Organometallics
Gilman Reagents (Organocuprates): What They’re Used For
Last updated: May 28th, 2026 |
So What Are Gilman Reagents Used For, Anyway?
Last time we talked about how to make Gilman reagents (organocuprates). In this post, we’ll talk about what they’re actually used for.
Gilman reagents have some useful contrasts to Grignard reagents.
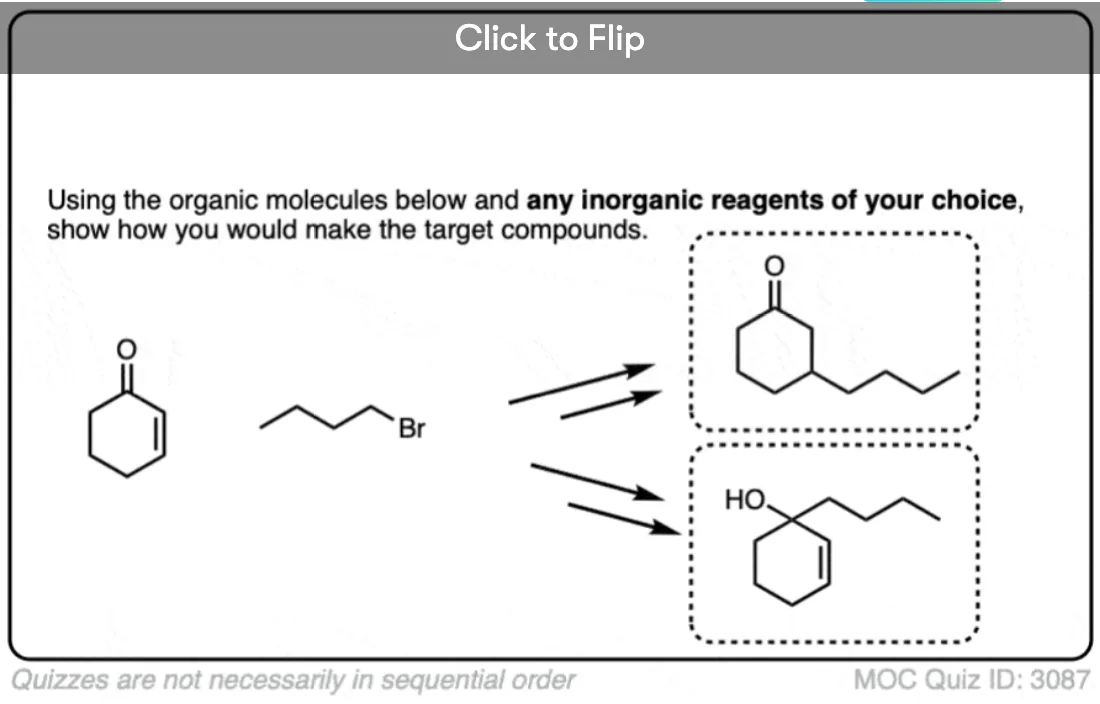
- First of all, they can be used to perform conjugate addition reactions (“1,4-addition”) on alpha,-beta unsaturated ketones. In contrast, Grignard reagents tend to only add to the carbonyl carbon.
