Enols and Enolates
Reactions of Enols: Acid-Catalyzed Aldol, Halogenation, and Mannich Reactions
Last updated: May 29th, 2026 |
Reactions of Enols – Aldol Condensation, Halogenation, Mannich Reaction
In a previous post on keto-enol tautomerism, we described the keto and enol forms. In this post we explore the reactivity of the enol tautomer, which can act as a nucleophile in several important reactions.
In this post we’ll have lots to say about the role of acid catalysis in helping to promote keto-enol tautomerism, as well as more discussion on how acid accelerates reactions along the lines of “the conjugate acid is a better electrophile” and “the conjugate acid is a better leaving group“.
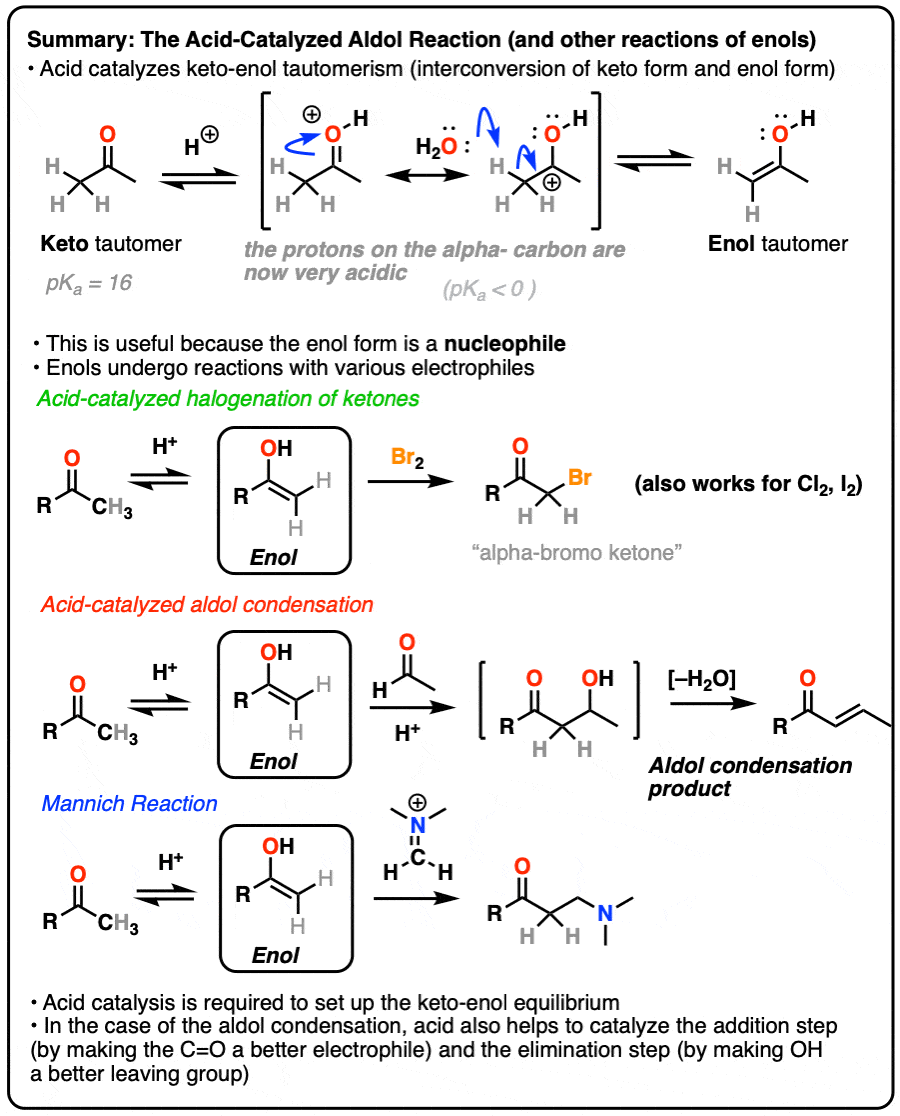
Table of Contents
- Reactions of Enols
- Reactions on the Alpha-Carbon: Epimerization
- How Is Keto-Enol Tautomerism Catalyzed By Acid?
- Bromination of Ketones
- The Acid-Catalyzed Aldol Reaction – And Its 3 Paths For Acid-Catalysis
- The Mannich Reaction
- Summary
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Reactions of Enols
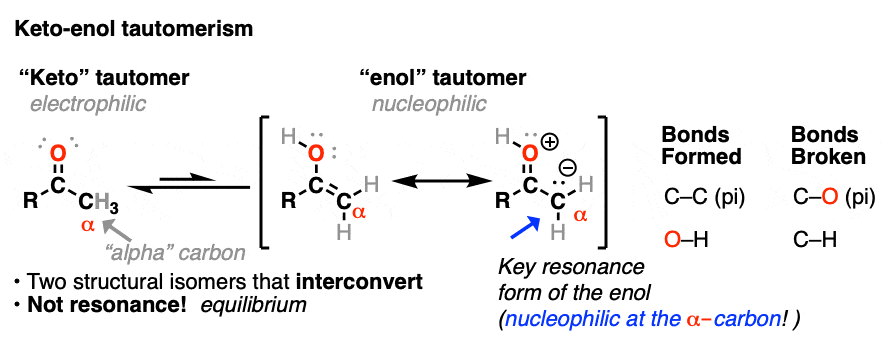
In a previous post we explored keto-enol tautomerism [See post: Keto-Enol Tautomerism] and described how the enol tautomer is the nucleophilic alter-ego of the electrophilic keto tautomer. In this article we’ll get into some more practical aspects, namely: what kinds of reactions do enols undergo?
Although not nearly as nucleophilic as enolates [Note 1], enols do play a key role in several important organic reactions.
In this article we’ll look at three important examples:
- Halogenation of ketones
- The acid-catalyzed aldol condensation.
- The Mannich reaction (which is a lot like the aldol except that the enol adds to a C-N pi bond instead of a C-O pi bond)
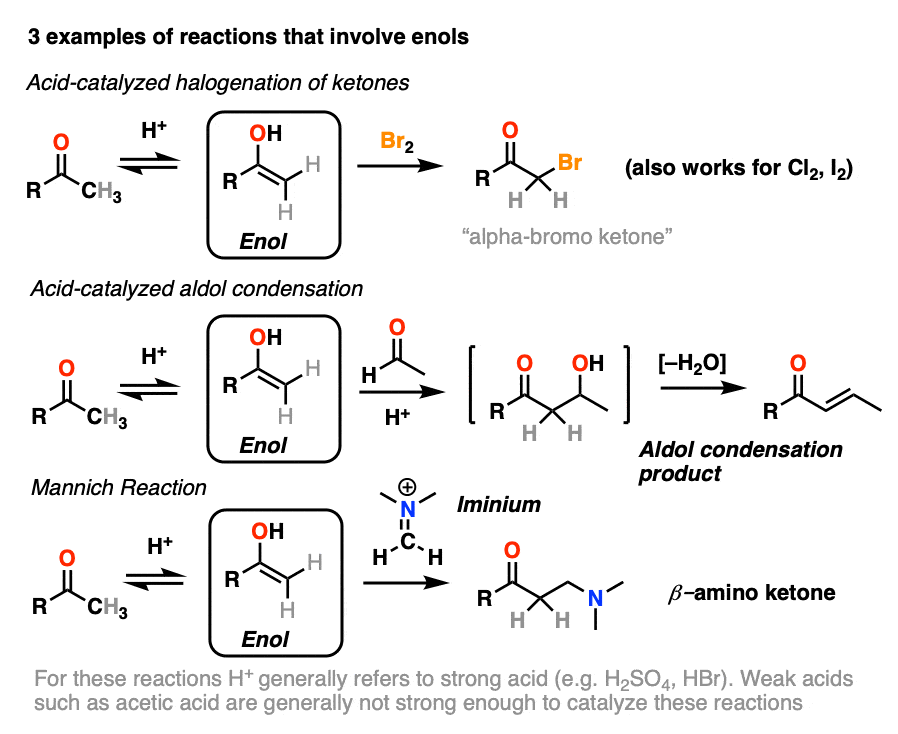
If you look closely at each of the three reactions above, you will note that each of them are catalyzed by acid.
Why is that? Let’s have a closer look.
2. Reactions On The Alpha Carbon: Epimerization
Let’s set the stage with a reaction that doesn’t really seem to accomplish all that much.
If you take a solution of an enantiomerically pure, optically active ketone like this one [(S)-2-methylcyclohexanone] add a few drops of strong acid and stir overnight, you’ll come back the next morning to find that its optical activity has completely disappeared. [Note 2 – acetic acid isn’t strong enough, but a few drops of HBr will do]
It’s still 2-methylcyclohexanone, but it’s become an equal mixture of the (R) and (S) enantiomers. A racemic mixture in other words. (See post: What’s a racemic mixture?)
