Alcohols, Epoxides and Ethers
Epoxide Ring Opening With Base
Last updated: May 28th, 2026 |
Ring-opening of epoxides with basic nucleophiles
- Epoxides are cyclic ethers with considerable ring strain (about 13 kcal/mol)
- Epoxides undergo reaction with nucleophiles under basic conditions at the least substituted end of the epoxide
- If a chiral center is at this carbon, its stereochemistry will be inverted.
- Nucleophiles that will add to epoxides include hydroxide ion (HO-) alkoxides, Grignard reagents, organolithium reagents, and LiAlH4 (among others)
- A simple way to remember this is to think of epoxide opening under basic conditions as proceeding much like an SN2 reaction.
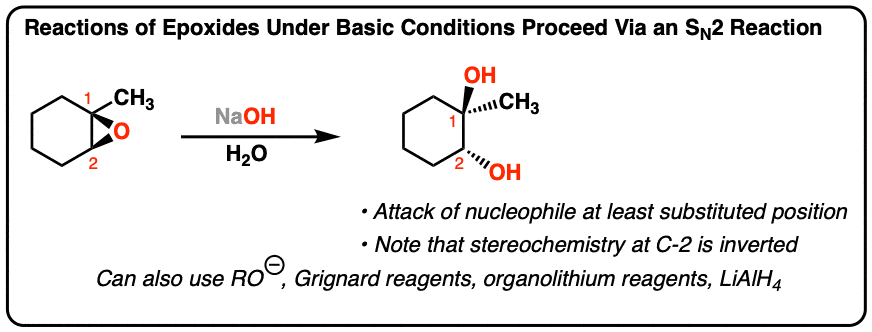
Table of Contents:
- Opening Epoxides With Base: How it Works
- The Role of Solvent
- Understanding Separate Quench Steps: 1) Add this 2) THEN add this!
- What Doesn’t Work: Poor Nucleophiles
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Opening Epoxides With Base: How It Works
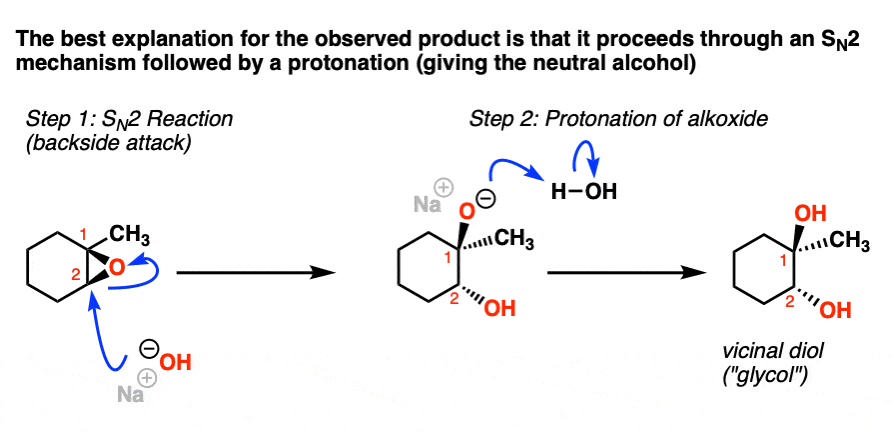
In the last post we discussed the reactions of epoxides under acidic conditions and saw how they resembled the “3-membered ring” family of alkene mechanisms. We left off by noting that the reaction of the epoxide (shown above) with NaOH in H2O gave a different product than that obtained through H3O+. Therefore it must go through a different mechanism.
Our question is, how does this reaction work?
Noting the fact that the nucleophile (HO- ) attacks the epoxide at the least substituted position (C2) and results in inversion of stereochemistry at this position, our best evidence is consistent with this reaction proceeding through the familiar SN2 mechanism followed by a transfer of proton from the weakly acidic solvent (H2O) to the alkoxide (RO – ) providing a neutral alcohol.
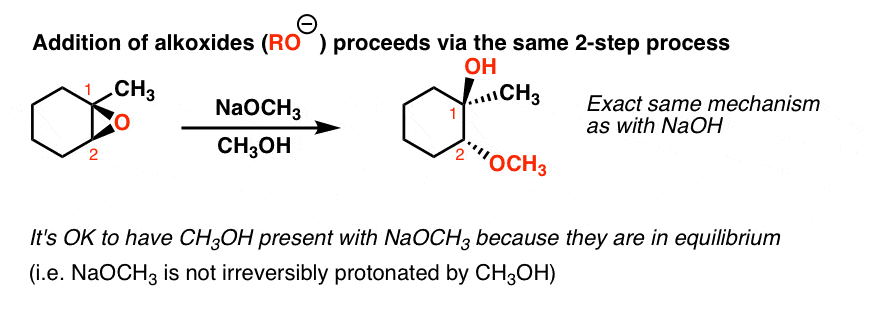
Along the same lines, if epoxides are treated with alkoxides (RO- ) in alcohol solvent, they lead to essentially the same reaction. Here’s an example with NaOCH3. Note that the proton must come from CH3OH here.