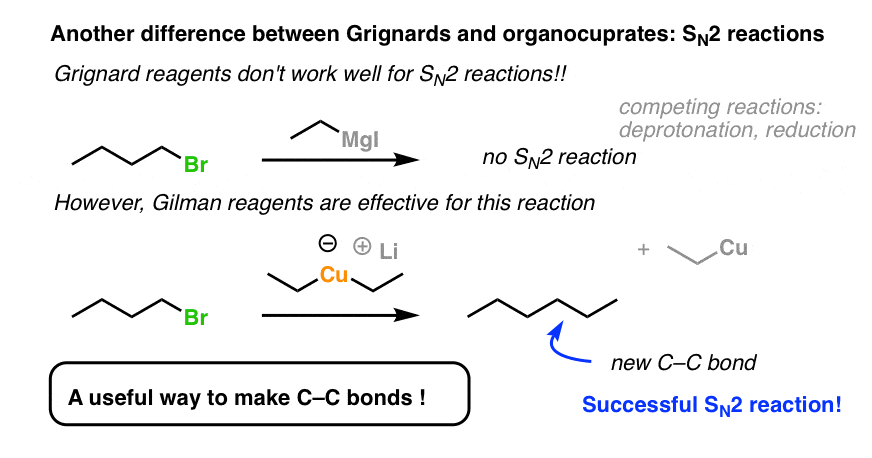
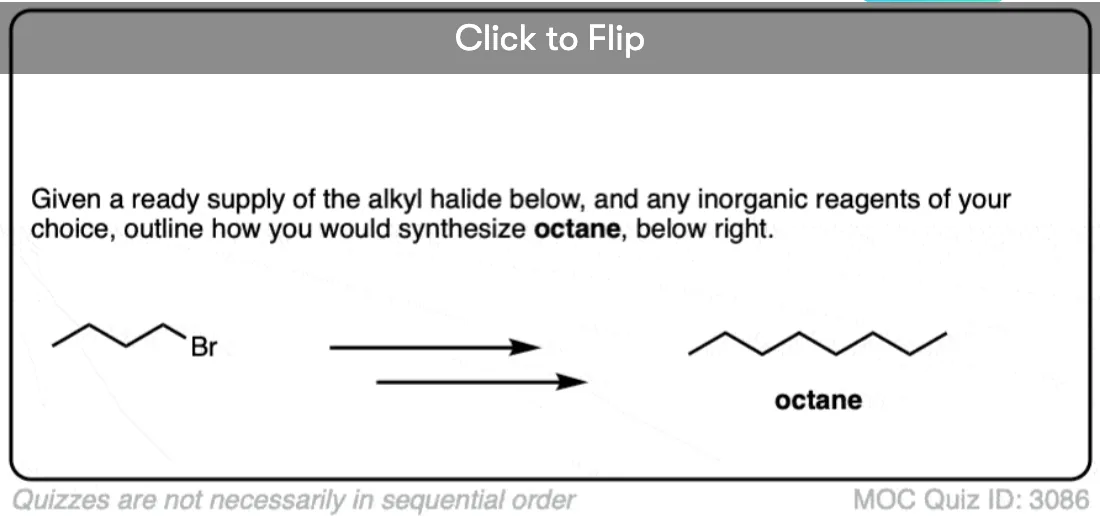
- Secondly, Gilman reagents are useful nucleophiles for SN2 reactions, forming C-C bonds with primary alkyl halides and sulfonates (e.g. tosylates, mesylates)
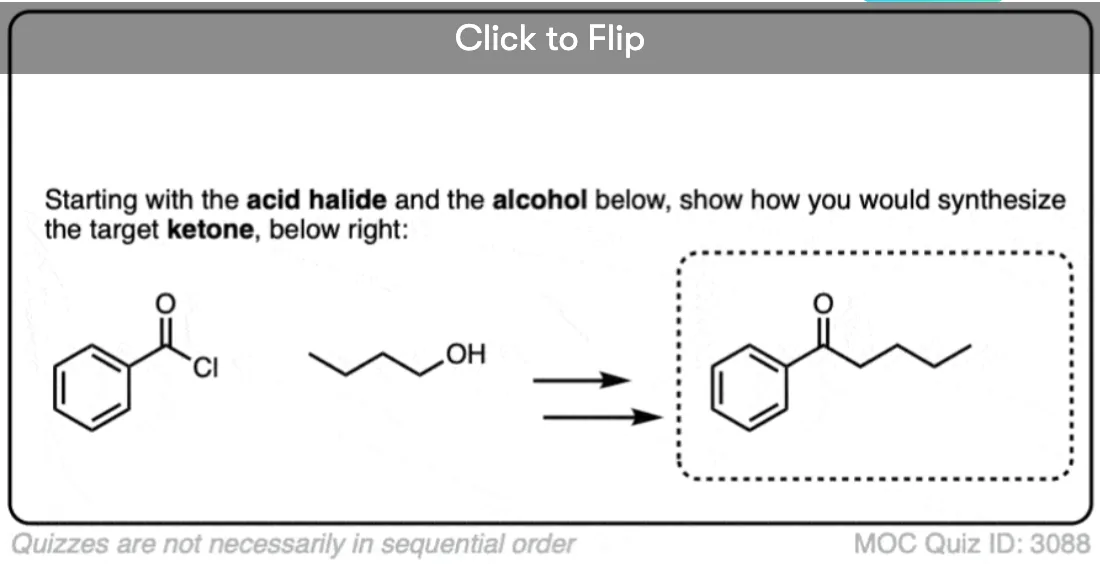
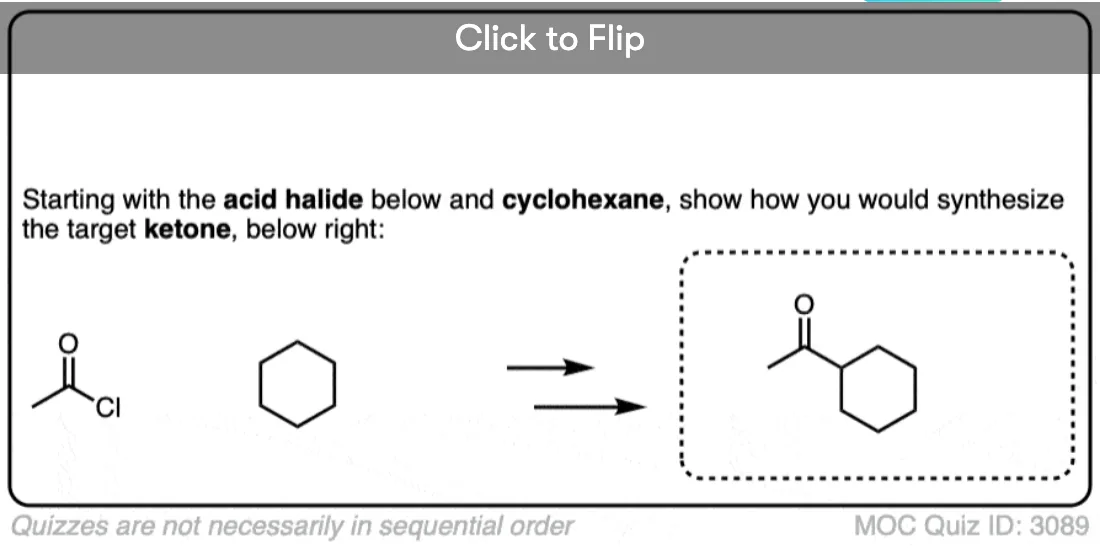
- Third, Gilman reagents will convert acid halides to ketones through nucleophilic acyl substitution. Grignard reagents will add twice to acid halides to give tertiary alcohols.