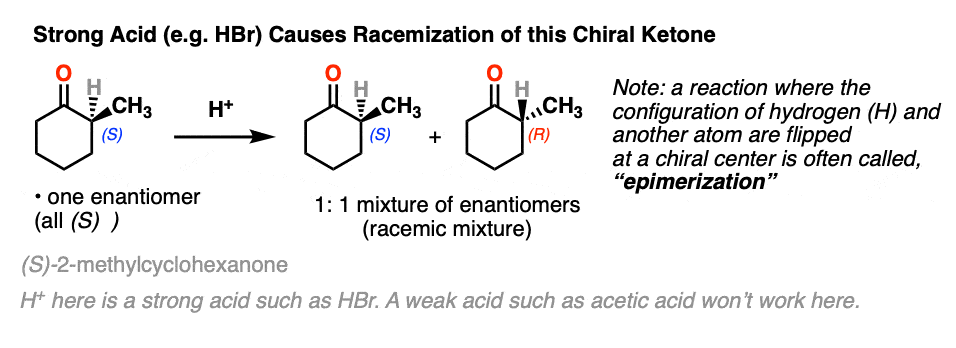
In contrast, if you take a very similar enantiomerically pure ketone (3-methyl cyclohexanone) and perform the exact same sequence, the optical activity will remain unchanged.
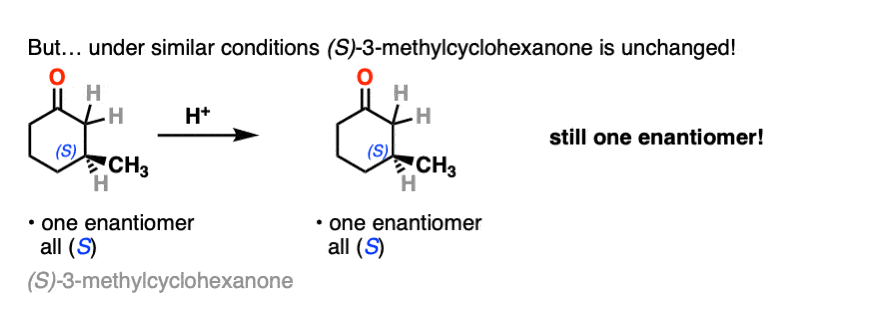
What’s going on here?
Both ketones can undergo tautomerism to give the enol form, where C-2 is trigonal planar (sp2 hybridized). However, the enol form of 2-methylcyclohexanone, no longer has a chiral center. The enol form is achiral.
When this flat, achiral enol is then protonated by acid to regenerate 2-methylcyclohexanone, it will do so with equal likelihood from the top and bottom face, resulting in an equal (i.e. racemic) mixture of the (R) and (S) enantiomers.
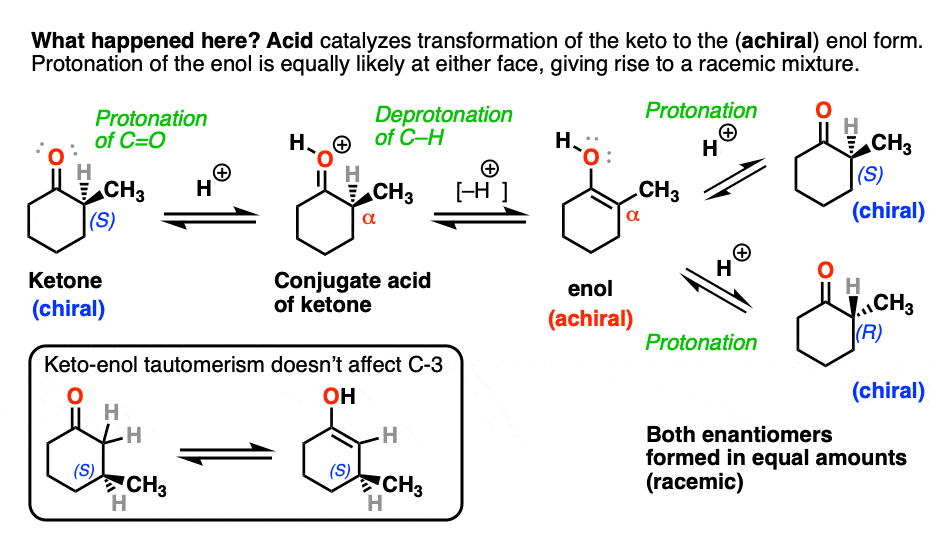
On the other hand, (S)-3-methylcyclohexanone retains its optical activity since its chiral center isn’t destroyed when the enol is formed.
A reaction where two substituents on a chiral center are interchanged (with one of them usually being H) is called epimerization.
Epimerization of the alpha-carbon of ketones and aldehydes can readily occur in the presence of acid and base. Carbons further down the chain are not affected since it is only the alpha carbon that is acidic.
3. How Exactly Is Keto-Enol Tautomerism Catalyzed By Acid?
Could the same experiment have been performed without acid? Yes – but it would have been much slower.
Acid helps to catalyze keto-enol tautomerism, allowing for rapid equilibrium between the keto and enol forms.
How specifically does acid help here?
If you go way back in time to Org 1, you might find a reaction buried deep in your memory banks called the E1 reaction. (See post: The E1 Reaction and Its Mechanism)
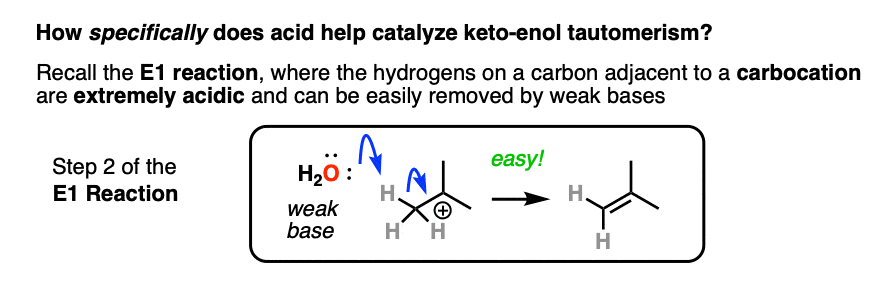
After the leaving group leaves, the next step in the E1 is deprotonation of the carbon next to the carbocation to give a new pi bond. The E1 requires only a weak base for this step (even water will do!) since the leaving group has already left and the carbon has an empty p-orbital available to form the new pi-bond. (Remember that the E2 requires a stronger base in order to get that leaving group to leave – See post: The E2 Mechanism).
The carbon of a protonated aldehyde or ketone is very similar to a carbocation. It has a significant resonance form where the formal charge is on carbon and the oxygen is neutral. [Note 3 ]
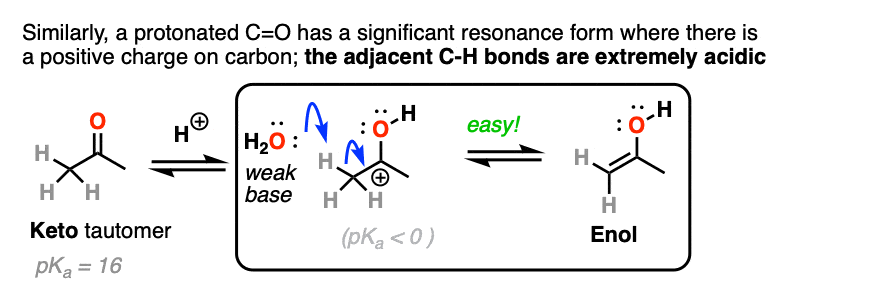
Deprotonation of the C-H next to the carbonyl (pKa <0 ) is therefore considerably easier than it would be in the case of the neutral aldehyde/ketone (pKa = 16)
TL: DR Adding acid makes it easier to deprotonate C-H and therefore makes formation of the enol much easier.
Acid also helps with the conversion of the enol back to the ketone, which involves (slow) protonation of the alpha-carbon followed by (fast) deprotonation of O-H.
For the acid-catalyzed conversion of enol to keto, hover here and an image will pop up or click on this link.
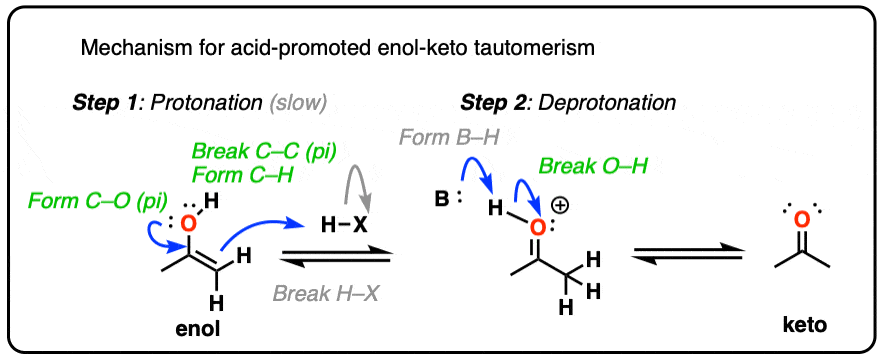{kind=link}
Since both the forward and backward reactions are accelerated, that’s what we mean when we say that acid “helps to establish the equilibrium”.
Now let’s look at some of the reactions of enols.