2. The Role Of Solvent
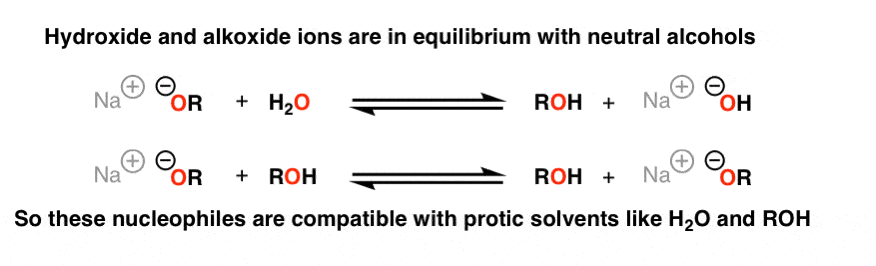
Now – at the end of the last post, we mentioned that acids are incompatible with some strong nucleophiles because they will be destroyed by irreversible protonation [classic example: Grignard reagents and water]. Should we be worried about NaOH in H2O or NaOCH3 in CH3OH? No! These are examples of bases in equilibrium with their conjugate acids.
It’s fine to have NaOH in the same reaction vessel with H2O since the reaction is reversible.
However, if we move to more strongly basic nucleophiles, such as Grignard reagents (RMgX), organolithium reagents (R-Li) or hydrides (H-) we should be concerned, because the conjugate acids of each of these species (pKa of about 50 for alkanes, and about 38 for H2) are much weaker acids than ROH (pKa 16-18).
[A good rule of thumb for “reversibility” of an acid base reaction is a pKa difference of 8 or less – see A Handy Rule of Thumb for Acid-Base Reactions].
That means that if these strong bases come into contact with ROH, an irreversible acid-base reaction will occur. We thus obtain an alkoxide (RO-) which is insufficient in strength to deprotonate alkanes or H2.
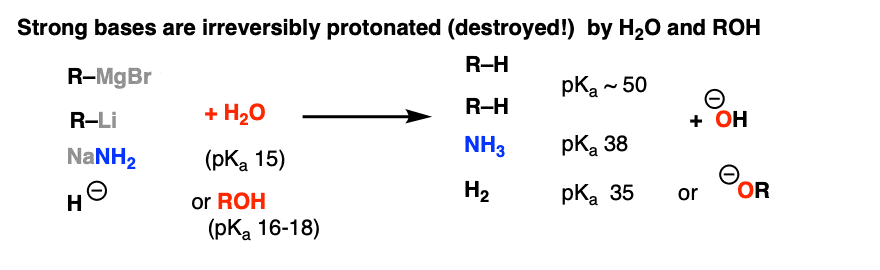
Bye-bye nucleophile!
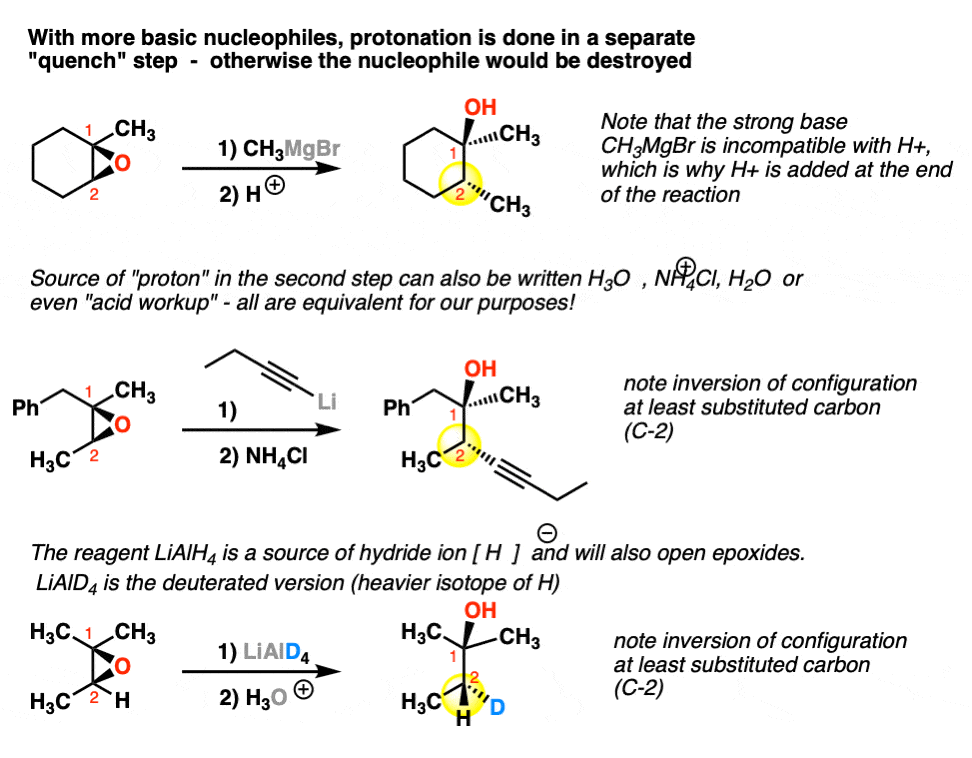
3. Understanding Separate Quench Steps: 1) Add this 2) THEN, add this!
What this means in practice is that we just move the protonation step to the end, in a process sometimes called a “quench”. Note how each of the following reactions has a Step 1) and a Step 2).