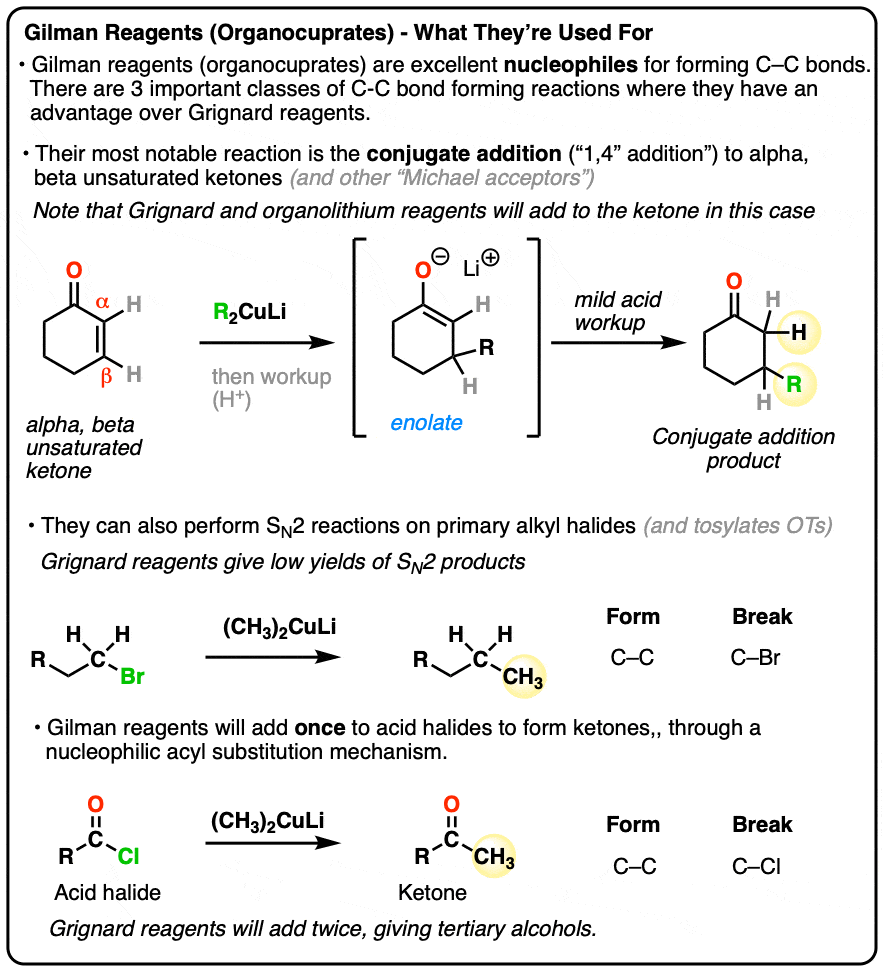
Table of Contents
- Gilman Reagents vs Grignard Reagents
- Conjugate Addition: A Key Reaction of Gilman Reagents
- How do you know whether “normal” or “conjugate” addition will occur?
- Conjugate Addition Mechanism
- Gilman Reagents Are Great Nucleophiles For SN2 Reactions
- Conclusion: Gilman Reagents
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Gilman Reagents vs. Grignard Reagents
As I hinted at last time, Gilman reagents provide an interesting contrast with Grignard and organolithium reagents.
