Dienes and MO Theory
Electrocyclic Reactions
Last updated: June 2nd, 2026 |
Electrocyclic Reactions And 4-Membered Rings: Conrotatory and Disrotatory Ring Opening And Closure
In this article, we investigate electrocyclic ring-opening and ring-closing in systems with 4 pi electrons (“4π” systems).
Electrocyclic reactions under “thermal” conditions (i.e. just heating, no UV light) result in different products than are obtained under “photochemical” conditions (i.e. irradiation with UV light).
Look closely at the stereochemistry below!
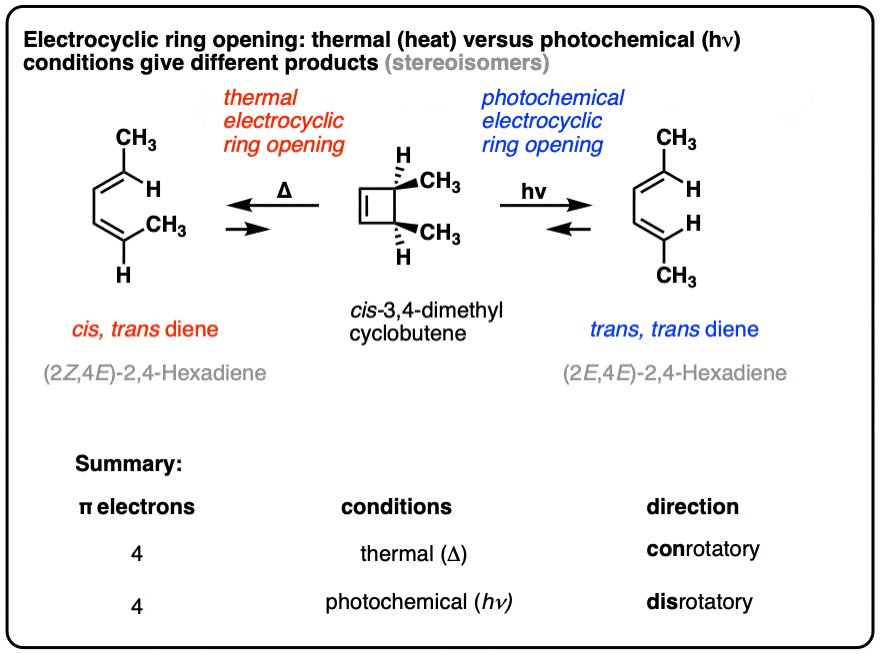
The reason for these different outcomes is that the highest-occupied molecular orbital (HOMO) in the thermal case has different orbital symmetry than the highest-occupied molecular orbital (HOMO) in the photochemical case.
The change in orbital symmetry leads to a change in how the end carbons must rotate to form a new sigma bond, which can be either “conrotatory” (both ends rotate in the same direction) or “disrotatory” (both ends rotate in opposite directions), as we’ll see below.
Much more detail below. In a future post we’ll cover the rules for systems with six pi electrons ( “6π “) and higher.
Table of Contents
- Electrocyclic Ring-Opening of Cyclobutene To Give Butadiene
- Electrocyclic Ring Closure Of 1,3-Butadiene to Give Cyclobutene
- Electrocyclic Reactions Are Stereospecific
- Orbital Symmetry And “Thermal” Electrocyclic Reactions
- Orbital Symmetry In “Thermal” Electrocyclic Ring Opening
- Applying “Conrotatory” Rotation Predict The Stereochemistry Of Substituents From Electrocyclic Ring-Opening
- Predicting The Products From Conrotatory Ring-Closing
- Electrocyclic Reactions Under “Photochemical” Conditions
- How Do “Photochemical Conditions” (UV light) Affect Electrocyclic Reactions?
- The Orbital Symmetry Of Electrocyclic Ring Opening Under “Photochemical” Conditions
- Applying “Disrotatory” Rules To Electrocyclic Ring-Opening Under Photochemical Conditions
- Summary: Thermal vs Photochemical Ring Opening and Closing
- Notes
- Quiz Yourself!
- References and Further Reading
1. Electrocyclic Ring-Opening of Cyclobutene To Give Butadiene
The year was 1905. Chemist Richard Willstätter and his collaborator Wolfgang von Schmaedel were heating a reaction mixture in their laboratory at the University of Munich. They were performing a fairly routine elimination reaction that has now become known as the Hofmann elimination.
The expected product was cyclobutene.
When the reaction mixture was analyzed, cyclobutene was indeed formed. Mysteriously, however, considerable amounts of 1,3-butadiene were also isolated.
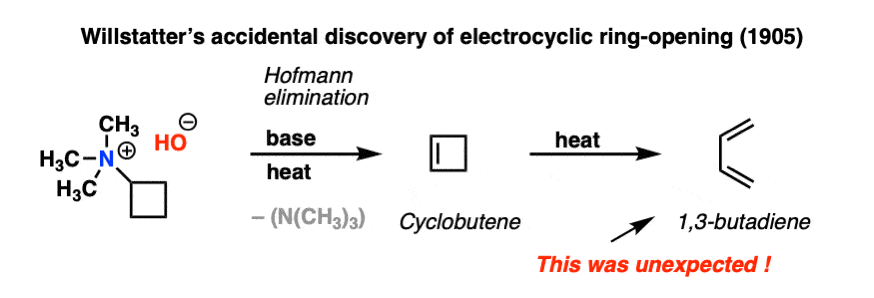
This was a strange and unexpected result! Willstätter had no clear understanding of how 1,3-butadiene had formed at the time, and it would remain thus. He would go to his grave (in 1942) without knowing that he had accidentally discovered what would later be called an electrocyclic reaction; specifically, the electrocyclic ring opening of cyclobutene. And it’s not as if Willstätter was a chump; a few years later he was the first to synthesize cycloöctatetraene, and a few years after that he would win the Nobel Prize in Chemistry for determining the structure of chlorophyll and other plant pigments. [Note 1]
In the following decades, the reaction (and its many variants) was studied in more detail. It was verified that heating cyclobutene did indeed form 1,3-butadiene.
Interestingly, however, heating cyclobutane did not lead to any ring-opening reaction.