Step 1 is addition of nucleophile. After the reaction is done, we add our source of proton (Step 2) to quench. Note that there are many reagents commonly written for this process – H2O, H3O+, H+, NH4Cl, “acid workup” just to name a few. They all mean the same thing!
Here are some examples of reactions of epoxides with strongly basic nucleophiles. Grignard reagents , organolithium reagents, and hydride (e.g. LiAlH4 , a source of H- ).
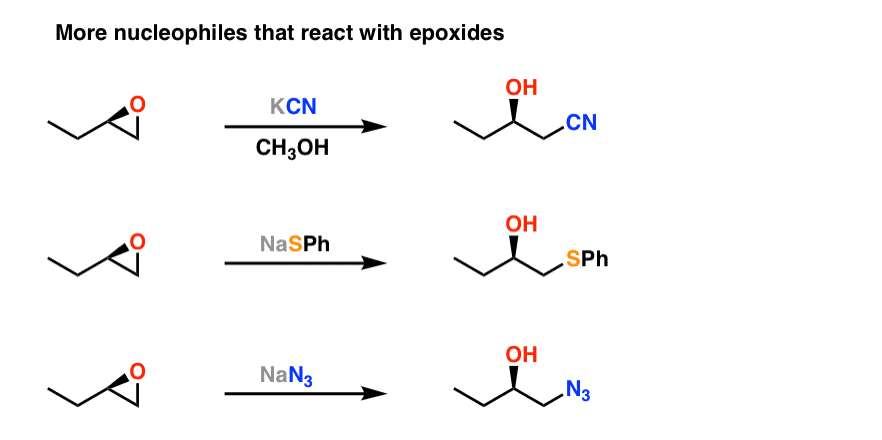
Is there anything else? [yes – there are other nucleophiles which will react with epoxides, but they are seldom seen at this level. However I have added them in Note 1 at the bottom].
4. What Doesn’t Work: Poor Nucleophiles
Just like any SN2 reaction, for this pathway to work, we really must be dealing with a decent nucleophile. Poor nucleophiles, especially neutral species like H2O, ROH, and RCOOH simply won’t cut it.
In order for epoxides to react we need either to use strongly acidic conditions (good for weak nucleophiles) or basic conditions (HO- , RO- , RMgBr, RLi, LiAlH4).

This concludes our exploration of epoxides and ethers. However we still have a lot to talk about regarding alcohols – and certainly we aren’t done with examples of SN1 and SN2 reactions. That’s coming up next.
Next Post – Making Alkyl Halides From Alcohols
Notes
Note 1. Some other nucleophiles which react with epoxides but are rarely seen in introductory organic chemistry. Note that the second and third examples don’t show the source of proton (H+) that adds to the oxygen, but it can be added afterwards in a “quench” step.
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
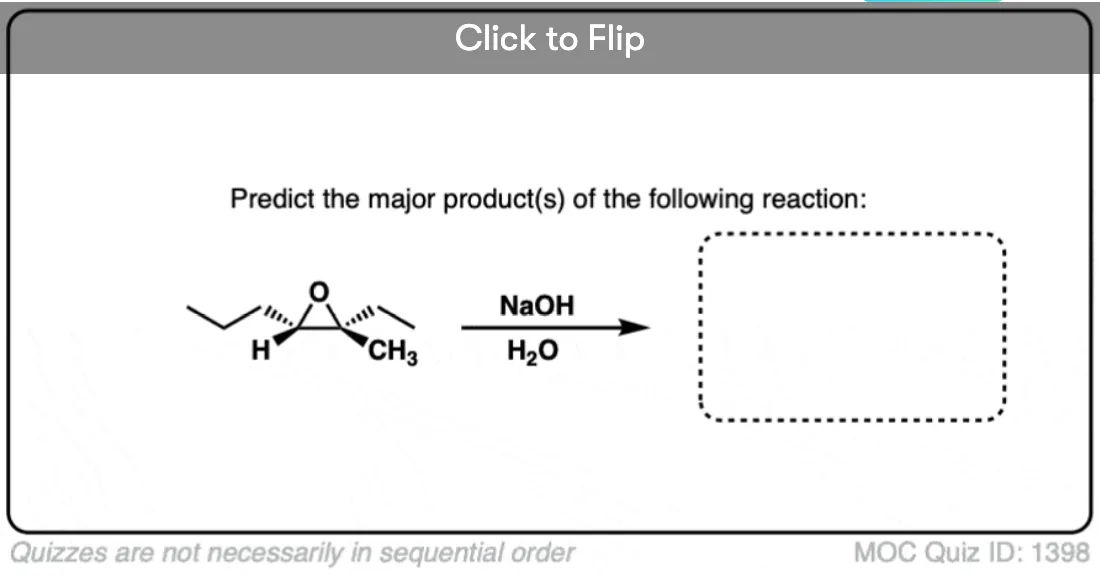
Become a MOC member to see the clickable quiz with answers on the back.