Remember all those examples of Grignard reagents adding to aldehydes, ketones, and esters? Well, Gilman reagents don’t generally do that (they will add to acid chlorides, but I digress)
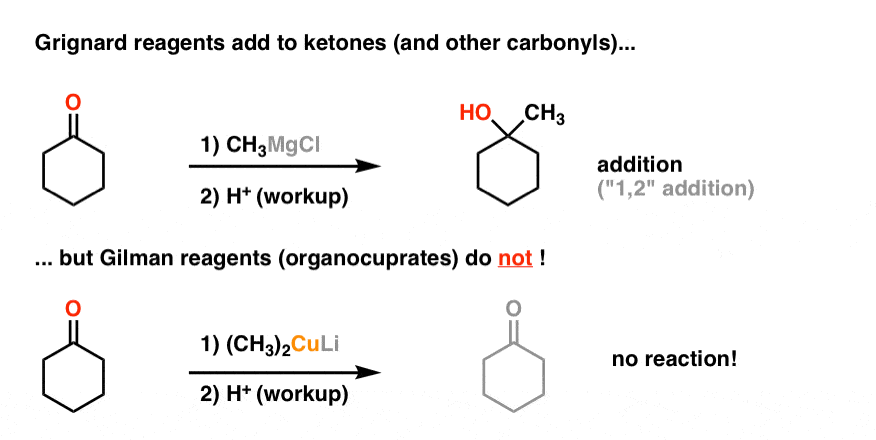
You might find yourself wondering, “So what?”. Why do we have to bother learning about these things if they’re not even very reactive?
Well, dear reader, let me fill you in on a second example. We’ll change one small thing, and everything changes.
Let’s put a double bond next to the ketone and run the reaction again.
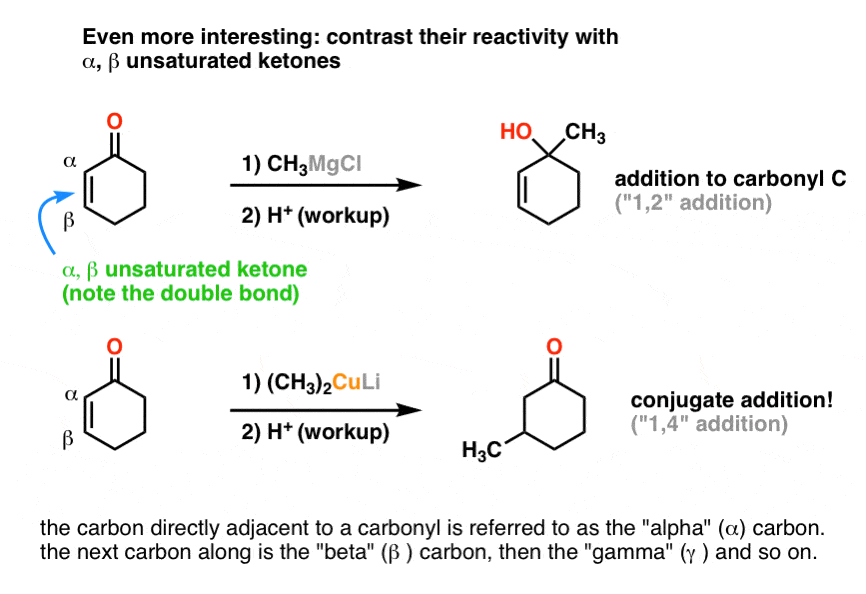
Whoa. What just happened there?
2. Conjugate Addition: A Key Reaction of Gilman Reagents
The Grignard reagent reacted the same way (to the carbonyl) but for the organocuprate, see that we’ve broken the C-C π bond (double bond) and formed a new C-C bond ?
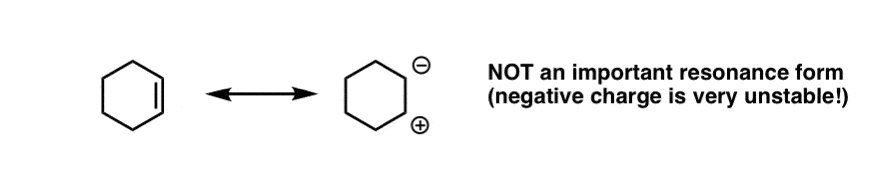
If that seems strange to you, it should! Isolated alkenes, such as cyclohexene, for instance, don’t do this reaction.
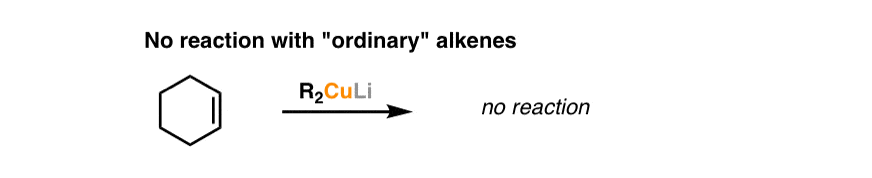
So there must be something important about the fact that the alkene is next to a carbonyl. Why might that be important?
Look at the resonance forms and you will see a clue.