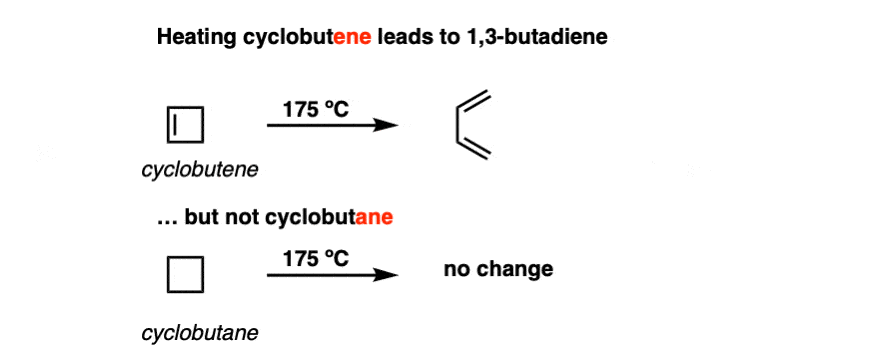
The ring-opening of cyclobutene (and other cyclic molecules, as we will see) have been come to be known as electrocyclic reactions. An electrocyclic reaction is a reaction where a single bond is formed between the termini of a pi system. The converse process (i.e. ring-opening, breaking a single bond to form a new pi system) is also an electrocyclic reaction.
Note 2. Official definition of electrocyclic reaction.
2. Electrocyclic Ring Closure Of 1,3-Butadiene to Give Cyclobutene
In time, it was also found that 1,3-butadiene was also capable of undergoing the converse reaction: a ring closure to give cyclobutene.
Due to ring strain [see post: Calculation of ring strain in cycloalkanes] the closure of 1,3-butadiene to butadiene is usually unfavorable, but certain substituted dienes can be coaxed into closing the ring [see below].
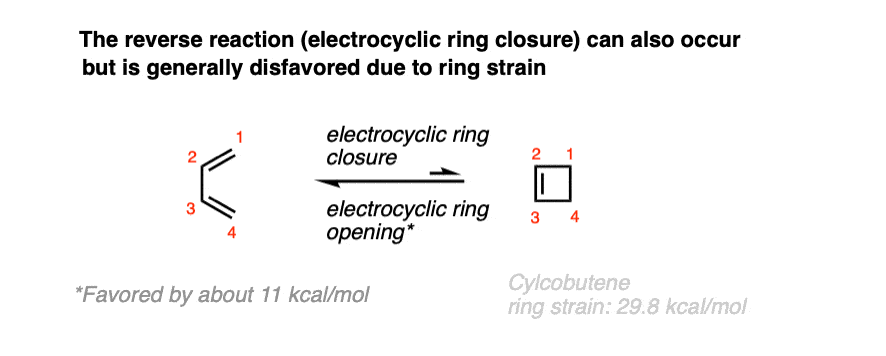
(Note: The ring strain in cyclobutene is about 29.6 kcal/mol, compared to 26.3 kcal/mol for cyclobutane)
Since cyclobutene and 1,3-butadiene can interconvert, this implies that electrocyclic ring-opening and electrocyclic ring-closing goes through a common transition state, and there is an equilibrium between these two molecules at high temperatures.
It’s helpful to examine the bonds that form and break.
- In the forward direction, a C–C pi bond and a C–C sigma bond break, and two C–C pi bonds form.
- In the reverse direction, two C–C pi bonds break, and a C–C pi bond and a C–C sigma bond are formed.
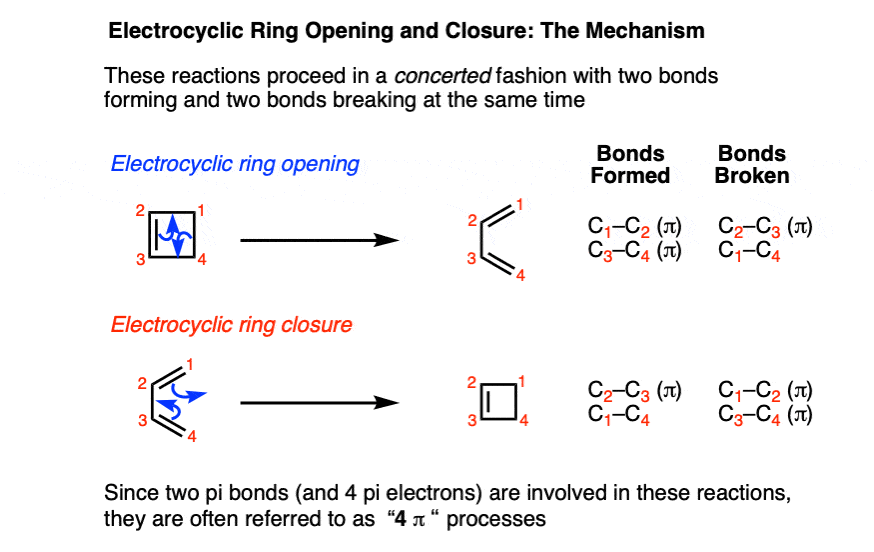
Electrocyclic ring-opening and ring-closing also share the same transition state.
3. Electrocyclic Reactions Are Stereospecific
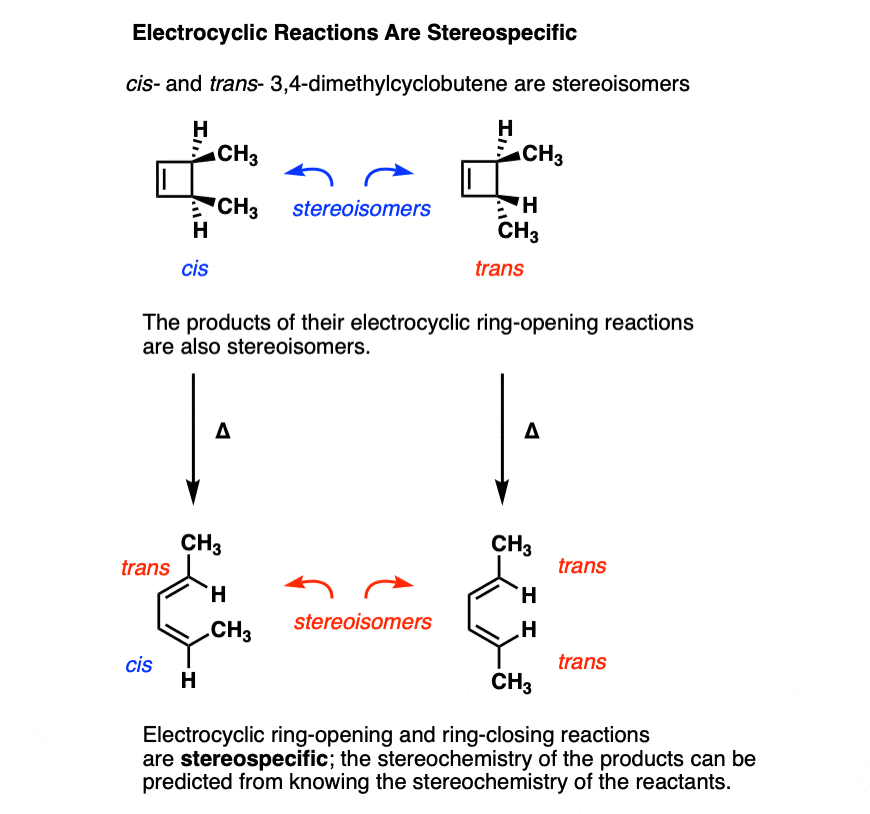
Moving beyond cyclobutene to substituted cyclobutenes, a very interesting pattern is observed: the stereochemistry in these reactions follows a well-defined pattern.