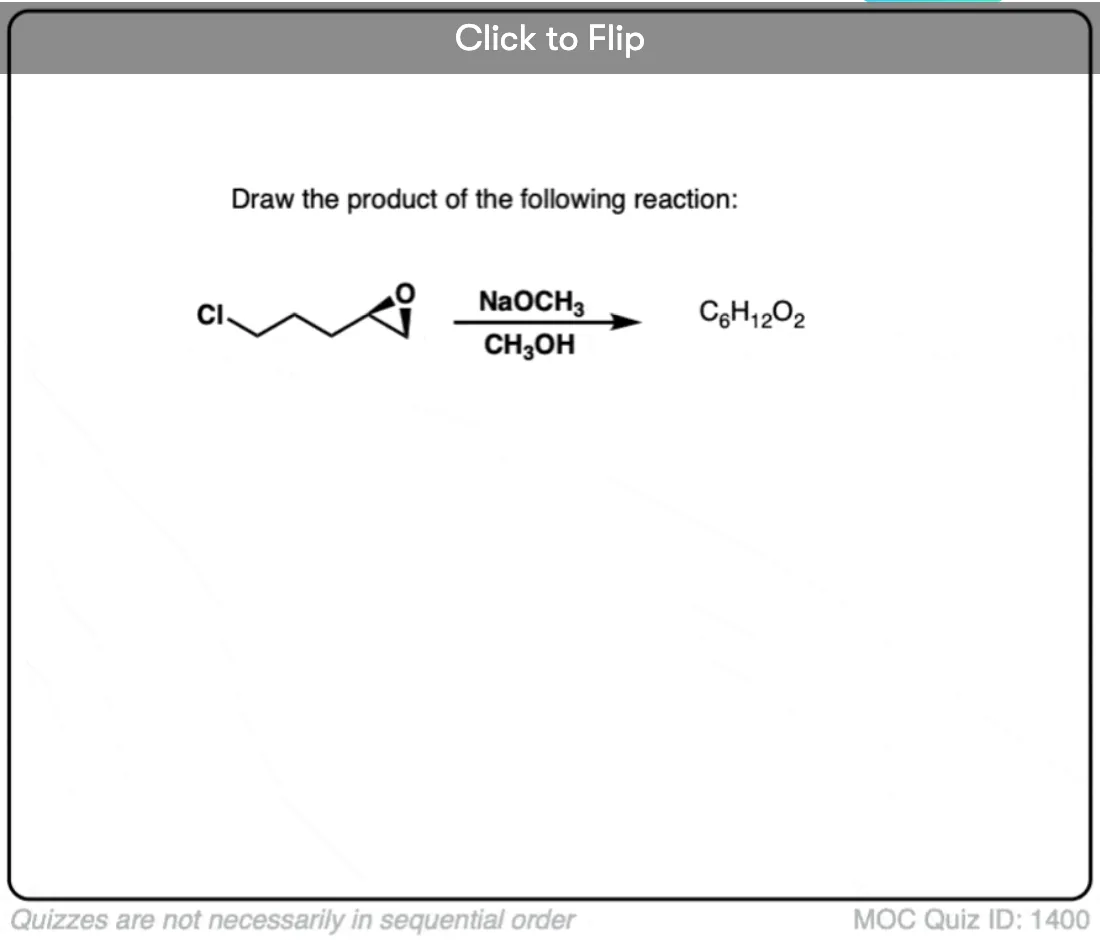
Become a MOC member to see the clickable quiz with answers on the back.