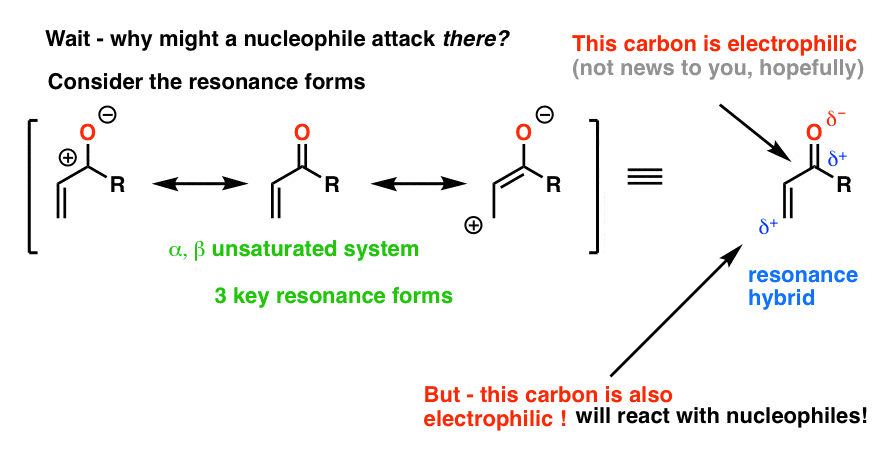
There is an important resonance form where the carbon two carbons away from the carbonyl carbon (we call this the “beta” (β) position) bears a positive charge. In the resonance hybrid, therefore, that carbon bears some partial positive charge.
In other words, that carbon is electrophilic. It can react with nucleophiles! (Such as organocuprates).
Contrast that with ordinary alkenes, where the resonance form with a carbon bearing a negative charge is not an important resonance form. The fact that the charge is placed on oxygen in the resonance form of an α,β unsaturated system is the key to the relative importance of that resonance form. [Compare the basicity of alkoxides (RO-) and alkyllithiums (R-) and that will give you an idea of their relative stabilities].
[For more, see article – The Michael Addition and Conjugate Addition]
3. Wait: How Do You Know Whether “Normal” Addition or “Conjugate” Addition Will Occur?
This brings up an important question: How do you know whether a nucleophile will attack at the carbonyl carbon (sometimes called “1,2 addition” in our jargon) or at the beta position (“1,4 addition” or “conjugate addition”).
Simple question. Very difficult to answer succinctly, and too big a topic for this post.
Short answer: memorize that Grignards add to carbonyls, while organocuprates do conjugate addition.
(ducks while people throw things at the screen)
I only say “memorize” because in order to adequately understand this phenomenon, we’d have to go into some molecular orbital theory to get at the key concept of “Hard Soft Acid Base (HSAB) Theory“, and at this point, we’re not going to cover it.
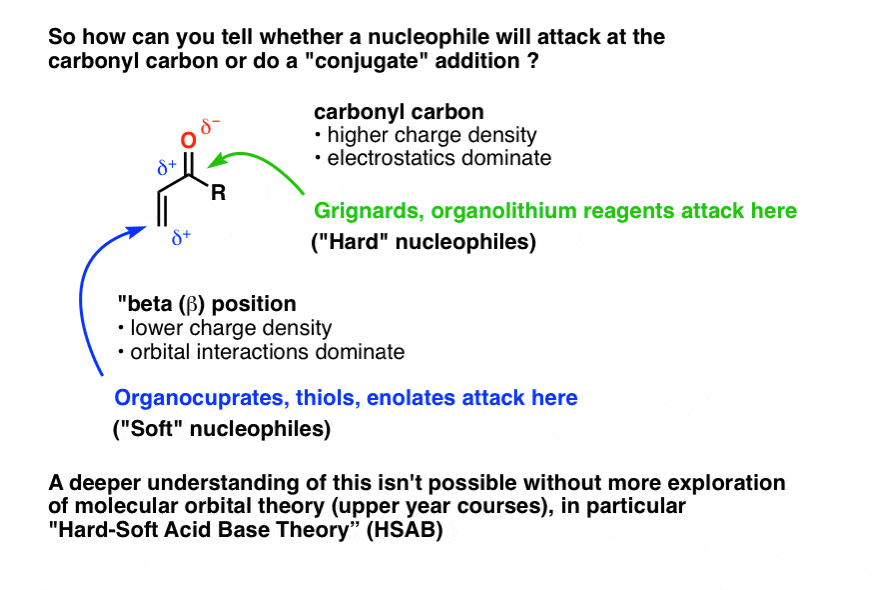
What about the mechanism of the reaction? Now that’s something we can cover.