Electrocyclic ring opening is a stereospecific process. A reaction is termed stereospecific if starting materials differing only in their configuration are converted into stereoisomeric products.[Note 3]
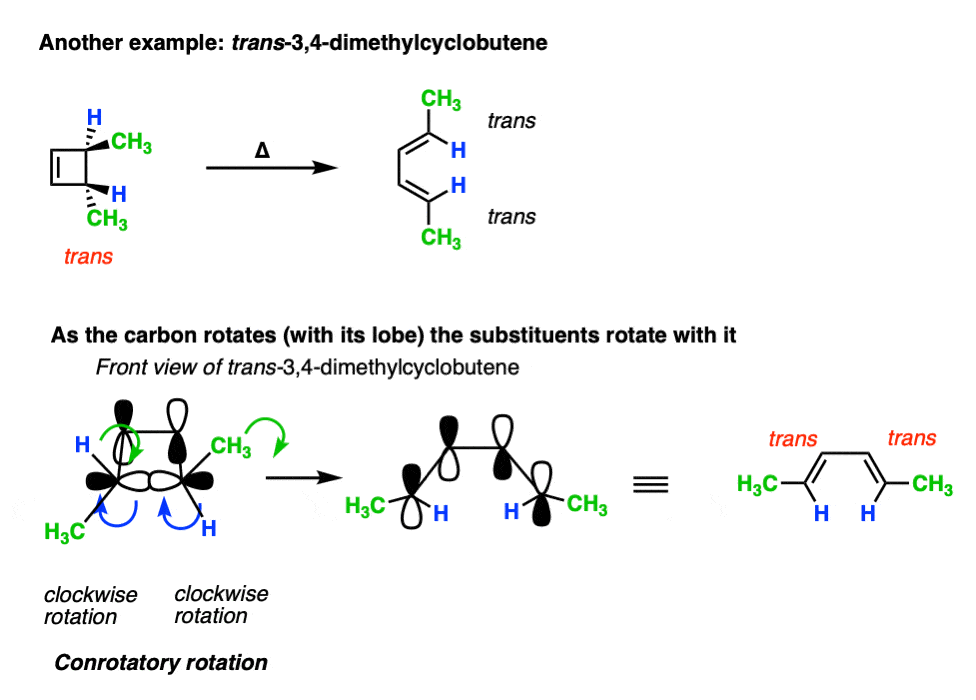
A classic example is cis- and trans– 3,4-dimethylcyclobutene , which are stereoisomers that differ only in their configuration. [For a review, see: Types of Isomers]
- Heating cis-3,4-dimethylcyclobutene gives the trans, cis diene.
- Heating trans-3,4-dimethylcyclobutene gives the trans, trans diene.
The trans, cis diene and the trans, trans diene are stereoisomers.
In other words, if if you know the configuration of your starting material, and you know the “rules” that the reaction follows, you can predict the product with 100% accuracy.
Why are these reactions so predictable? Because these reactions follow the rules for conservation of orbital symmetry (the “Woodward-Hoffmann Rules”).
Another way to put it is that we can understand the course of these reactions by examining the frontier orbitals (the “highest-occupied molecular orbital” [HOMO] and “lowest unoccupied molecular orbital” [LUMO] of the pi system), as we’ll see below.
[Electrocyclic ring closure is stereospecific too – hover for image or click this link.
{kind=link}
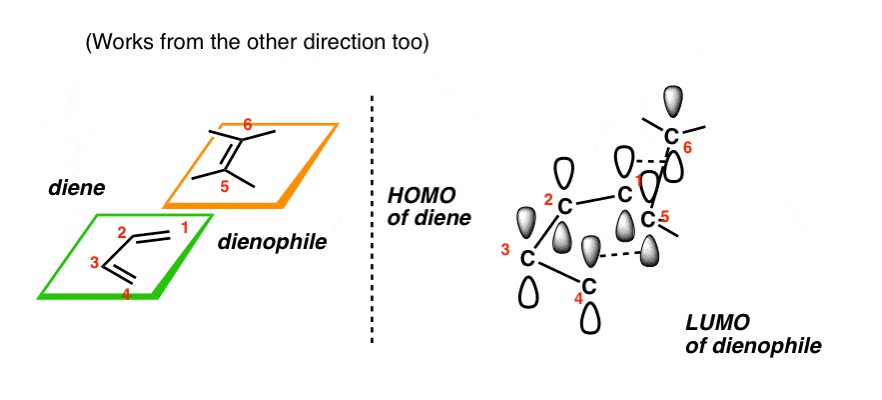
[We’ve previously seen the Woodward-Hofmann rules at work in the Diels-Alder reaction [see: HOMO and LUMO in the Diels-Alder Reaction], where constructive orbital overlap between the highest-occupied molecular orbital (HOMO) of the diene and the lowest-unoccupied molecular orbital (LUMO) of the “dienophile” is responsible for the Diels-Alder being an “easy” reaction. They are also at work in the Cope and Claisen rearrangements, among many others. ]
4. Orbital Symmetry And “Thermal” Electrocyclic Reactions
Like the Diels-Alder reaction, the patterns governing electrocyclic reactions are also understood by analyzing the molecular orbitals. In this case since the reaction occurs within only one pi system, we only have to worry about the HOMO.
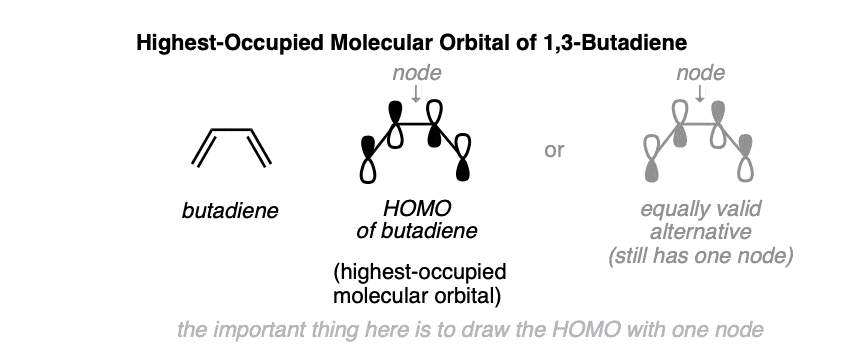
What does the highest-occupied molecular orbital (HOMO) of 1,3-butadiene look like? We’ve seen how to build up the pi molecular orbitals here. [see: The Pi Molecular Orbitals of Butadiene and How To Draw Them] so we’ll skip right to the punch line.