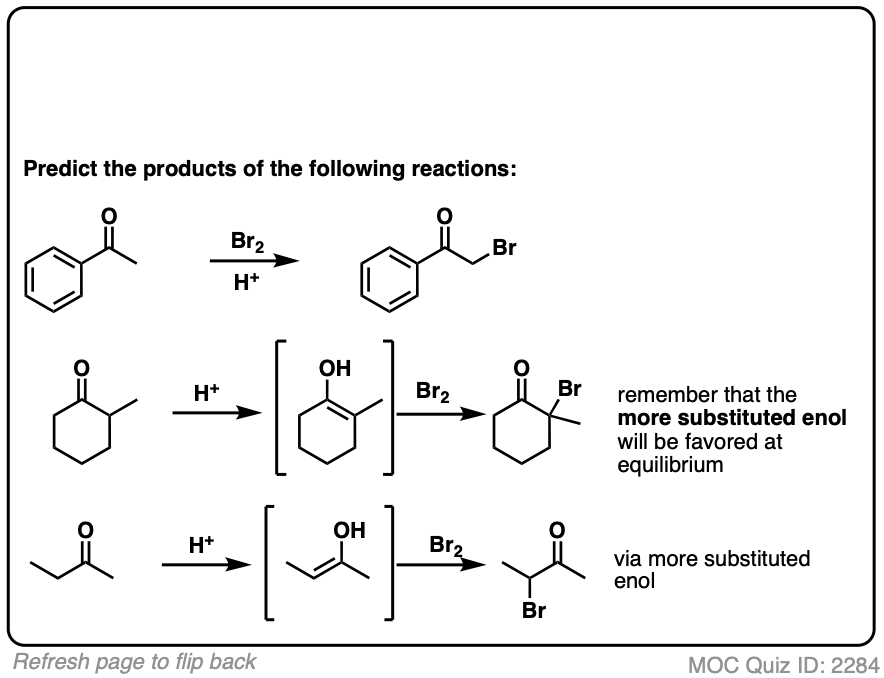
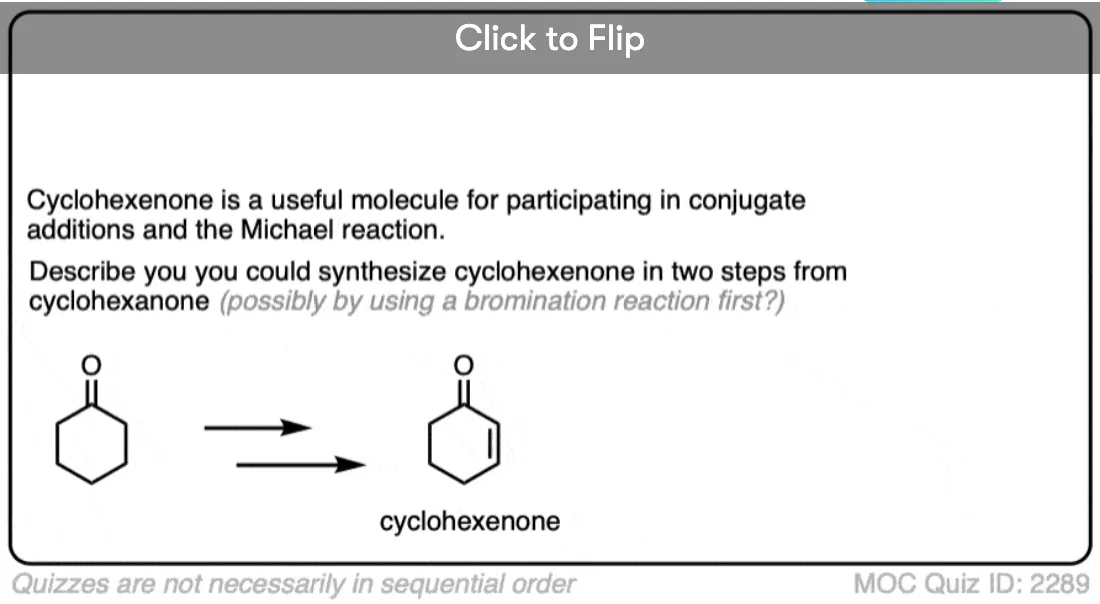
4. Bromination of Ketones
Enols are really just electron-rich alkenes, and will undergo many of the same reactions. You might recall that alkenes will readily attack halogens such as Br2 to give dihalides, for example. (See post: Bromination of alkenes)
If you add strong acid to a ketone, and mix in a halogen (Cl2, Br2, I2), you obtain a new alpha-halo ketone.
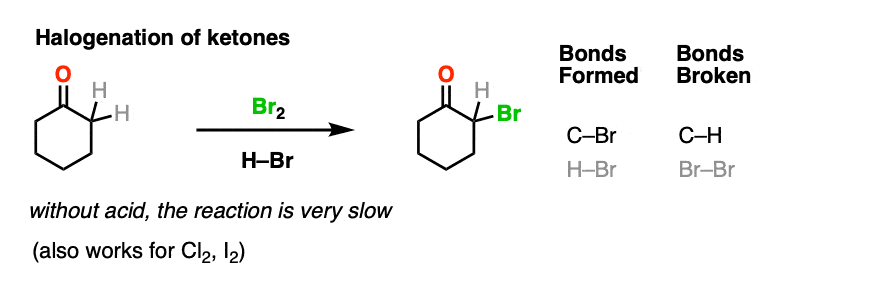
In the absence of acid, the reaction is “exceedingly slow”. [Note 4] Furthermore, careful measurements show that adding an excess of Br2 does not increase the reaction rate.
Knowing this, a mechanism that makes sense is a (slow) conversion of the ketone to its enol form, followed by (fast) halogenation.
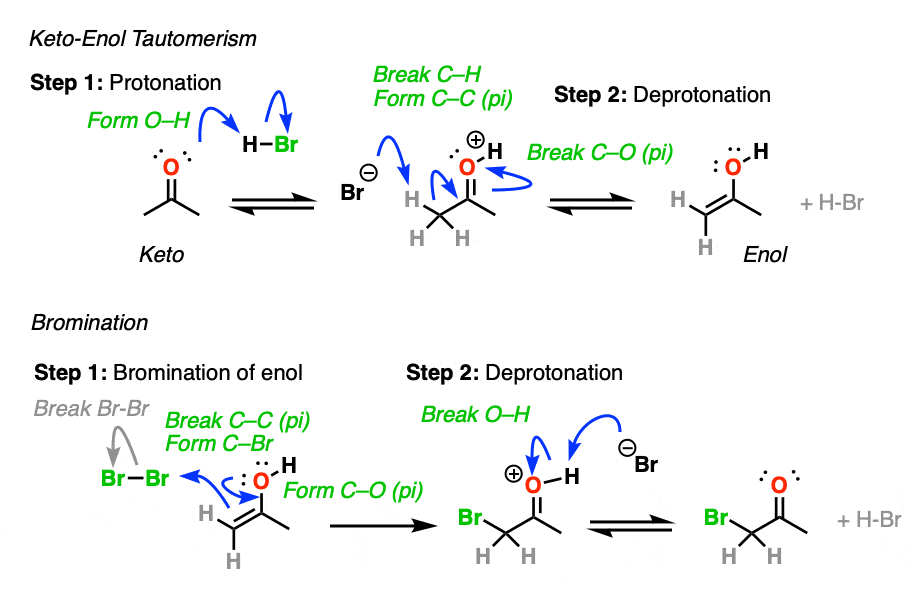
When more than one enol can be formed from the same ketone, the halogen will add to whichever enol form is the most favored at equilibrium (generally the most substituted – see previous post on keto-enol tautomerism for more details).