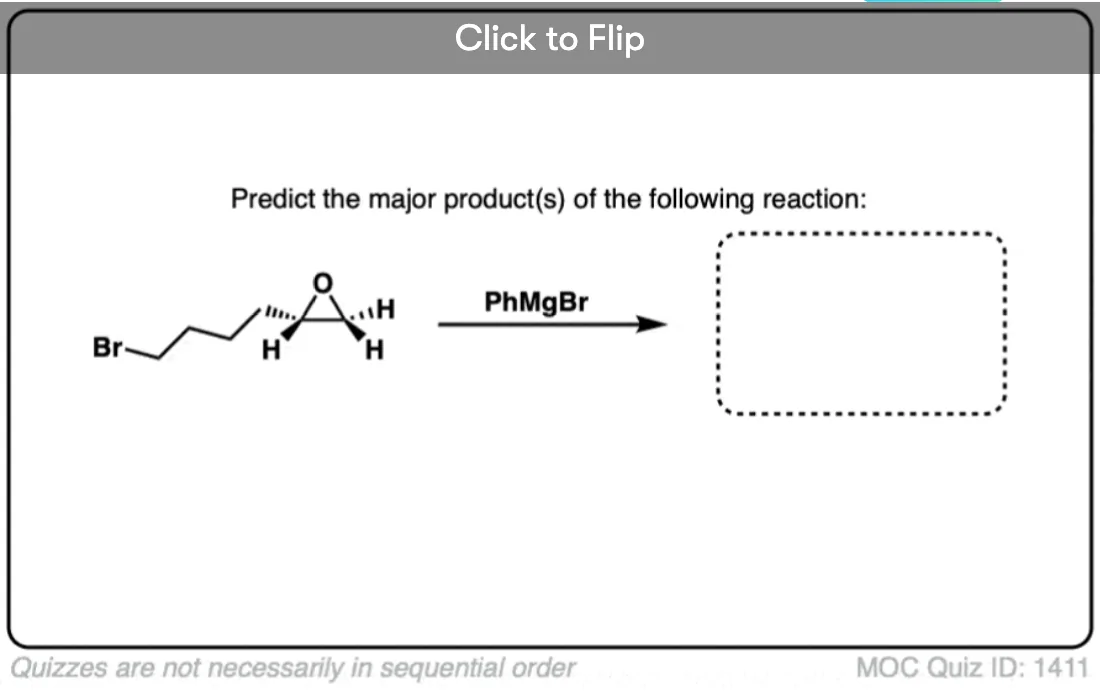
Become a MOC member to see the clickable quiz with answers on the back.
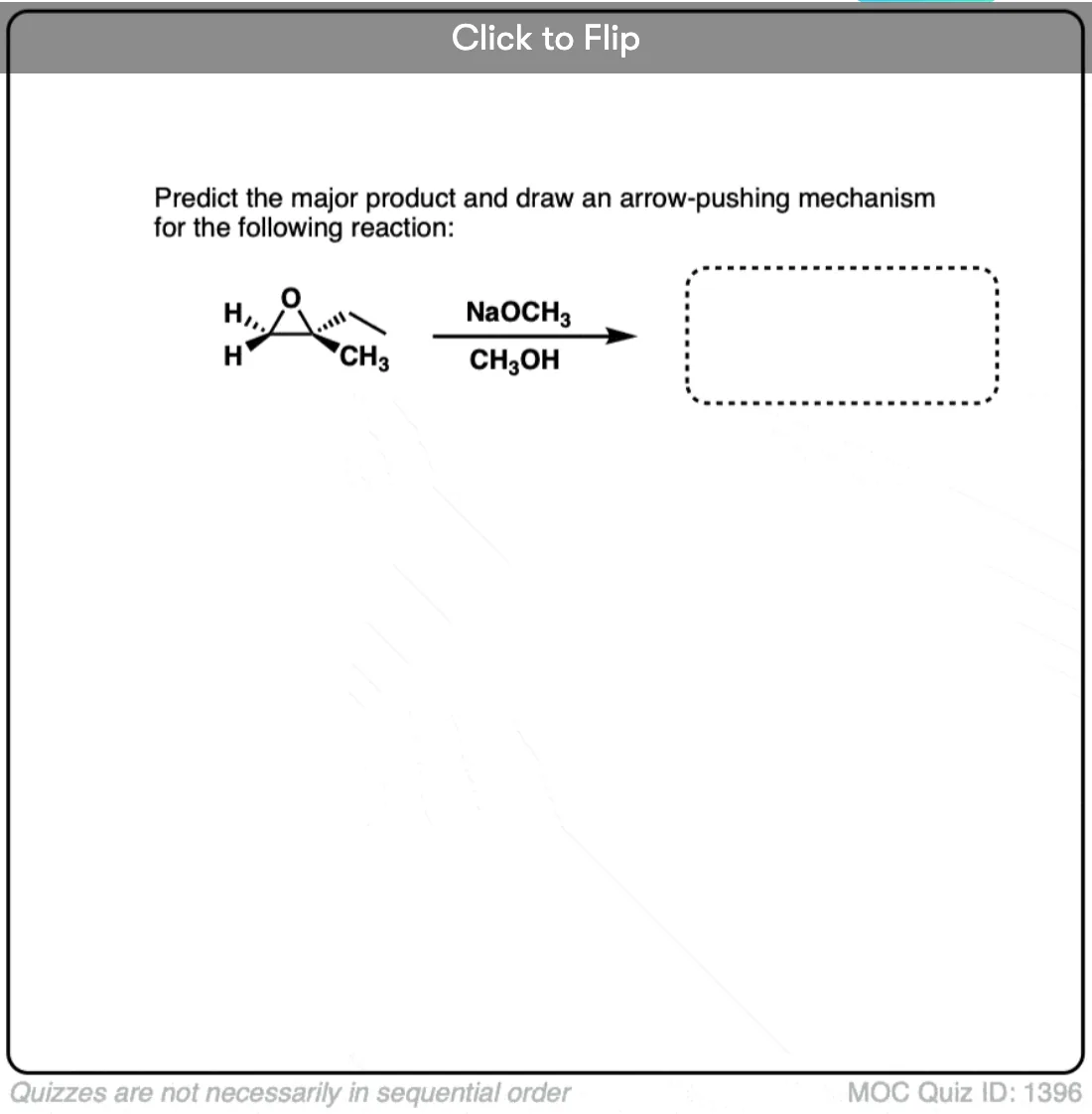
Become a MOC member to see the clickable quiz with answers on the back.