The highest-occupied molecular orbital of butadiene is drawn below. Note how it has only one node (i.e. where there is a change of sign), between C-2 and C-3. (It doesn’t matter which of the two ways you draw the orbitals, so long as it only has one node!)
In the reaction of 1,3-butadiene –> cyclobutene, a new sigma bond is formed between C-1 and C-4 termini.
Where do the orbitals that form this sigma bond come from? From the p orbitals on C-1 and C-4 that are breaking their pi bonds with C2 and C3 respectively!
In order for a bond to form, there must be constructive overlap between these two orbitals. By “bonding interaction” I mean that they both have to have the same phase, either both shaded or both white.
This bonding interaction can happen if both orbitals turn in a clockwise motion, OR if they both turn in a counter-clockwise motion.
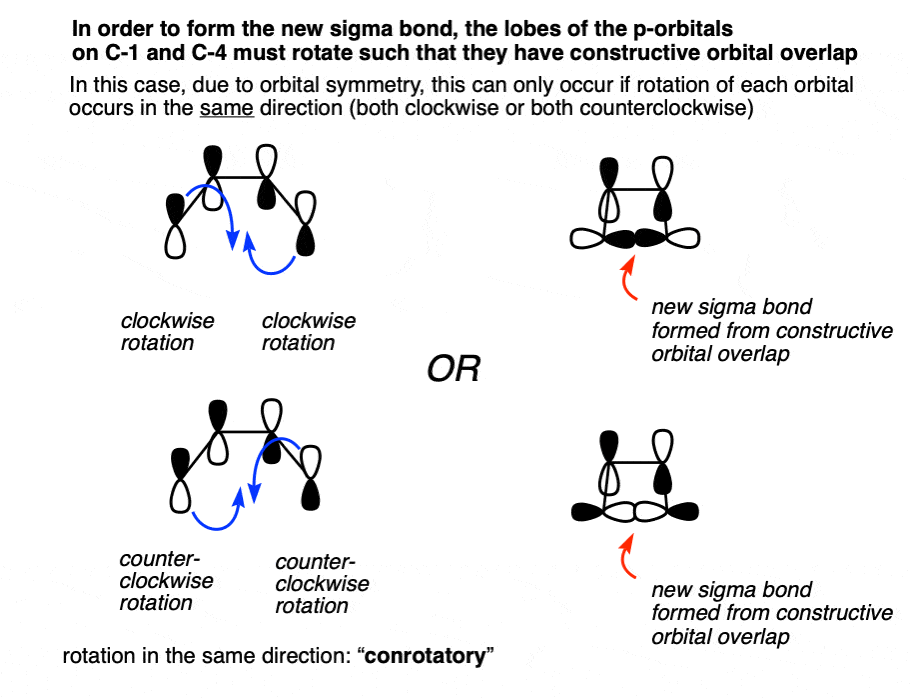
What happens if they don’t both turn either clockwise or counterclockwise? No bond forms, because the orbitals have the opposite phases and thus destructive orbital overlap! Hover for image or click this link
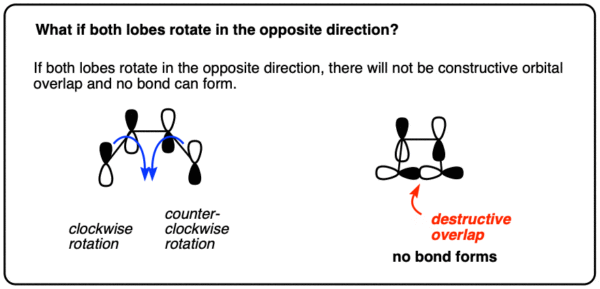{kind=link}
5. Orbital Symmetry In “Thermal” Electrocyclic Ring Opening
We can also analyze the reaction in the reverse direction, where the sigma bond is being converted into a pi bond. In this case, the lobes also turn the same direction in order to be converted into the HOMO of butadiene.
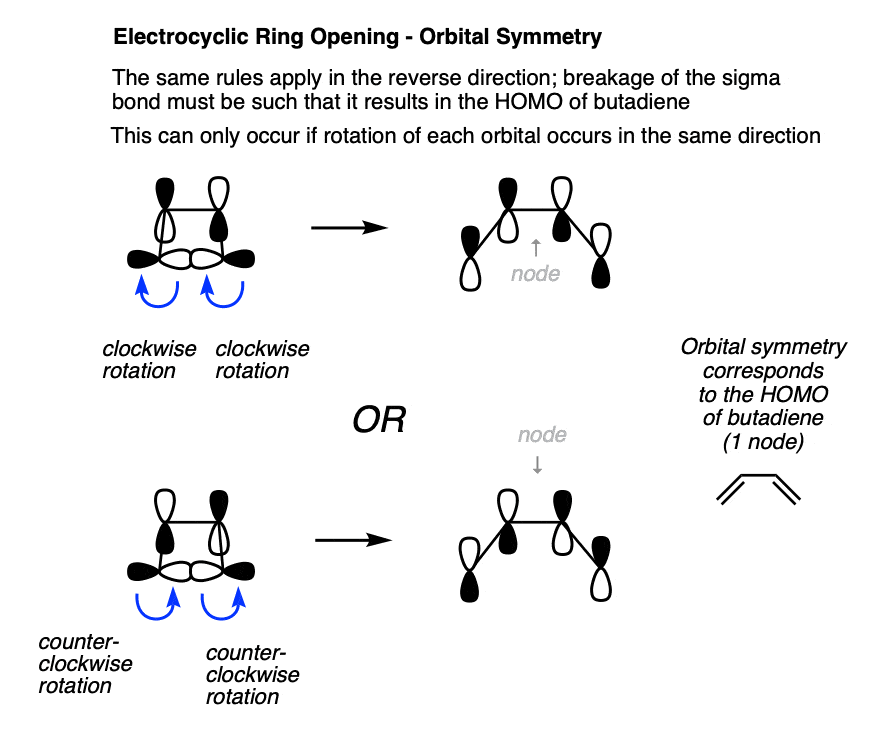
Note again that both of these lobes rotate in the same direction; either both clockwise or both counterclockwise.
Since the phrase “rotation in the same direction” comes up so much, it has been granted a special name. We call it “conrotatory” rotation, as exemplified in these “double egress” hospital doors. [Note 5]
6. Applying “Conrotatory” Rotation Predict The Stereochemistry Of Substituents From Electrocyclic Ring-Opening
It’s not just the p-orbitals that rotate in a conrotatory fashion – it’s the atom itself, which means that the two other substituents come along for the ride.