 Click to Flip
Click to Flip
5. The Acid-Catalyzed Aldol Reaction And Its Three Paths For Acid-Catalysis
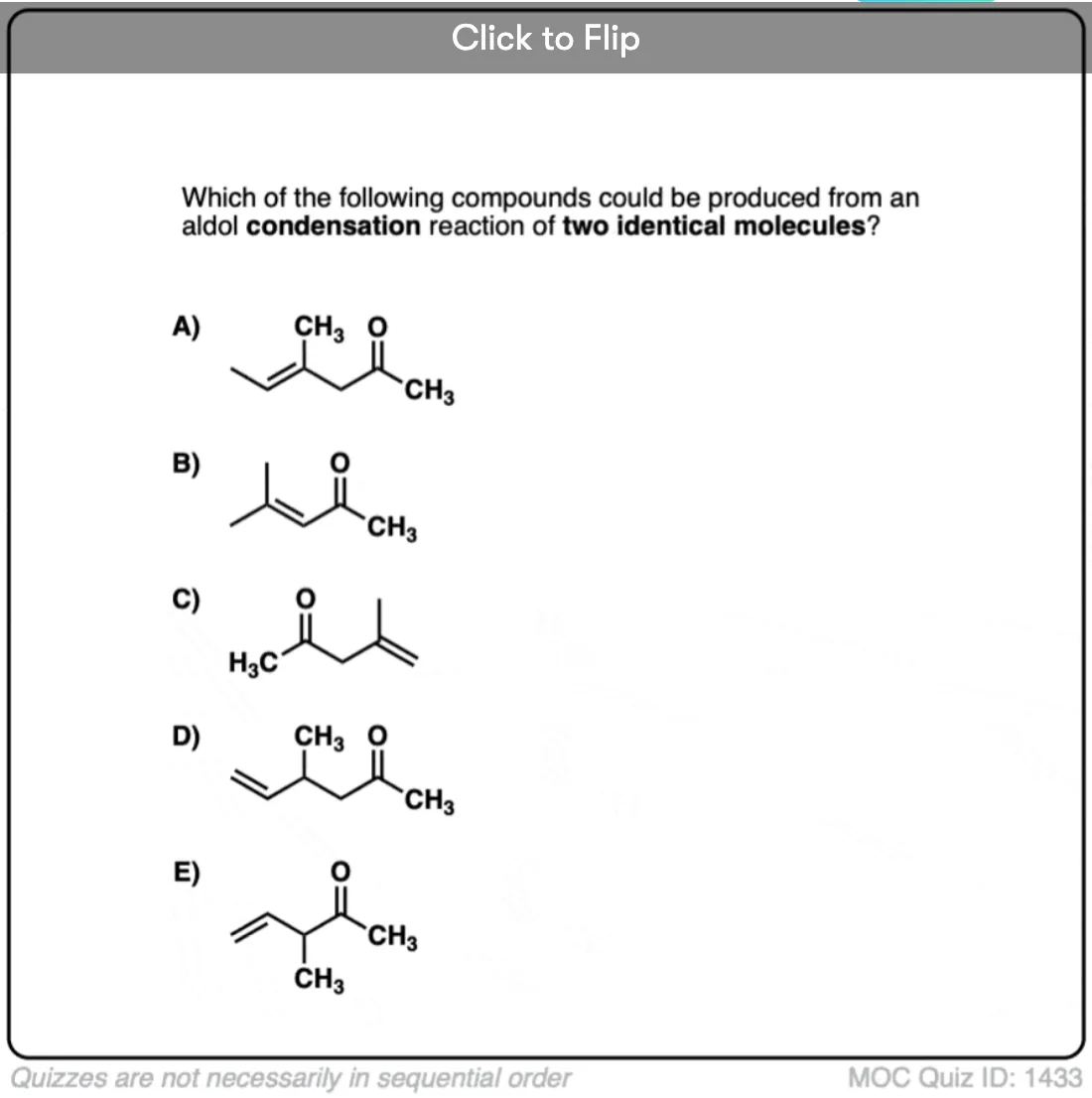
Previously we saw how the conjugate base of enols (known as “enolates“) can add to aldehydes and ketones, a reaction known as the aldol reaction. (See post: The Aldol Addition and Condensation)
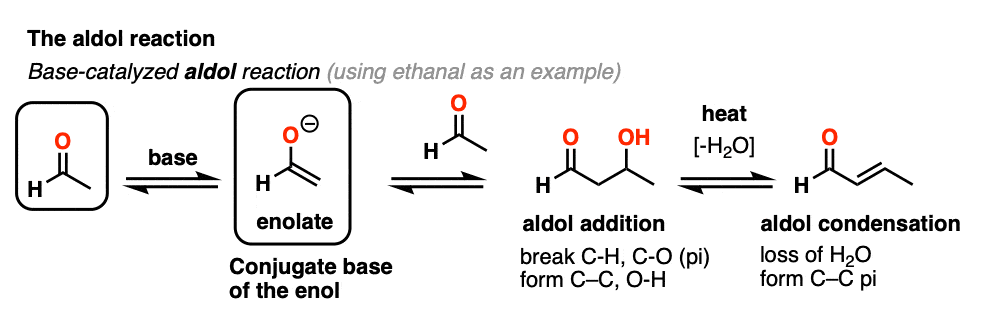
Enols can also perform the aldol reaction aldehydes and ketones, if an acid catalyst is present to help move things along. The acid-catalyzed aldol reaction almost always results in the condensation product (loss of H2O), even without heat.
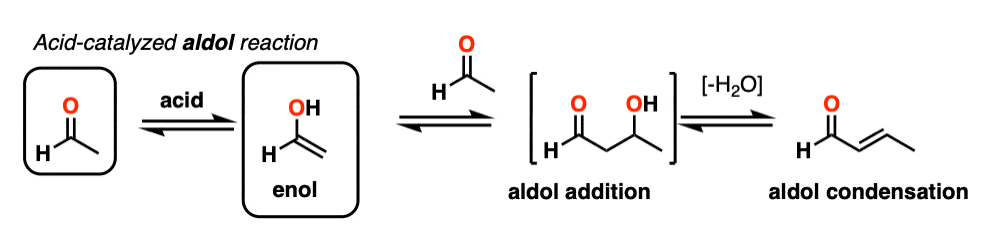
Being harder to control, the acid-catalyzed aldol tends to get less use than the base-catalyzed version. [Note 5] Still, it’s instructive because it actually highlights three key ways that acid can catalyze reactions, which roughly highlights the three stages of this reaction.
In the first stage, acid catalyzes keto-enol tautomerism, which provides us with our nucleophile (the enol) as seen previously.
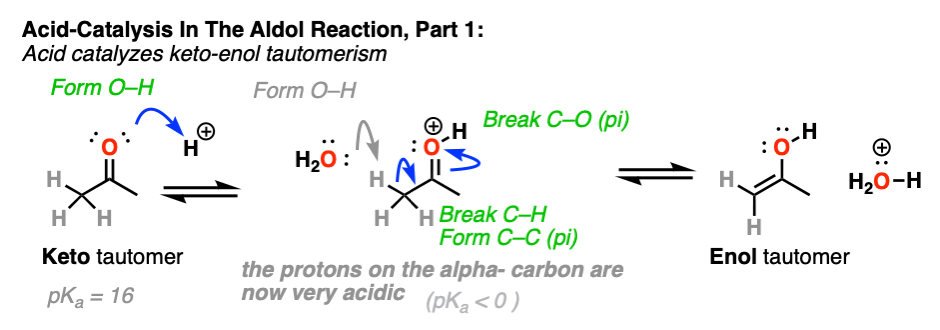
In the second stage, acid protonates the oxygen of an aldehyde or ketone. The resulting conjugate acid of the aldehyde/ketone is a much better electrophile at carbon, owing to a contribution from a key resonance form with a positive charge on carbon [Note 3].
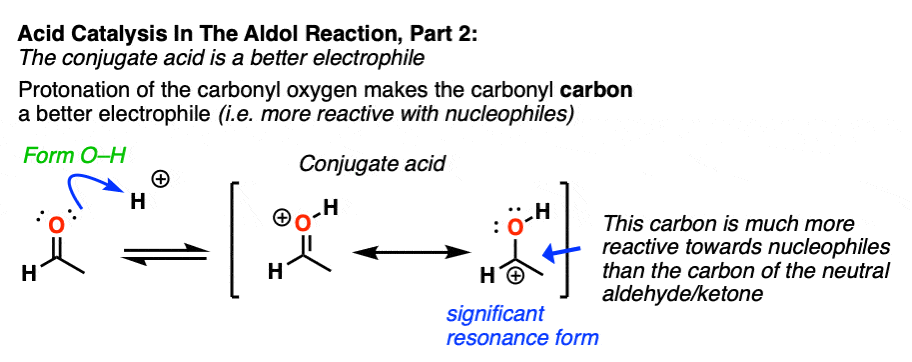
The protonated aldehyde/ketone then undergoes addition from the nucleophilic enol, resulting in the aldol addition product (after deprotonation).
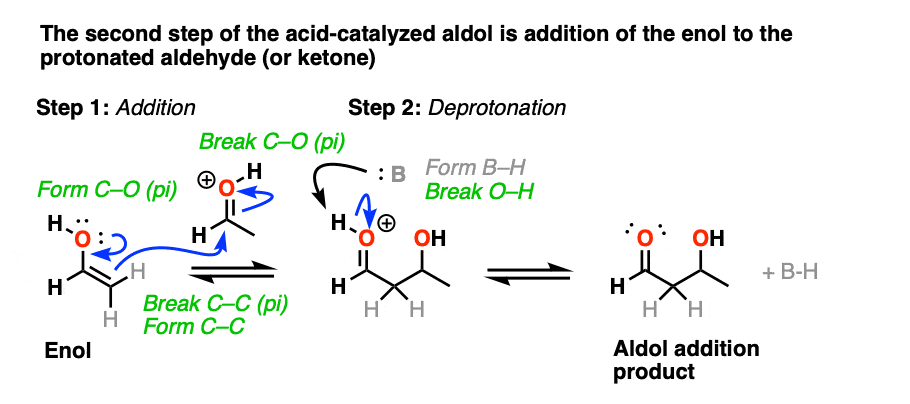
The third stage of the acid-catalyzed aldol highlights how the conjugate acid is a better leaving group. Protonation of the hydroxyl group (OH) of the addition product results in the much better leaving group H2O.
