amides (pka of NH2- = ~35). No reaction occurs.
3) Competing electrophilic sites. Sometimes reactions fail not because there is a lack of reactivity, but because there is too much reactivity, and it is unselective. One example is the reaction of Grignards with alkyl halides. In theory, this reaction should work like a charm – strong nucleophile (Grignard) plus an alkyl halide with a good leaving group (good electrophile) should provide a new carbon-carbon bond. In practice, the reaction usually fails, because the Grignard can also act as a base, by deprotonating the alkyl halide. When a number of different reactions can occur on a substrate – nucleophilic addition, deprotonation, and a whole bunch of other side reactions stemming from these two processes – the result (as my friend Jeff would say) is often something you write down in your lab notebook as B.F.M. The B stands for B and the M stands for mess. What F stands for usually depends on your mood.
you really dont know how much this site helps self studying students again another perfect explination
well, right now i m in class 12 preparing for iit and whatever stuff u give…..really develops a love (a bond) with ORGANIC chemistry…thank you for all this work…….thanking from the depth of my heart
I absolutely loathe the way conjugate addition is taught, think it is one of the very worst handled parts of organic chemistry. I’m taking the ACS National Organic Test, and they *love*, **love**, ***love*** conjugate addition, including enone.
I’ve read the pertinent sections in three or four of the major organic texts, and they all fail disastrously. They fling around useless terms like “strong” or “weak” bases, don’t provide any guidance on the criteria for achieving kinetic or thermodynamic products as major and/or in what proportions, then leave you to hang when it comes to solving problems. I’m absolutely livid.
This is about the best description I’ve seen, although I’m still confused. It seems like unless you’re laying on some serious heat (like a Wittig or a Grignard reagent) onto a conjugated enone, you might as well fall back on 1,4-addition products as being major?? I wouldn’t characterize methoxide as a “weak” base (try dropping a beaker down your shorts and tell me it is “weak”), but apparently it is in terms of conjugate addition…..