Understanding that the terminal carbons must rotate in a “conrotatory” fashion for 4pi electrocyclic reactions is the whole key to being able to predict the stereochemistry.
For these reactions, it helps if you imagine looking at the molecule from the front.
Here’s an example of 3,4-dimethylcyclobutene, showing how the two methyl groups each rotated the “same way” (conrotatory), resulting in the cis-, trans- product.
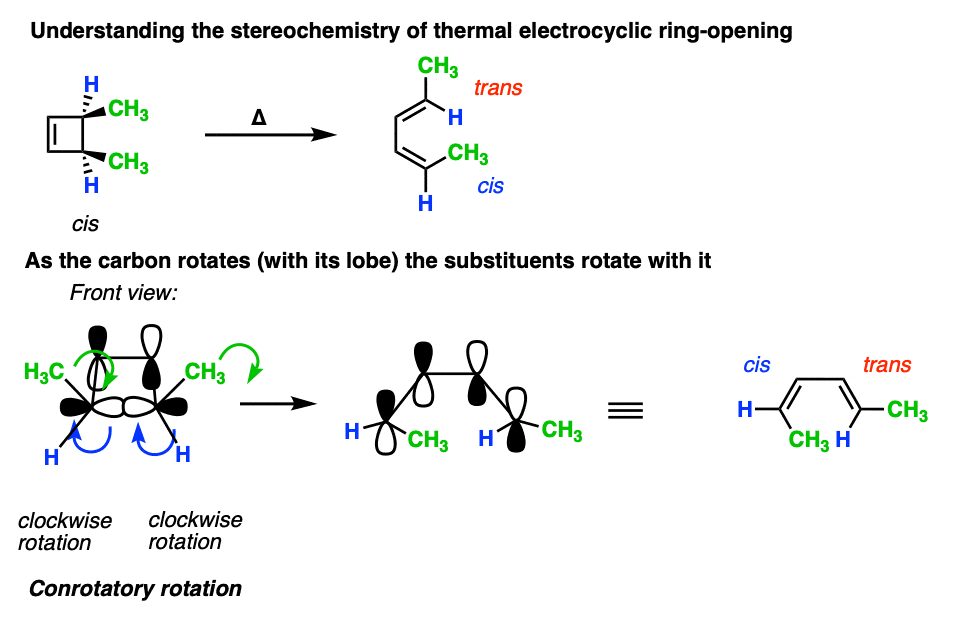
Applying the same rules to trans-3,4-dimethylcyclobutene results in the trans, trans diene. [Note 4]
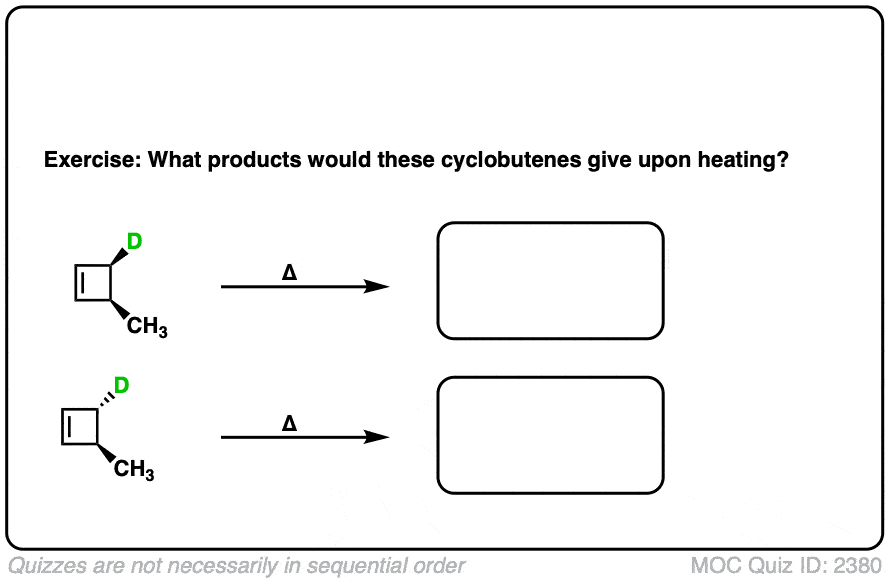
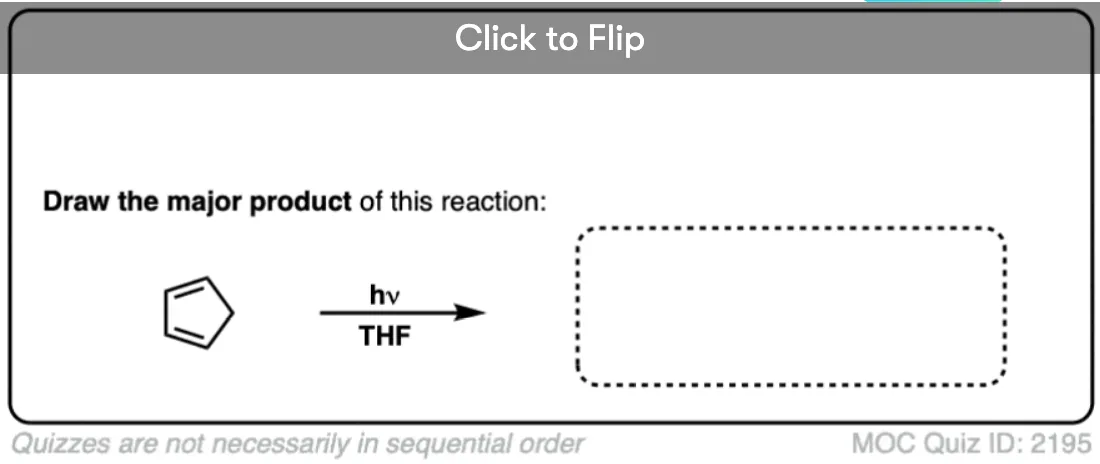
Ready to try it for yourself? Try this quiz.