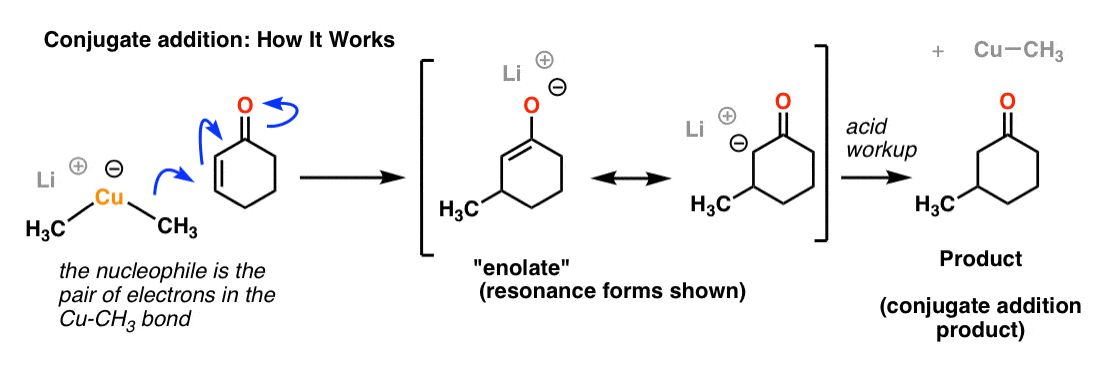
4. Conjugate Addition Mechanism
In the first step, the nucleophile (which is the pair of electrons in the Cu-CH3 bond, NOT the negative charge on copper!) forms a bond with the beta position of the ketone. The C-C π bond breaks, forming a negative charge on the alpha carbon. We can actually go further and draw a resonance form where we form a new C-C π bond and place the negative charge on oxygen. You’ll see this chemical species a lot in subsequent chapters – it’s called an enolate, and it’s very important. (See article: Enolates)
For now, the key takeaway is that the negative charge is on the oxygen, which is considerably more stable (less basic) than having a negative charge on carbon.
Adding acid will protonate the enolate (which is a base, after all) and result in our final product.
5. Gilman Reagents Are Excellent Nucleophiles For SN2 Reactions
But wait! Conjugate additions aren’t all organocuprates can do.
If you have a keen eye for the other posts in this series, you might have noticed that SN2 reactions were conspicuously absent on the list of reactions that Grignards are useful for. [Why’s that? Great question. The short answer is, we observe that a lot of side reactions tend to occur, like deprotonation and reduction. Using a Grignard reagent to do an SN2 reaction to form a C-C bond is generally not a great process].
However, once we switch to a Gilman reagent, the SN2 works well. This is a handy reaction to have in the toolbox for forming C-C bonds.
That about sums it up for Gilman reagents right now. We could add that they can be used to make ketones from acid halides , I hesitate to put that in at the moment, given that this post is long enough as it is. [The general mechanism is covered in this article on Nucleophilic Acyl Substitution. ]
6. Conclusion: Gilman Reagents
Gilman reagents (organocuprates) perform two reactions that Grignard reagents (and organolithiums) do not:
• They perform conjugate additions to α,β unsaturated ketones.
• They are effective nucleophiles for SN2 reactions.
In the next post, we’ll change to a more controversial topic – transition metal catalyzed reactions.
Next Post: Heck, Suzuki, and Olefin Metathesis Reactions
Notes
Note 1. Yet More Information, Because This Hasn’t Been A Long Enough Blog Post Already
Here’s a quiz for you. What would be the better nucleophile? An organocopper reagent or an organocuprate reagent?
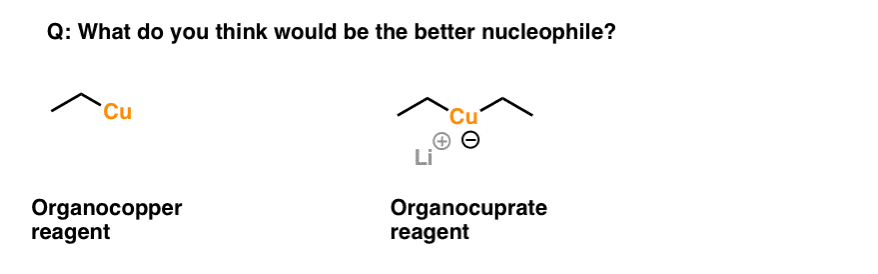
When thinking about this, analyze the leaving group. Therein lies the clue.
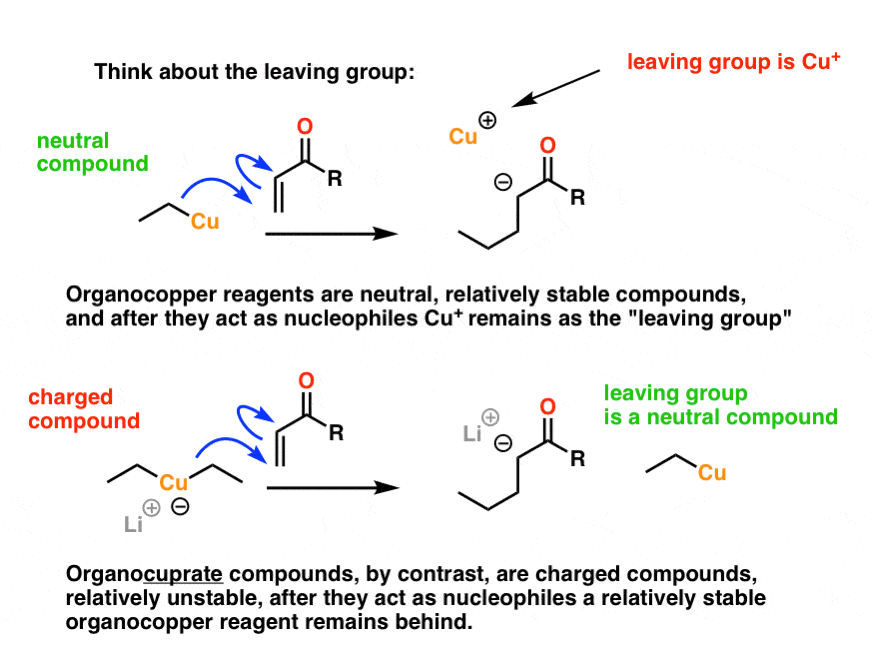
When organocopper reagents act as nucleophiles, they go from neutral, relatively stable compounds to ionic Cu+. Although this is a sweeping generalization, charge minimization is generally associated with greater stability in organic chemistry. We’re going from a neutral compound (organocopper) to a charged ion (Cu+). [I could also add that Cu+, being a soft ion, is not very effective in binding to O-, but that’s a pretty advanced point].