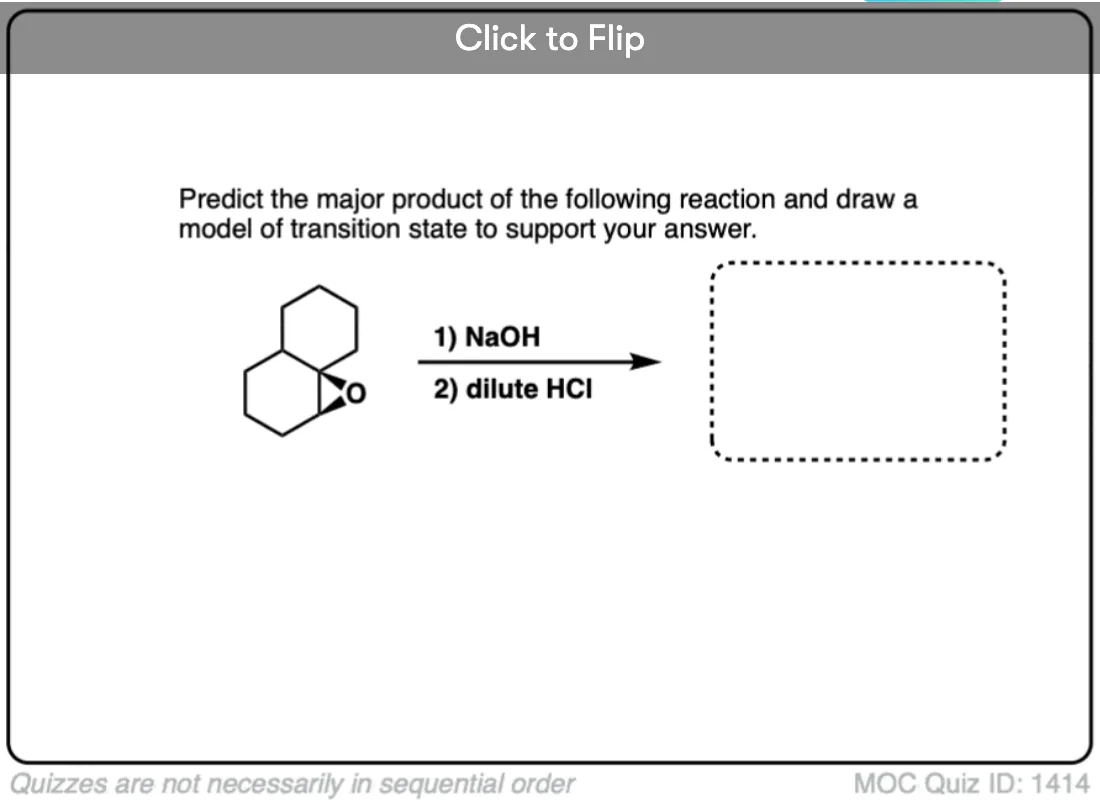
Become a MOC member to see the clickable quiz with answers on the back.