 Click to Flip
Click to Flip
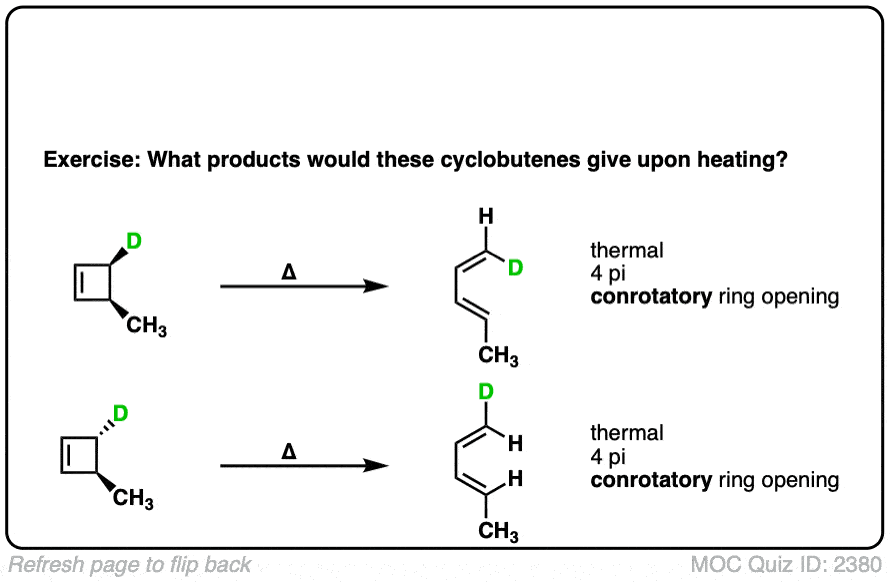
7. Predicting The Products From Conrotatory Ring-Closing
What about the going in the opposite direction, from a diene to cyclobutene? How do we predict the products in this case?
By applying the exact same methodology. The terminal carbons will rotate in a conrotatory fashion.
Again, it’s helpful to look at the molecules from the front. Here’s an example of a cis-, trans- diene undergoing conrotatory ring closure, either both-clockwise or both-counter-clockwise.
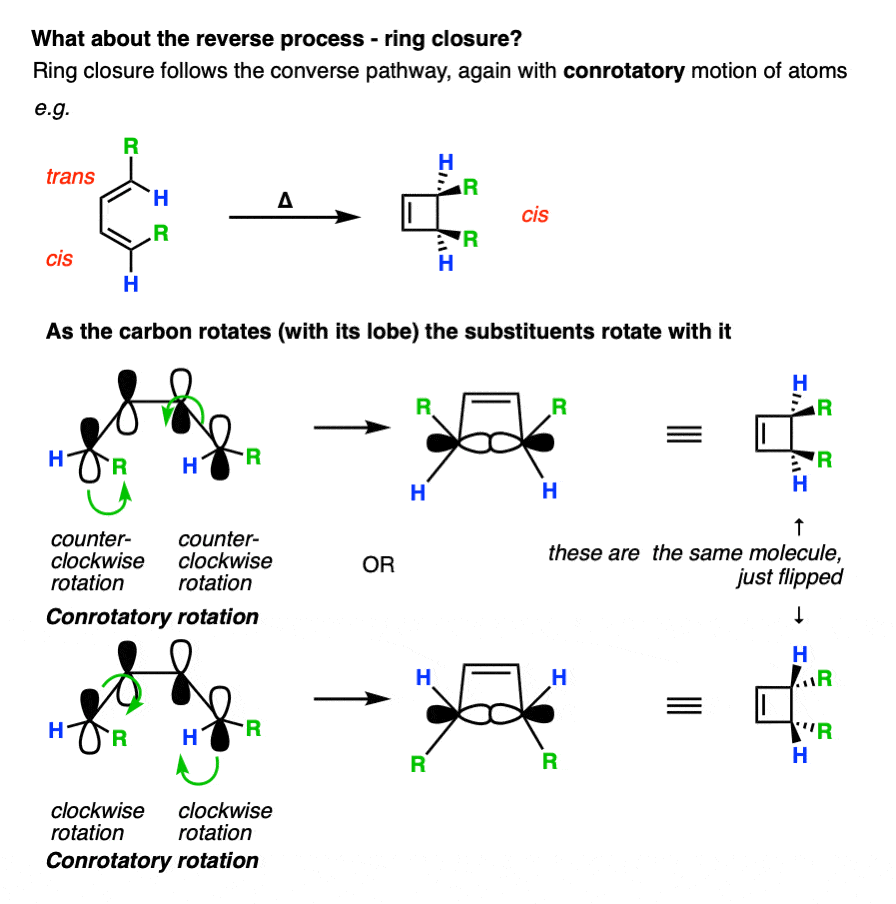
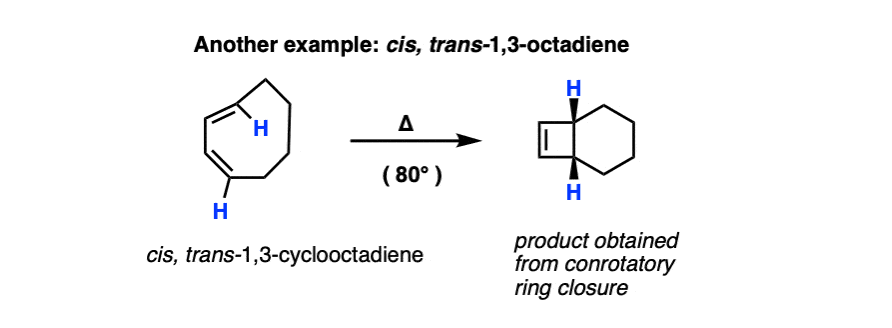
Remember that electrocyclic ring closure to give dienes is usually unfavorable due to ring strain? Here’s an example of a favorable electrocyclic ring closure, since it relieves the strain in a cyclic diene containing a trans– double bond.
8. Electrocyclic Reactions Under “Photochemical” Conditions
It turns out there’s another way to coax electrocyclic reactions to occur, other than heating the crap out of them.
Light (which we denote as hν ) can also do the trick. (In the cases of dienes, we’re generally talking about UV light here, since their absorbance maxima are deep into the UV.)
However, when one uses UV light, the stereochemistry of the products is different from those we see under “thermal” conditions. In electrocyclic ring-opening of cis-3,4-dimethylcyclobutene the trans,trans-diene is formed instead of the cis-, trans- product we observed under “thermal” conditions.
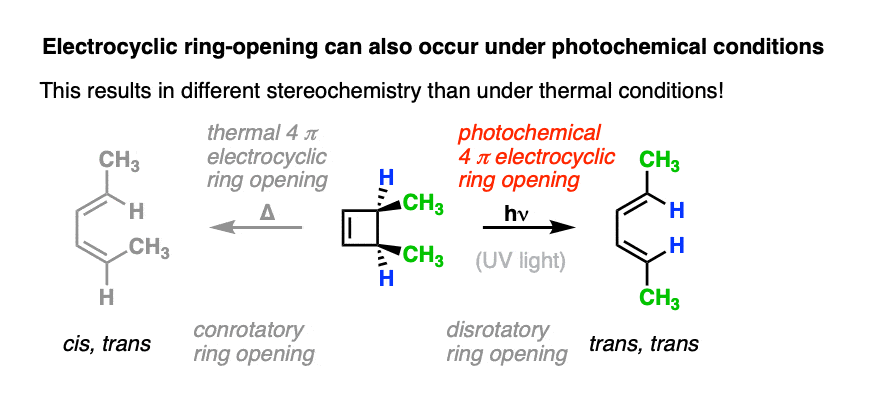
Also note that trans-3,4-dimethylcyclobutene gives the cis-,trans- product under photochemical conditions!