In one version of this mechanism, water is “pushed out” by the enol form of the addition product. Simultaneous formation of C-O (pi) and migration of C-C (pi) in what can be called a “1,4-elimination” or “conjugate elimination” step. There are other ways to show this occurring, which are detailed in the notes. [Note 6]
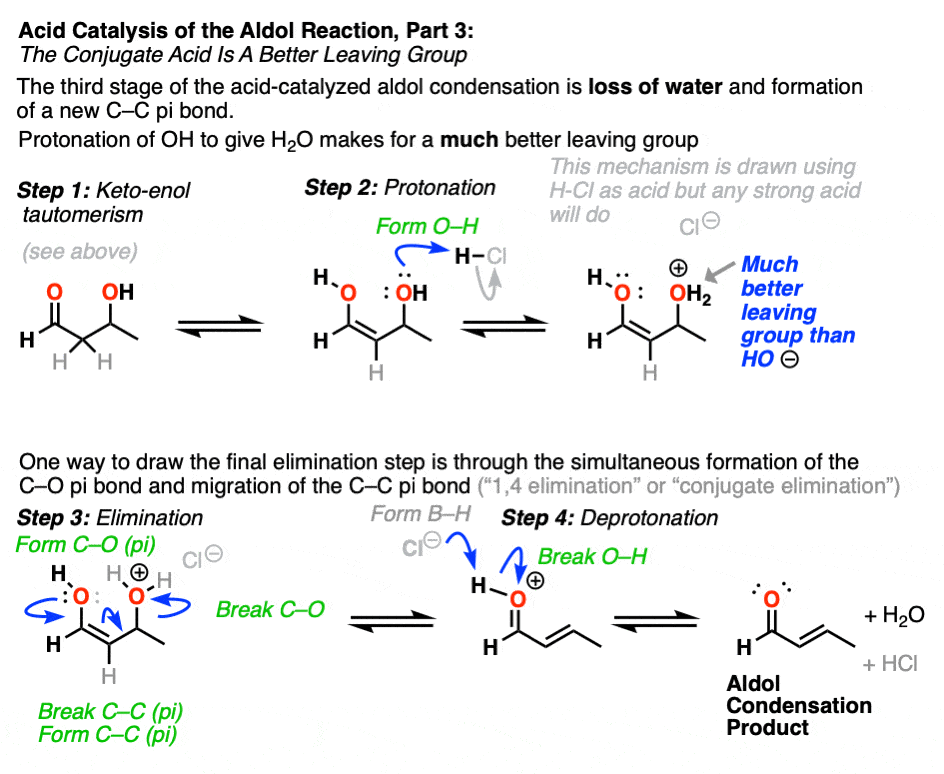
Deprotonation of oxygen then results in the neutral aldol condensation product.
Here are a few examples of aldol condensation reactions, including an example of an intramolecular acid-catalyzed aldol condensation.
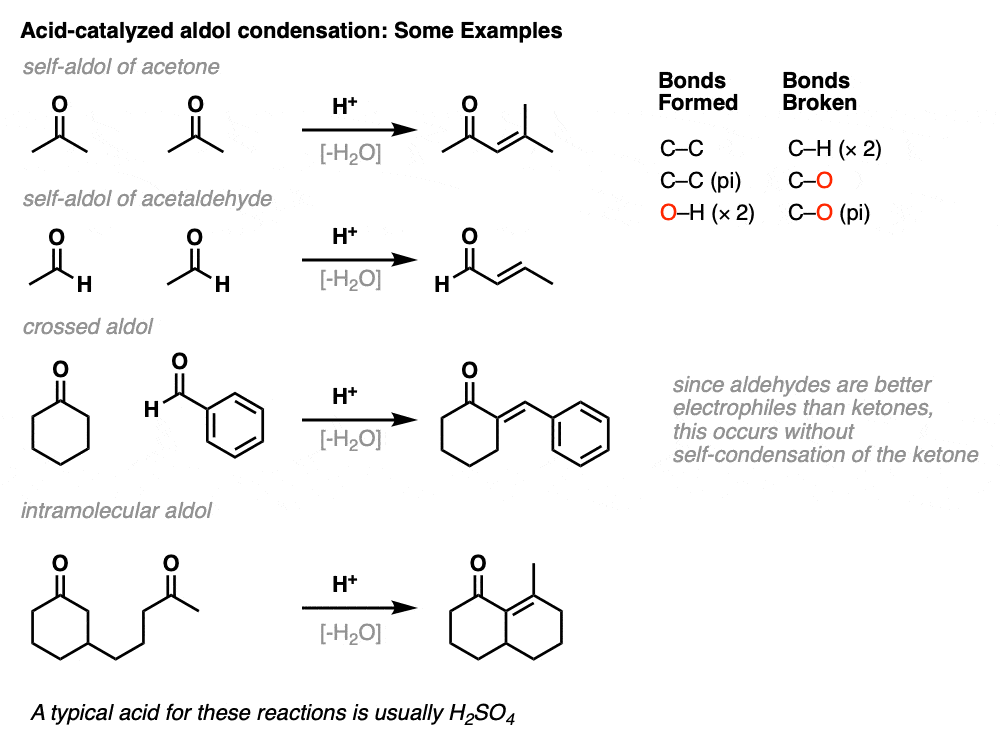
The acid-catalyzed aldol reaction can also happen intramolecularly. There is a variant of the Robinson Annulation that uses acid, for example. (See post: The Robinson Annulation)
6. The Mannich Reaction
The Mannich reaction is a close relative of the aldol reaction, except the electrophile is a C=N bond instead of a C=O.
The electrophile in the Mannich is generally not a neutral imine, but a positively charged iminium ion.
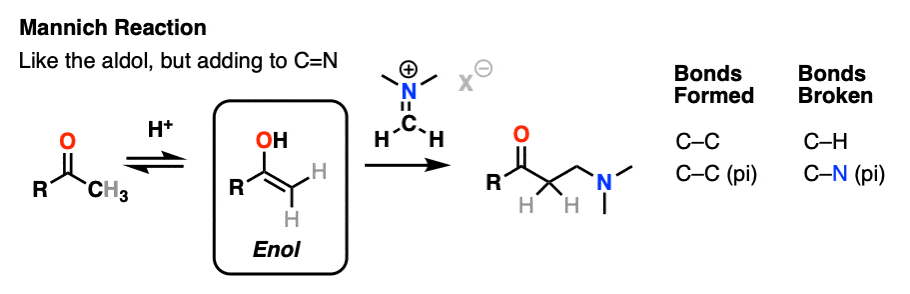
The first stage of the Mannich reaction is – you guessed it! – formation of the enol tautomer.
In the second stage, the enol adds to the iminium ion. Deprotonation then leads to the neutral product.
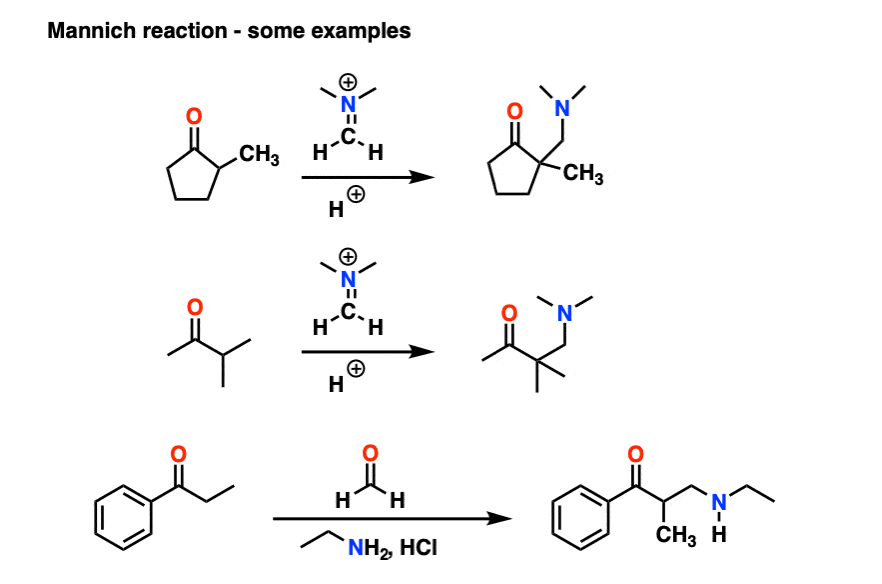
Like the aldol, if it’s done under acidic conditions, the reaction will proceed through the more-substituted enol.
7. Conclusion
So what have we learned? A few things.
- Enols are the key nucleophilic intermediate in halogenation, the acid-catalyzed aldol reaction, and the Mannich reaction.
- Acid catalyzes keto-enol tautomerism; the alpha-carbon of the protonated carbonyl is considerably more acidic than that of the neutral aldehyde or ketone.
- An aldehyde or ketone with a chiral center on the alpha carbon will undergo epimerization in the presence of strong acid.