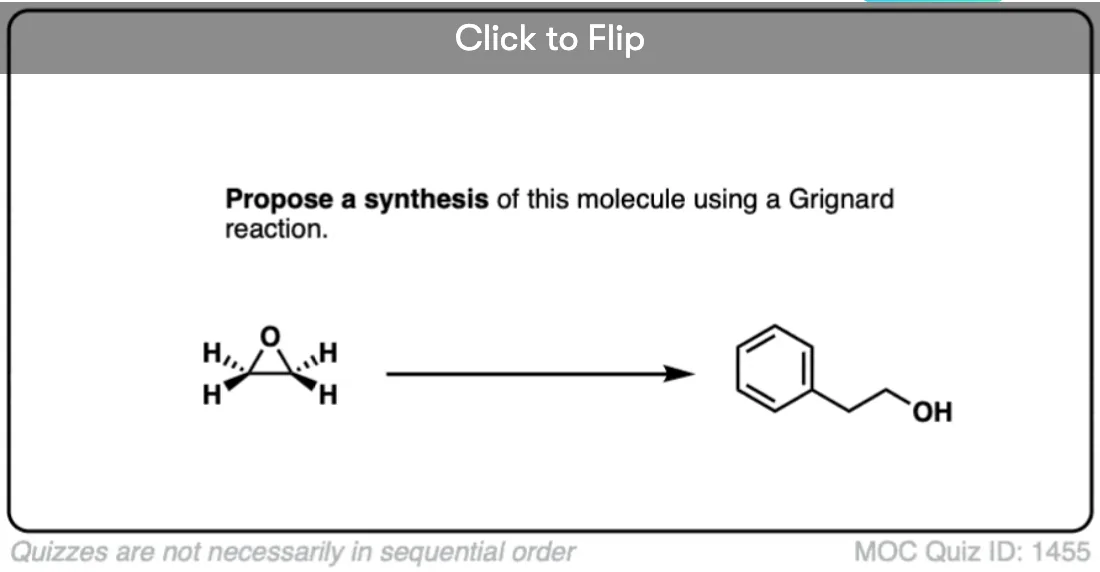
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- —The velocities of combination of sodium derivatives of phenols with olefine oxides
David Runciman Boyd and Ernest Robert Marle
J. Chem. Soc., Trans., 1914, 105, 2117-2139
DOI: 10.1039/CT9140502117
This and the accompanying paper are early examples of ring-opening of epoxides with alkoxides. - —The velocities of combination of sodium derivatives of phenols with olefine oxides. Part II
David Runciman Boyd and Miss Doris Feltham Thomas
J. Chem. Soc., Trans., 1919, 115, 1239-1243
DOI: 10.1039/CT9191501239 - CYCLOHEXENE IMINE
Iain D. G. Watson, Nicholas Afagh, and Andrei K. Yudin
Org. Synth. 2010, 87, 161
DOI: 10.15227/orgsyn.087.0161
The first reaction in this procedure from Organic Syntheses is the ring-opening of cyclohexene oxide with sodium azide. As Prof. Yudin shows, this reaction is a useful precursor for the synthesis of N-H aziridines, the nitrogen analog of epoxides. - The Reaction of Dimethylmagnesium and of Diethylmagnesium with Cyclohexene Oxide
Paul D. Bartlett and C. Manly BerryJournal of the American Chemical Society 1934 56 (12), 2683-2685DOI: 10.1021/ja01327a045This is an early example of ring-opening of epoxides with Grignard reagents.
- The stereochemical course of epoxide ring opening by allylic grignard reagents
Hugh Felkin, Georges Roussi
Tetrahedron Lett. 1965, 6 (46), 4153-4159
DOI: 10.1016/S0040-4039(01)99581-4
This is a more advanced (and perhaps less relevant) paper, attempting to resolve whether allylic Gringard reagents go through an SN2’ reaction. This could be resolved easily with isotopic labeling and 13C NMR. - Acid- and base-catalyzed ring-opening reactions or a sterically hindered epoxide
Herbert Mayr, Rainer Koschinsky, Elfriede Will, and Englbert Bauml
The Journal of Organic Chemistry 1987, 52 (7), 1342-1344
DOI: 1021/jo00383a033
A nice paper describing the different products obtained when opening a hindered epoxide under acidic vs. basic conditions. Prof. Mayr (LMU, Germany) is well known for his extremely rigorous work in physical organic chemistry, where he has developed scales for quantifying electrophilicity and nucleophilicity.
what is the epoxides ring opening with aluminum butoxide?
How does aluminum butoxide catalyze this reaction?
By aluminum butoxide, could you be more specific? Do you have an exact example you are thinking of, or are you referring to one or more of the examples in the post.
Hi, what would happen if an epoxide is reacted with NaBH4 in the presence of EtOH, would the NaBH4 act as a nucleophile and then the EtOH would attatch? Thanks
Generally NaBH4 is not a strong enough nucleophile to react with epoxides. You need a counter-ion that is more strongly coordinating to oxygen. LiAlH4 will work.
Would NaBr work in basic conditions?
Generally not. Even though it would release ring strain, you’d be going from a very weak base (Br-) to a much stronger base (O- ) if bromide ion opened the epoxide. Furthermore, even if it did, the reverse reaction would easily occur – recall that epoxides are formed from the closure of deprotonated halohydrins!
https://www.masterorganicchemistry.com/2015/01/26/epoxides-the-outlier-of-the-ether-family/
You really need the presence of an acid to stop the reaction at the halohydrin stage.
When using a strong base in the presence of an epoxide such as the one in the example presented would it be possible for an E2 reaction to occur as opposed to an Sn2? Couldn’t an alkoxide deprotonate a carbon adjacent to the epoxide, kicking off the electrons to the oxygen and forming a pi-bond?
That can indeed happen! However you tend to need a very bulky amide base for that to occur. The work of Hodgson is important in this regard, see here: https://onlinelibrary.wiley.com/doi/10.1002/0471264180.or029.03
THANK U FOR THIS BEAUTIFUL WEBSITE honestly this is a life saviour
If it’s useful to you – great!