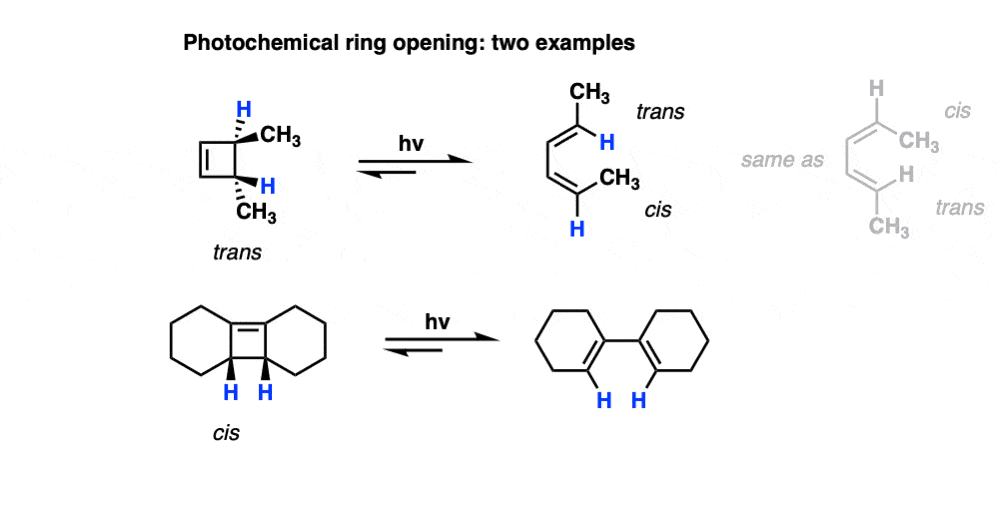
9. How Do “Photochemical Conditions” (UV light) Affect Electrocyclic Reactions?
So what’s going on? The first thing to note is that UV light will promote an electron from the highest-occupied molecular orbital (HOMO) of the diene pi system to the lowest-energy unoccupied molecular orbital (LUMO). See: Introduction to UV-Vis Spectroscopy
If we examine the appearance of the HOMO of butadiene with the appearance of the next-highest orbital (π3), you’ll note that there are two nodes in the pi system instead of one, but more importantly, the lobes on the end of the pi-system have a different relationship.
In the HOMO of butadiene (π2) the lobes on C-1 and C-4 are aligned in opposite directions, whereas in the LUMO the lobes on C-1 and C-4 are aligned in the same direction.
So when UV light promotes an electron from the HOMO of butadiene (π2) to the LUMO of butadiene (π3), that orbital becomes the “new” HOMO.
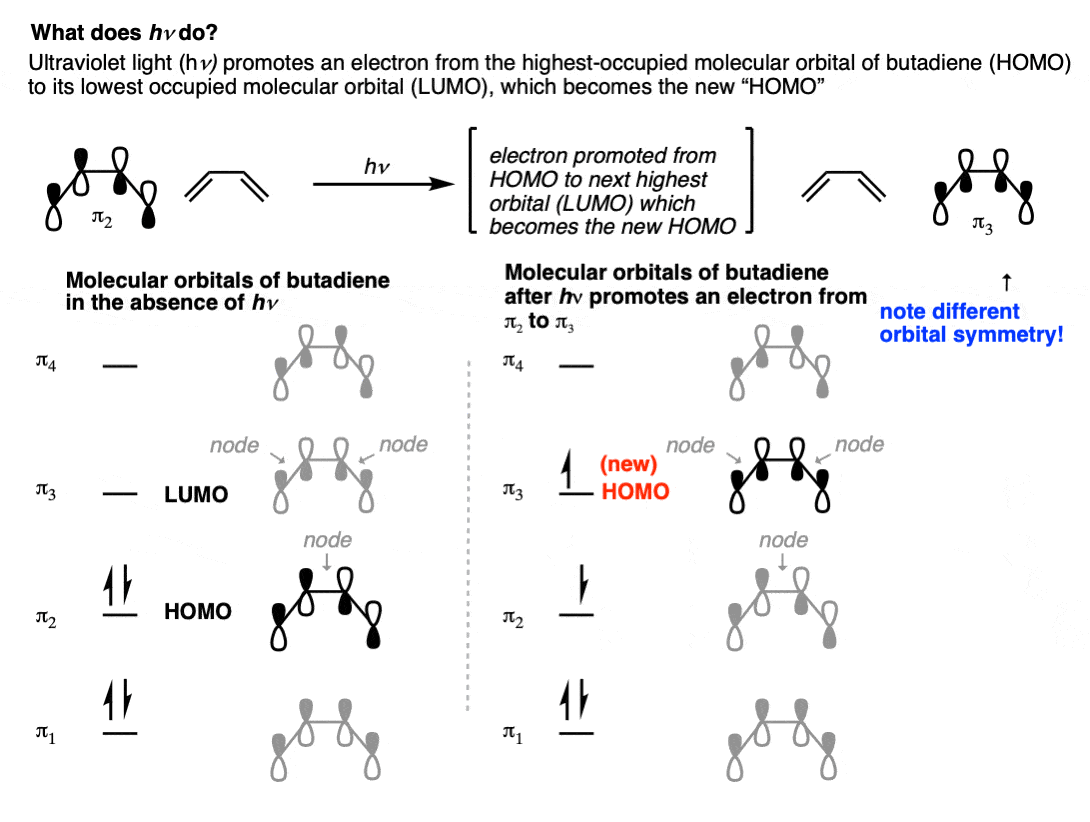
Since the symmetry of the lobes on C-1 and C-4 are different between π2 and π3, the terminal carbons have to rotate in opposite directions in order to form a bond, in contrast to “thermal” conditions where they needed to rotate in the same direction.