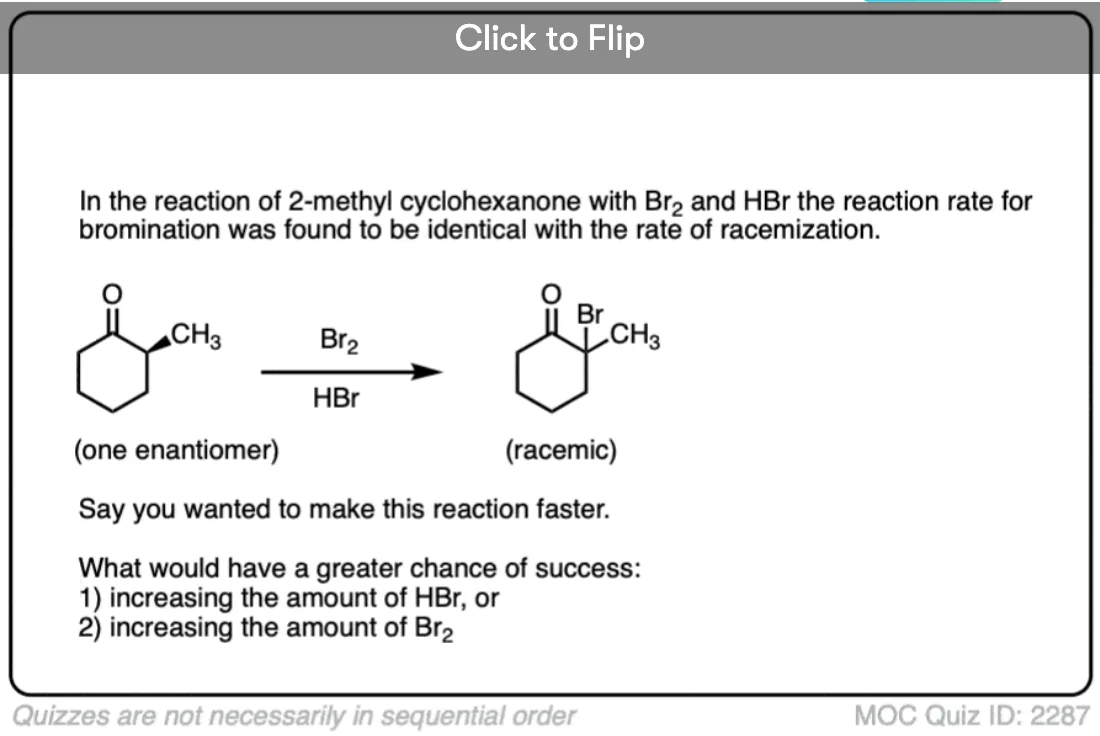
- Treating an aldehyde or ketone with Br2 in the presence of strong acid (HBr) will form a new C-Br bond on the alpha carbon. No reaction occurs in the absence of strong acid.
- Acid can catalyze the aldol condensation. Acid assists in 1)promoting keto-enol tautomerism, 2) in activating the carbonyl towards attack by the enol nucleophile, and 3) facilitates elimination through making the resulting OH group into a better leaving group.
- The Mannich reaction is similar to the aldol reaction but involves addition of an enol to a C=N bond instead of a C=O bond. It is also catalyzed by acid.
Notes
Note 1 – “Enols not as nucleophlic as enolates.” The conjugate base is always a better nucleophile than the corresponding neutral species. One quantification for RO(-) versus ROH, for example, has CH3CH2O(–) at about 10 orders of magnitude more nucleophilic than CH3CH2OH [see Mayr’s website here for the source – you might have to dig around].
There isn’t a similar treatment for comparing the nucleophilicity of similar enolates versus enols (they are harder to study) but 10 orders of magnitude is probably a good guess.
Note 2 – “acetic acid isn’t strong enough, but a few drops of HBr will do”. In one study, it was found that the speed of racemization of an enantiomerically pure ketone was extremely slow in acetic acid / chloroform but very rapid in the presence of traces of HBr. (the ketone was 2-o-carboxybenzylindan-1-one, see J. Chem. Soc. 1934 773).
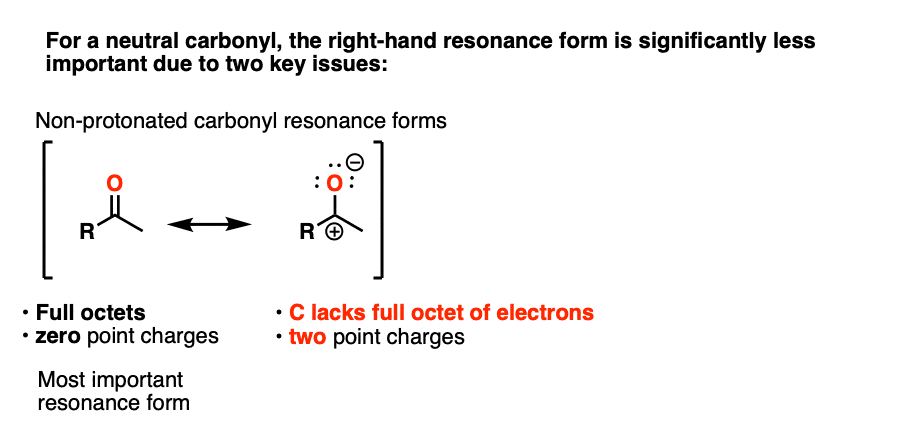
Note 3 – “It has a significant resonance form where the formal charge is on carbon and the oxygen is neutral” .
One intuitive way of gauging how much “carbocation” character the ketone has is to compare the relative contributions made by the carbocation-containing resonance form to the overall resonance hybrid. (The intuitive rules for gauging the relative importance of resonance forms is covered here, 4 Rules For Evaluating Resonance Structures). Two key items relevant here are the number of point charges (zero is preferred) and the number of atoms with full octets (zero is preferred).
In the case of the neutral ketone, the resonance form with the carbocation has two point charges (versus zero for the C=O form) in addition to the lack of a full octet on C.
Once the carbonyl is protonated, both resonance forms have the same number of point charges (one). Therefore the resonance form where carbon lacks a full octet (the carbocation) will be less bad overall, and therefore the carbocation will make a greater contribution to the resonance hybrid, relative to non-protonated carbonyl.
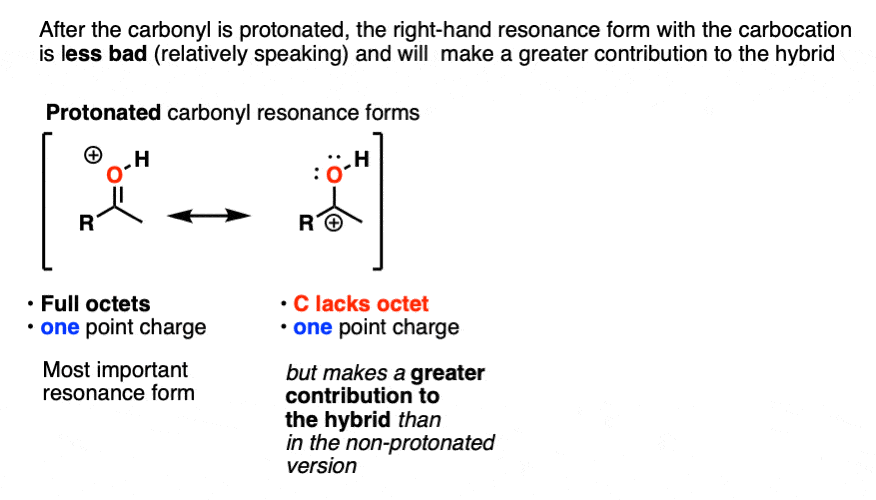
One way to quantify this effect is to examine C=O stretches in IR spectroscopy, since the stronger the bond, the higher the wavenumber. Neutral acetone has a C=O stretch at 1765 cm-1 whereas protonated acetone has a C=O stretch at 1580 cm-1. That demonstrates a significant weakening of the C-O pi bond.
The same argument can be made for why the positively charged iminium ion is a better electrophile than a neutral imine.
Note 4 – “in the absence of acid, bromination is exceedingly slow”. This is a direct quote from Lapworth (ref) in his seminal study on acid-catalyzed bromination.
Note 5 – “the acid-catalyzed aldol tends to get less use than the base-catalyzed version.” The base-promoted aldol gets more use because it’s much easier to control which enolate you can form through use of the appropriate base. In the acid-catalyzed aldol you are held hostage to whichever enol form is more thermodynamically stable.
However there is a version of the aldol reaction called the Mukaiyama aldol reaction which proceeds with an enol ether as the nucleophile, and employs strong Lewis acids (e.g. TiCl4, which smokes in the presence of air) to assist their reactions with aldehydes.
Note 6 – “There are other ways to show this occurring, which are detailed in the notes. “
The elimination going through a conjugate elimination process is was determined in this study of the self-condensation of acetaldehyde (see ref).
There is some variation on how this step is shown in textbooks, and furthermore, there is also variation on how this happens in real life.