Excellent work sir. I want to ask that LiAlH4 in the presence of AlCl3 , will it be able to break the epoxide ?(as we don’t have bronsted protonating source for the last step! )
Is LiAlH4 even compatible with AlCl3 ? I’ve never seen those two reagents used together.
Hi nicely explained. I have one query what if i want to attach a cysteine to glycidyloxypropyltriemthoxysilane using just sodium hydroxide as the solvent ? will it work?
For base-promoted openings of epoxides with other nucleophiles I recommend you read Myers’ handout. http://hwpi.harvard.edu/files/myers/files/25-transformations_of_23-epoxy_alcohols.pdf
Hi James, thanks for your post! I was wondering if you could help me solve this problem: I am a rookie in synthetic chemistry and I was trying to get rid of some hemiacetals in my epoxy product. I tried hydrolysis in acid solution. It turned out the epoxide hydrolyzed as well. Would you suggest anything that could help?
Hemiacetals should go away by themselves unless you have a very electronegative group adjacent to the carbonyl or if you are forming a cyclic hemiacetal. This doesn’t answer your question but hydrolysis of an acetal in the presence of an epoxide is probably not going to work. You might need to reconfigure your synthetic scheme, perhaps protecting the alcohol so that it doesn’t form the hemiacetal in the first place.
How would you open an epoxide with NaOH that has two chirality centers that are trans?
I’m not certain how to answer your question without a diagram. If you’re looking for selectivity between opening two epoxides, that will be difficult to achieve with NaOH unless there is a clear steric or electronic preference favoring one attack versus the other.
What about reactions of epoxides like 1,2 epoxy cyclohexane with PCl5, say ?? How to approach them, plz do tell..
That’s not really the opening of an epoxide with a base. There, you’re trying to coordinate the epoxide oxygen to the electrophilic phosphorus, forming a transient ion pair that will release chloride ion and open the epoxide. Kind of beyond the scope of what we deal with here.
Hi,
I have a product from a oxido-reductase enzyme that I suspect has an epoxide group, rather than another type of oxidation. I have done NMR and mass spectrometry, both fit with the epoxide hypothesis, but I would really like to use another method to confirm. This webpage has been helpful. What I would like to do is a methanolysis reaction. My plan is to dissolve the compound after lyophilizing in dry methanol. Then prepare a solution of 1 N H2SO4 in dry methanol, and add enough to result in a final concentration of 0.2 N H2SO4. That should label the epoxide with a OCH3 group. According to the paper below it should be done after 1 h at room temperature.
I can then analyse the result by GC-MS, and/or NMR.
As I am not a hard-core organic chemist, I want to know if I am overlooking something.
Hello,
I’m a mechanical engineer and my chemistry knowledge is poor, so sorry in advance if my question is silly. I’m conducting an experiment and attach some polymer chains with amine groups to a surface with epoxide group without doing anything special and just in potassium phosphate buffer (PH 7.4). Now imagine, after a point, I want to disable the epoxide groups and stop any reaction between my polymer chains and the surface (perhaps by closing the epoxide ring ??) while my polymer chains are still there. So my question is if it’s possible and how?
Thanks
thanks
Actually James, my doubt is how would the oxirane ring open with LAH and AlCl3 ( electrophile).
Typo in “The Role of Solvent” first example… The first equation should have NaOH and not NaOR.
This is probably a stupid question. From my understanding, for an epoxide, the more substituted carbon would bear more positive charge. Why wouldn’t the nucleophile attack the more substituted carbon instead? Is it something has to do with steric hindrance?
Hi – not a stupid question at all. You are right that the more substituted carbon would bear more positive charge. However, when we have a relatively strong nucleophile (such as RO- , HO-, Grignard reagents, or LiAlH4) what turns out to be more important is steric hindrance.
Think of it like an SN2 reaction, where the least substituted carbon will react most quickly.
When a weaker nucleophile is involved (such as H2O) we need to add an acid catalyst. In this case, the reaction will primarily occur on the carbon best able to stabilize positive charge. In this respect it is somewhat like an SN1 reaction, although it still occurs with inversion of configuration in this case.
Hope this helps! James
Would the nucleophile be RO- or OH- ?
In which case? It could be either, depending on conditions.
Excellent post!
I’ve a doubt though. What if I tried to add a tertiary alcohol to an asymmetrical epoxide. Since they are the most weakly acidic alcohols, would they follow a basic mechanism (sn2)? would the outcome depend on the solvent or medium used? Would they even react in the first place? If they did, would they prefer to undergo a C-O bond cleavage thereby forming a stable carbocation? I’m really confused on this, it would be a lot of help if I could get this cleared…
Thanks
This post covers opening epoxides with base. Adding a neutral tertiary alcohol does not fall into this category. You’d need to form the conjugate base of the tertiary alcohol – say, for example, sodium t-butoxide. Sodium t-butoxide can act as a nucleophile to attack the least substituted position of an epoxide and open it, just like other alkoxides do.
Carbocations are not relevant to this discussion because they don’t form under these basic conditions.