Compare that to organocuprates. There, we’re starting as a relatively unstable charged species, and our final copper product is the neutral organocopper reagent. This is definitely downhill in terms of stability. It’s reasonable to expect that the organocuprate will be more reactive, and hence be a better nucleophile.
The same principle can be used to explain why NaBH4 is a better reducing agent than BH3, and LiAlH4 is a better reducing agent than AlH3.
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
Gilman reagents, or Lithium organocuprates (R2CuLi), are useful nucleophiles in organic synthesis. These have a different reactivity from Grignard reagents and organolithiums, since Gilman reagents are softer.
Gilman reagent formation:
- The Preparation of Methylcopper and some Observations on the Decomposition of Organocopper Compounds
Henry Gilman, Reuben G. Jones, and L. A. Woods
The Journal of Organic Chemistry 1952 17 (12), 1630-1634
DOI:1021/jo50012a009
One of the first papers by Prof. Henry Gilman (U. of Iowa) on ‘Gilman Reagents’ – diorganocopper compounds. In this case he describes the preparation of dimethylcopper.
Conjugate addition of Gilman reagents: - Chemistry of carbanions. XVII. Addition of methyl organometallic reagents to cyclohexenone derivatives
Herbert O. House and William F. Fischer Jr.
The Journal of Organic Chemistry 1968 33 (3), 949-956
DOI: 1021/jo01267a004 - Conjugate Addition Reactions of Organocopper Reagents
Gary H. Posner
React. 1972, 19, 1-114
DOI: 10.1002/0471264180.or019.01
Organic Reactions is a series of reviews maintained by the ACS Division of Organic Chemistry, and this review by Prof. Posner, a specialist in cuprate chemistry, covers everything you’d want to know about conjugate additions with copper, as of 1972. Detailed experimental procedures are given towards the end.Ketones can be synthesized by the addition of Gilman reagents to acyl halides: - Methyl and -alkyl ketones from carboxylic acid chlorides and organocopper reagents
G. H. Posner, C. E. Whitten
Tetrahedron. Lett. 1970, 11 (53), 4647-4650
DOI: 10.1016/S0040-4039(00)89398-3
One of the original papers on this reaction. Prof. Posner has done a lot of work studying organocopper chemistry in his career. - Organocopper chemistry. Halo-, cyano-, and carbonyl-substituted ketones from the corresponding acyl chlorides and organocopper reagents
Gary H. Posner, Charles E. Whitten, and Paul McFarland
Journal of the American Chemical Society 1972, 94 (14), 5106-5108
DOI: 1021/ja00769a066
An extension of the previous paper, expanding the substrate scope of the reaction.The SN2 reaction of Gilman reagents with alkyl halides is also known as the Corey-Posner-Whitesides-House reaction: - Selective formation of carbon-carbon bonds between unlike groups using organocopper reagents
Elias J. Corey and Gary H. Posner
Journal of the American Chemical Society 1967, 89 (15), 3911-3912
DOI: 1021/ja00991a049
Nobel Laureate Prof. E. J. Corey first describes the reaction of Gilman reagents from CH3Li ((CH3)2CuLi) with alkyl and aryl halides to form new C-C bonds. - Carbon-carbon bond formation by selective coupling of n-alkylcopper reagents with organic halides
Elias J. Corey and Gary H. Posner
Journal of the American Chemical Society 1968, 90 (20), 5615-5616
DOI: 1021/ja01022a058
Prof. Corey extends this method to n-butyl and ethyl (from n-BuLi and EtLi) as well. - Reaction of lithium dialkyl- and diarylcuprates with organic halides
George M. Whitesides, William F. Fischer Jr., Joseph San Filippo Jr., Robert W. Bashe, and Herbert O. House
Journal of the American Chemical Society 1969, 91 (17), 4871-4882
DOI: 1021/ja01045a049
Classic paper by Prof. G. M. Whitesides (MIT, now Harvard) on the coupling of Gilman reagents (R2CuLi) with aryl iodides. This reaction is exhaustively studied, and features a thorough mechanistic investigation, which is why his name is included in the reaction.