One way this is sometimes shown is as an E1-type reaction where water leaves, and there is a subsequent deprotonation event. This can happen; it’s most correct in cases where that would lead to a resonance stabilized carbocation (e.g. adjacent to an electron-rich aromatic ring). One prominent example can be found here. [Ref]
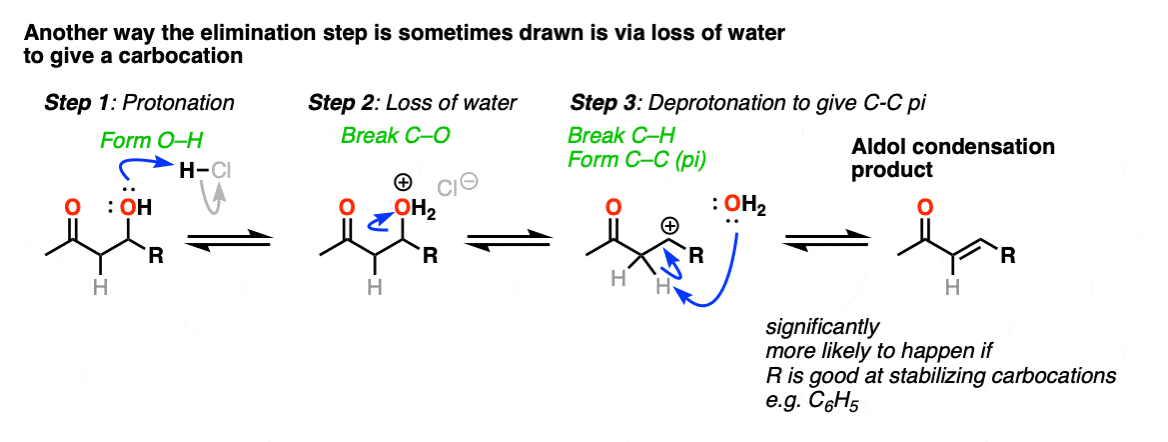
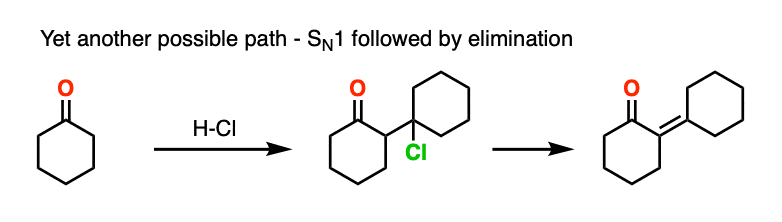
This goes way beyond what anyone reading this would likely care about, but there is also precedent for the OH group to undergo substitution (likely via SN1) to a good leaving group, which subsequently is removed with weak base (or via E1). This is known for the self-aldol condensation of cyclohexanone with HCl, for example.
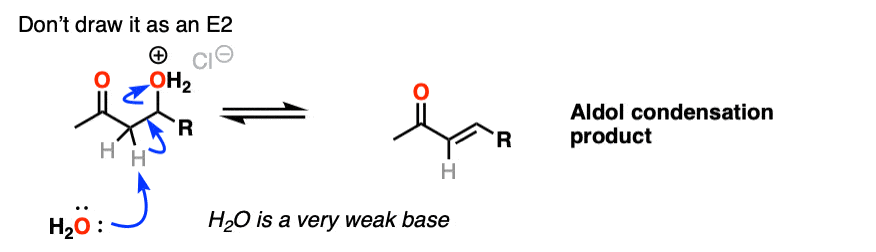
One thing I can feel comfortable saying is that you should not draw this as an E2 type reaction. It requires an anti-periplanar arrangement of the C-H and C-OH2 bonds, and I’m not aware of any evidence that this process depends on stereochemistry (the same way you see in the E2).
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
Volume 2 of Comprehensive Organic Synthesis was useful for this article, as were March’s Advanced Organic Chemistry, Carey & Sundberg, and Ingold’s Structure and Mechanism in Organic Chemistry.
The acid-catalyzed aldol reaction has been known since 1838, which makes it one of the longest-known organic reactions of all time. The first example was the dimerization of acetone, from J. Prakt. Chem 1838, 15, 129.
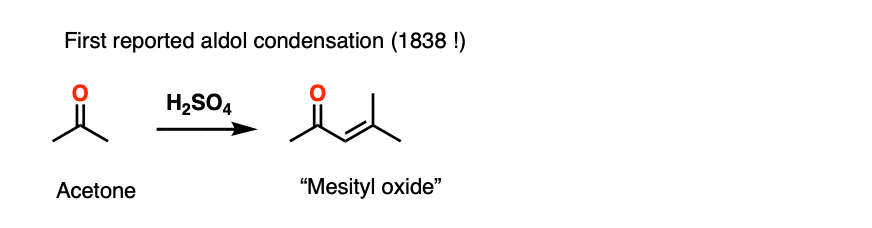
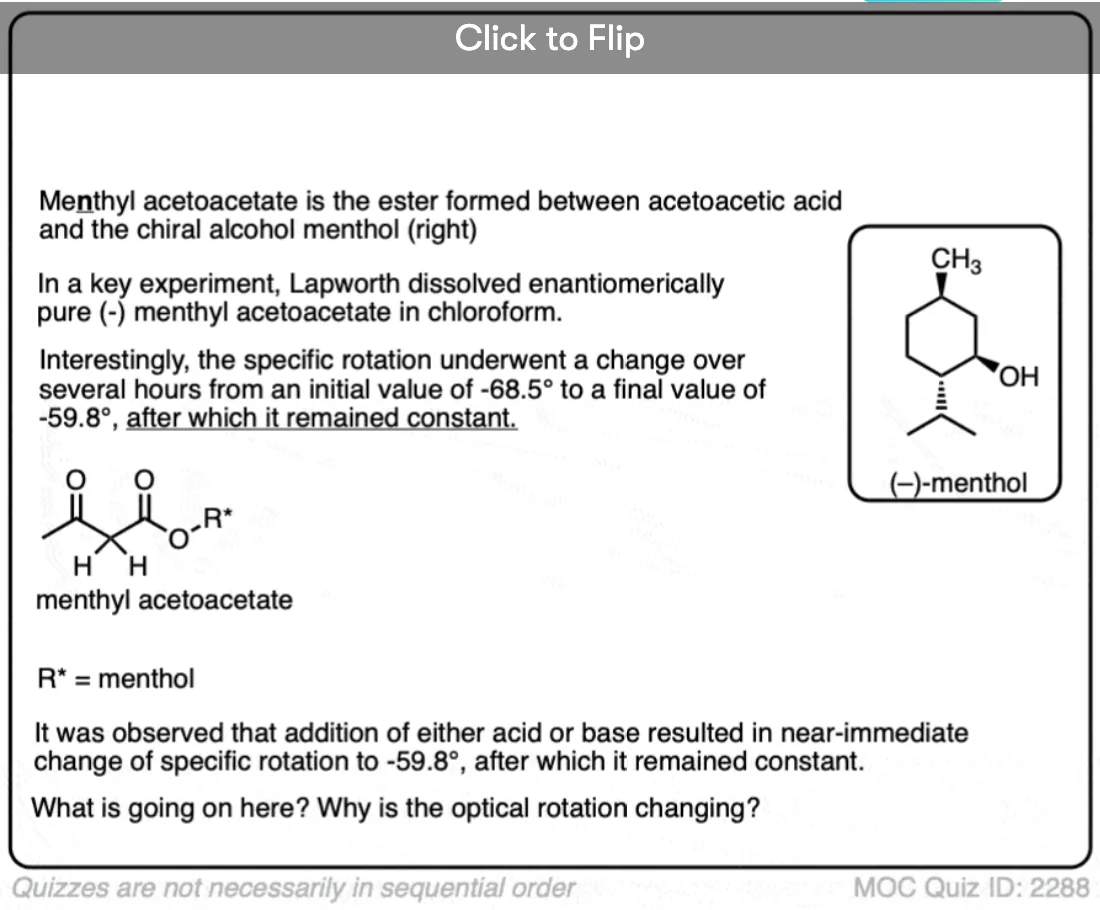
1. Optically Active Esters of B-Ketonic and B-Aldehydic Acids. Part II. Menthyl Acetoacetate
Arthur Lapworth and A. C. Osborn Hann.
J. Chem. Soc. , Trans. 1903, 83, 1114-1129
DOI: 10.1039/CT9038301114
A very instructive paper and one of the earliest examples containing a definitive proof of the role of acid in accelerating keto-enol tautomerism. The authors prepared the menthyl (not methyl) ester of acetoacetic acid [(+)-menthol is an optically active alcohol readily available from natural sources] and measured its optical activity. They found that the menthyl ester underwent mutarotation to an extent that was dependent on the solvent, with the rate of mutarotation being highly dependent on acid. This demonstrates the establishment of an equilibrium between the keto and enol forms.
2. The Action of Halogens on Compounds Containing The Carbonyl Group
Arthur Lapworth
J. Chem. Soc., Trans., 1904,85, 30-42
DOI: 10.1039/CT9048500030
Pioneering study on halogenation of carbonyl compounds.