What happens if there is already an R group on the beta carbon?
Still works! Plenty of examples in this volume: https://onlinelibrary.wiley.com/doi/abs/10.1002/0471264180.or041.02
Beyond acid chlorides, will Gilman reagents add to alkylsulfonyl chlorides like MsCl? I don’t have access to SciFinder or Reaxys and am having a hard time checking to see whether this would be a valid reaction.
Primary alkyl sulfonates work well. SN2 reactions between Gilman reagents and secondary alkyl sulfonates don’t work well. For that matter there are not many good examples of organocuprates reacting with secondary alkyl halides or tosylates.
https://pubs.acs.org/doi/pdf/10.1021/ja00804a039
Hey there, First of all amazing content up there. I am Tirth from India preparing for JEE. I have this doubt in the comparision of leaving group strength which you said. If organocuprate is going from more stability to less stability, it will not tend to perfom that reaction. Then shouldn’t it be less nucleophilic?
I don’t think I understand your question. What reaction are you thinking of doing with organocuprates?
the DOI is correct but the hyperlink is wrong
Fixed, thanks
reference 4 links to the wrong URL, here is the correct one:
https://www.sciencedirect.com/science/article/abs/pii/S0040403900893983
Thank you!
Which one is more reactive Gilman or organolithium? And why??
Reactive towards what, specifically?
Hi sir, can gilmann’s reagent react with all acid derivatives..what about R2Cd ‘s reactivity with them?
Thank you sir..
Gilman reagents can react with acid halides, anhydrides, and (sometimes) thiol esters. Everything else forget about it. Cadmium I believe to be even less reactive. See: https://pubs.acs.org/doi/pdf/10.1021/jo00134a015
Hi James,
Can Gilman reagent act as a nucleophile on reaction with vinyl halides to produce higher alkenes ?
Sort of. For example lithium dimethylcuprate will react with vinyl halides and you can substitute halogens with methyl groups. I’m not sure how well it works with other Gilman reagents.
This is not an SN2 reaction, it’s insertion of Cu into the C-(halogen) bond to get a transient copper(III) species which undergoes reductive elimination to give the substituted alkene.
What you are describing is usually best achieved through a cross-coupling reaction (e.g. check out the Suzuki, Negishi, and Kumada reactions)
Is negative charge on Cu?? Or is Gilman reagent is a co ordination complex???
There is a formal negative charge on Cu, but saying as carbon is more electronegative than Cu, the actual negative charge resides mostly on the carbon. Hence, this is a source of nucleophilic carbon.
Hello!
Can (Br-CH2)2CuLi be used in conjugate addition? I am struggling with determining basicity of this R-group.
Thank You.
No, it wouldn’t be stable at all. The Br would react with the carbanion.
why would the grignard and organolithium reagents readily add to ketones, but diorganocopper lithium reagents do not?
That’s an excellent question. One clue is that Cu is much more electronegative than Li and Mg, so the Cu-C bond has more of a covalent character.
In the example where CH3MgCl reacts with cyclohexanone, won’t it pull out the acidic h at alpha carbon and form CH4?
Thankyou in advance!
You are describing the Grignard reagent acting as a base. This can occur in some cases, such as when a ketone is quite sterically hindered, but under normal conditions the addition reaction tends to be faster.
One subtle point about the deprotonation reaction is that the C-H bond is only reasonably acidic when it is lined up close to 90° relative to the C=O bond, which allows for stabilization of the resulting lone pair by resonance.
Is it the pi star orbital of C=O which stabilizes the lone pair?
Yes, it’s donation of the pair of electrons in the C-H bond into the pi* orbital on C.
I thought that organocuprates could add to an acyl halide and replace the halide with the R group?
Yes, they can. Thanks for pointing out the omission – need to update this.
Yes, they certainly can, and form a new ketone.
Greetings! In the second and third example schemes you have the Gilman reagent as R2CuBr. Is that different from R2CuLi? Why would a halogen be in that species? Thanks!
Fixed. Thanks!
Can the R in a organocuprate compound be any type of compound? Can for example (CH2NO2)2CuLi be used? Or will the NO2 part change or affect the reaction somehow?
Thank you so much for your time.
Hi Janna – cuprates are excellent choices when the R group is strongly basic, since the Cu helps to decrease their basicity. In the case of CH2NO2(-) the parent molecule (nitromethane) is already quite acidic and the conjugate base is quite accessible through deprotonation. You could probably just deprotonate nitromethane with a strong base (e.g. LDA) and see how that works!
Is HSAB theory enough to explain why in the case of the unconjugated cyclohexanone there is no addition at all? I understand why 1,4 addition is preferable to 1,2 addition, but for an electrophilic carbon, and a nucleophile that is basically a carbanion, even a “softened”, with no other viable attack sites, “no reaction” seems a little extreme?
Thanks again for your time.