“Bromination of acetone under the conditions maintained is best regarded as the result of a slow, reversible change effected in the acetone by the hydrogen ions, followed by an almost instantaneous bromination of the product, a change which is not appreciably reversible. This intermediate product is perhaps the enolic form of the ketone, as it has been already shown that in many cases the rapid attainment of equilibrium between the tautomeric forms of carbonyl compounds is brought about by acids.”
3. Ueber die Condensation von Aethylmethylketon mit Benzaldehyd (Condensation of Ethyl Methyl Ketone With Benzaldehyde)
C. Harries, G. Hans Muller
Ber. 1902, 35(1), 966-971
DOI: 10.1002/cber.190203501156
Condensation of ethyl methyl ketone with benzaldehyde under acidic conditions results in the more-substituted aldol condensation product in high yield!
4. Optical Activity In Relation to Tautomeric Change. Part IV. Comparison of the Rates of Racemization and of Bromination of a Ketone
C. K. Ingold and C. L. Wilson
J. Chem. Soc. 1934, 773.
DOI: 10.1039/JR9340000773
Rate of bromination is essentially the same as the rate of racemization, consistent with enol formation as the rate-limiting step. “In chloroform and glacial acetic acid, racemization of the d-ketone was extremely slow but became very rapid in the presence of traces of hydrogen bromide”.
5. Carbonyl Reactions. VI. Evidence for Alternate Mechanisms for the Dehydration of β-Hydroxy Ketones1
Donald S. Noyce and Wilmer L. Reed
Journal of the American Chemical Society 1958 80 (20), 5539-5542
DOI: 10.1021/ja01553a056
This paper studies the acid-catalyzed aldol reaction of methyl ethyl ketone and various substituted benzaldehydes. The key nugget here is that the final dehydration step is proposed to occur through the enol form of the addition product when the aromatic ring has an electron-neutral (Ph) or electron poor (NO2) group in the para position, but through ionization (i.e. benzyl carbocation formation) with an electron-rich (OCH3) group in the para position. Stated reason is that there is only a 3fold change in rate for elimination step from p-methyoxyl to 3-nitro, inconsistent with what one would see if it involved a benzyl carbonium ion.
Worth noting that Donald Noyce’s brother was physicist Robert Noyce, the co-founder of Intel.
6. Carbonyl Reactions. VIII. The Kinetics of the Acid-catalyzed Condensation of Benzaldehyde and p-Nitrobenzaldehyde with Methyl Ethyl Ketone. Some Observations on ρ-σ Correlations1
Donald S. Noyce and Lloyd R. Snyder
Journal of the American Chemical Society 1959 81 (3), 620-624
DOI: 10.1021/ja01512a029
Part 8 of a series of papers from 1958-59 from the Noyce laboratory studying the acid-catalyzed aldol reaction between methyl ethyl ketone and substituted benzaldehydes.
7. Acid-catalyzed enolization and aldol condensation of acetaldehyde
Lynn M. Baigrie, Robin A. Cox, Henryka Slebocka-Tilk, Michal Tencer, and Thomas T. Tidwell
Journal of the American Chemical Society 1985 107 (12), 3640-3645
DOI: 10.1021/ja00298a039
Extensive physical-organic-chemistry study on the acid-catalyzed aldol condensation of the simple aldehyde, ethanal (acetaldehyde). The rate-limiting step is addition of the enol to protonated acetaldehyde. Notably their work is also consistent with the elimination of water occurring from the enol form of the addition product.
Two other useful snippets: “acetaldehyde is completely protonated to its conjugate acid in H2SO4 stronger than 64%. “Enolization of ketones is known to involve two water molecules acting together as a base”.
8. Acid-catalyzed Robinson Annelations
Clayton H. Heathcock, John E. Ellis, John E. McMurry, Anthony Coppolino,
Tetrahedron Letters, 12(52), 1971, 4995-4996
DOI: 10.1016/S0040-4039(01)97609-9.
This study contains several examples of acid-catalyzed Robinson annulations.
9. The Mannich Reaction with 2-Methylcyclopentanone and 2-Methylcyclo hexanone1a
Herbert O. House and Barry M. Trost
The Journal of Organic Chemistry 1964 29 (6), 1339-1341
DOI: 10.1021/jo01029a016
Several useful examples of Mannich reactions which show that they tend to go through the more substituted enol.
Please double check the “Acid-Catalysis In The Aldol Reaction, Part 1: Acid catalyzes keto-enol tautomerism” reaction scheme. The centre structure (protonated acetone) is uncharged and features a ten-electron oxygen atom. Replacing one electron pair at the oxygen atom with a positive charge will sort the problem out.
Fixed! Thanks for the spot!
James,
I liked very much this post! Thank you for your work.
In the Clayden book 2012 (pg 616), and in the Karty book (pg 912) the acid catalyzed dehydration is claimed as an E1, from the carbonyl, and not from the enol.
My feeling is that this mechanism involves an E1, but from the enol. Indeed, in this case, when water leaves, the carbenium ion formed benefits of three resonance contributors: one with the positive charge on the beta carbon, another one with the positive charge on the carbonyl carbon (the double bond has shifted), plus the oxycarbenium ion. So, this carbocation has many reasons to be a stable intermediate, and if you formally remove H+ from the oxycarbenium ion you have the final product.
Thanks for writing!
Noyce studied this: https://pubs.acs.org/doi/10.1021/ja01553a056
and concluded that while the E1 can certainly occur, especially where a relatively stable carbocation that can be formed (e.g. p-MeOC6CH4).the elimination generally goes through the enol. This is also the case for the self-condensation of acetaldehyde. see: https://pubs.acs.org/doi/10.1021/ja01553a056
Dear Sir,I am a research scholar,my area organic synthesis,I am doing aldehyde ketone reaction with acid catalyst,I used 2 acetyl benzofuran and 2, amino 5,chloro benzaldehyde,but my expected product is not come, but product came 2,amino,5, chloro accetophenone, single crystal xrd confirmed the structure,but how it is formed in which mechanism I want to try find mechanism please help any suggestions,
I would double check your starting materials.
In a crossed aldol reaction between 2-butanone and 2,2-dimethylbutanal, 2-butanone would be the carbanion/nucleophile considering 2,2-dimethylbutanal lacks acidic hydrogens. But aren’t there technically two different possible nucleophiles that 2-butanone could form, and wouldn’t that lead to two separate products? I understand that the more acidic hydrogen on 2-butanone is on carbon 3, but is hydrogen on carbon 1 of 2-butanone acidic at all? Can it not become the nucleophile, or is it just so unlikely that we don’t consider it?
Sir, why doesn’t oxygen of enolate ion attack the carbonyl carbon ? I have read that carbonyl carbon is a hard electrophile and oxyanion is a hard nucleophile, so why this doesn’t happen ?
It might happen to some extent but the product is a hemiacetal, and hemiacetals easily revert to starting materials. So it’s usually a cul-de-sac.
Professor, which is the slow/rate determining step in the aldol reaction?
Can you please reply soon??…. It’s urgent…
Thanks
In the acid-catalyzed aldol addition, the slow step is attack of the enol on the aldehyde/ketone. But for more information than you probably want, I recommend: https://pubs.acs.org/doi/10.1021/ja01553a056
I believed that a base is necessary to abduct alpha hydrogen to perform enolization. is such not the case? since in the second pathway you show that introducing acid increases rate of enolization.
yes, keto-enol tautomerism can be assisted through the use of either acid or base.
Why mostly aldol condensation reaction occurs In the presence of base
dear Sir,
i am research fellow and my research area is organic chemistry. the present site provides excellent information about organic reactions and mechanism. i am very much impressed and would like to mention lot of thanks for this presentation. by following this site, i am getting expected information and knowledge.
thanking you,
yours sincerely,
uttam patil
thank you.
The 1,4-elimination mechanism is new to me and appears to be a good idea. But , the usual 1,2-elimination mechanism is not so bad. Of course water as a base is not good if water is not used as the solvent [March (6th Ed page1349) says water acts as a base even in concentrated H2SO4]. The -OH2+ is a good leaving group and the H2O (or ROH when an alcohol is used) accepts the proton to form an oxonium ion. This may be viewed as an E1 elimination rather than the E2 elimination. Actually I find a discussion of the two mechanisms in Ingold (page1004-1005). Some people think that Cl- ion as the base instead of water to capture the proton to form HCl. But this is rather unlikely in hydroxylic solvents. I do not know which mechanism is the most probable one. Unfortunately, no chemists today will do experiments to elucidate the mechanism, because such project will not attract money.
If it was a straight E1 one might expect to see rearrangements, especially where a primary alcohol is concerned. Since the elimination of water from those substrates is facile, a mechanism where enolization occurs first followed by donation of a pair of electrons from the enol seems more reasonable to me rather than an E1 type pathway, but depending on the substitution pattern more than one mechanism might be operational. For a tertiary allylic alcohol, for example, this might be the dominant mechanism.
Thanks for taking the time to comment and also to add the annotations from March and Ingold.