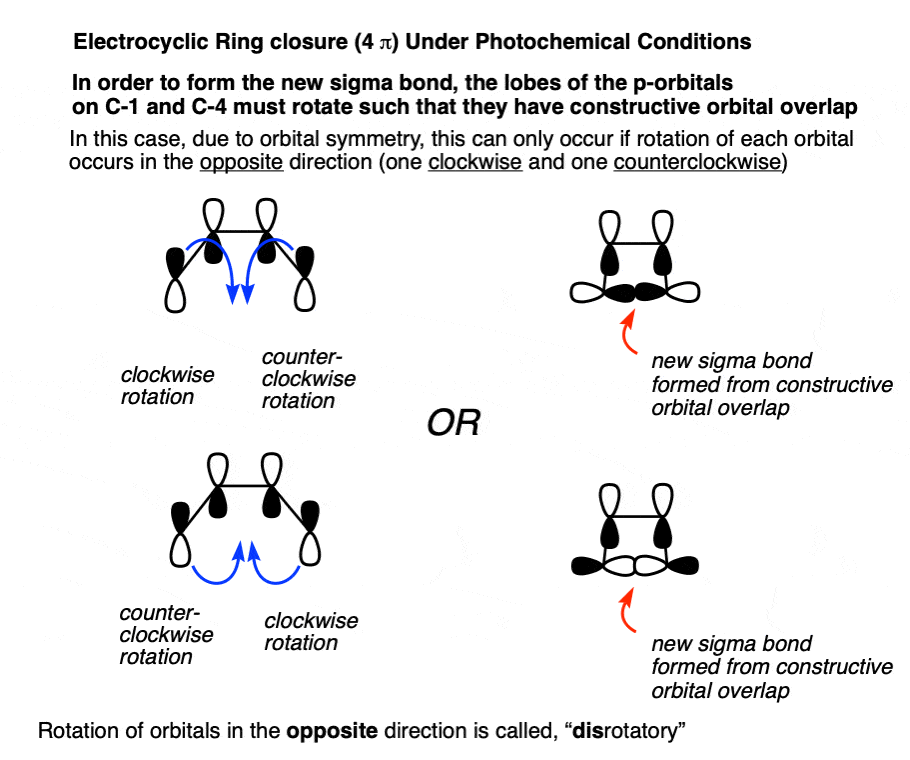
This term, “rotation in opposite directions” goes by the name, “disrotatory”, as demonstrated by the opening and closing of these double doors.
10. The Orbital Symmetry Of Electrocyclic Ring Opening Under “Photochemical” Conditions
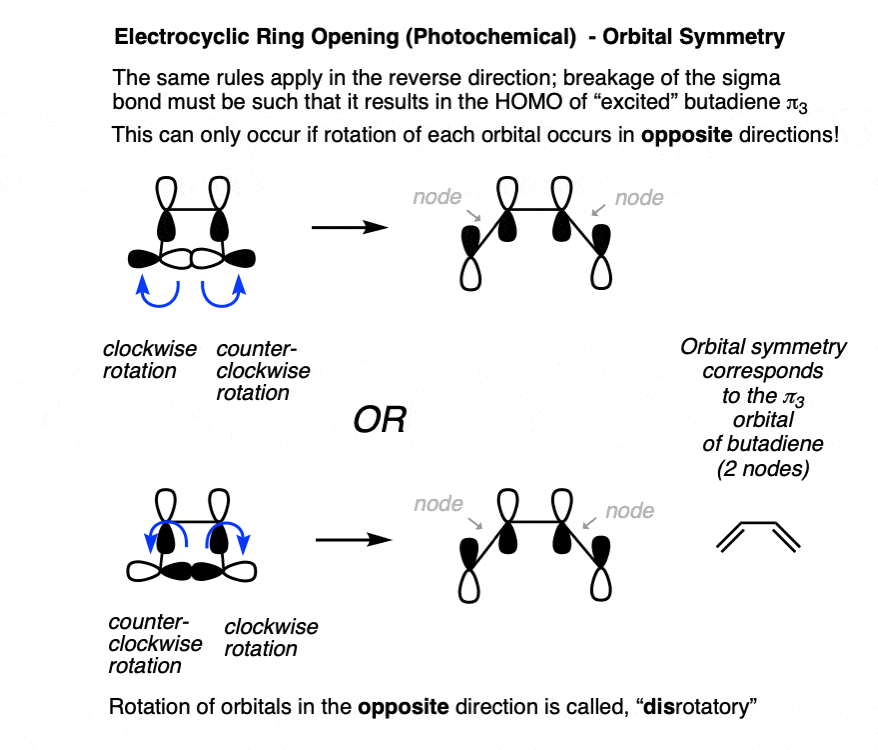
Since electrocyclic ring-opening follows the reverse pathway from ring-closing, we can apply the same rules in the opposite direction.
When breaking the C-C sigma bond, the lobes must break such that they align to re-form the pi bond, which occurs through adopting the orbital symmetry of the π3 orbital (i.e. with two nodes). That can only happen if the carbons rotate in opposite directions. [Note 6]
11. Applying “Disrotatory” Rules To Electrocyclic Ring-Opening Under Photochemical Conditions
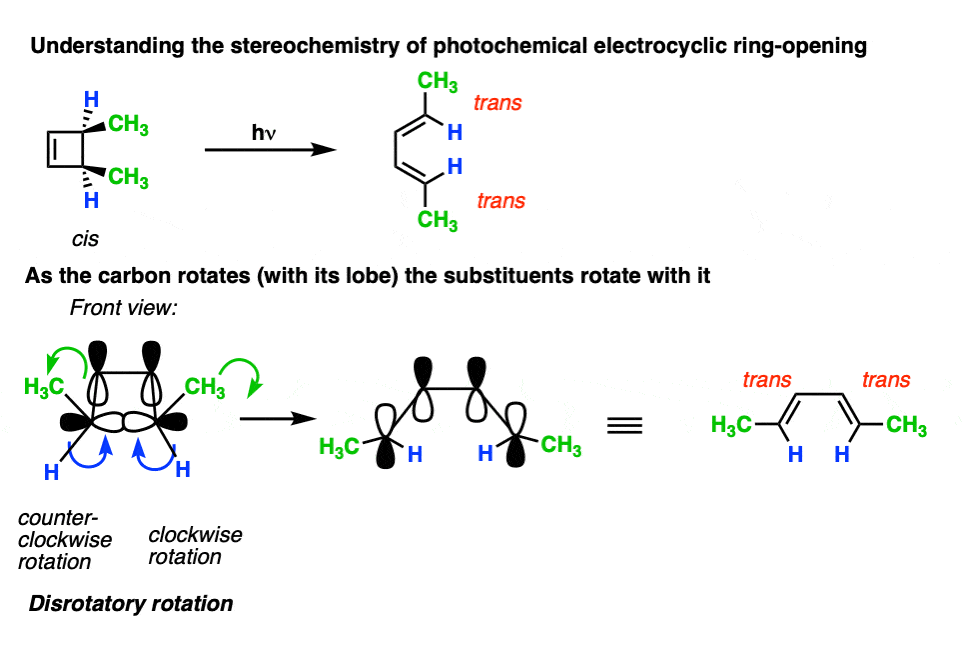
Let’s now apply these rules and see what happens to the stereochemistry of the substituents under photochemical conditions.
When cis-3,4-dimethylcyclobutene is irradiated, the trans,trans-diene is obtained.
Again, it usually helps to look at the cyclobutene end-on to visualize the lobes as a set of double doors that you push or pull open.
Here’s another example.
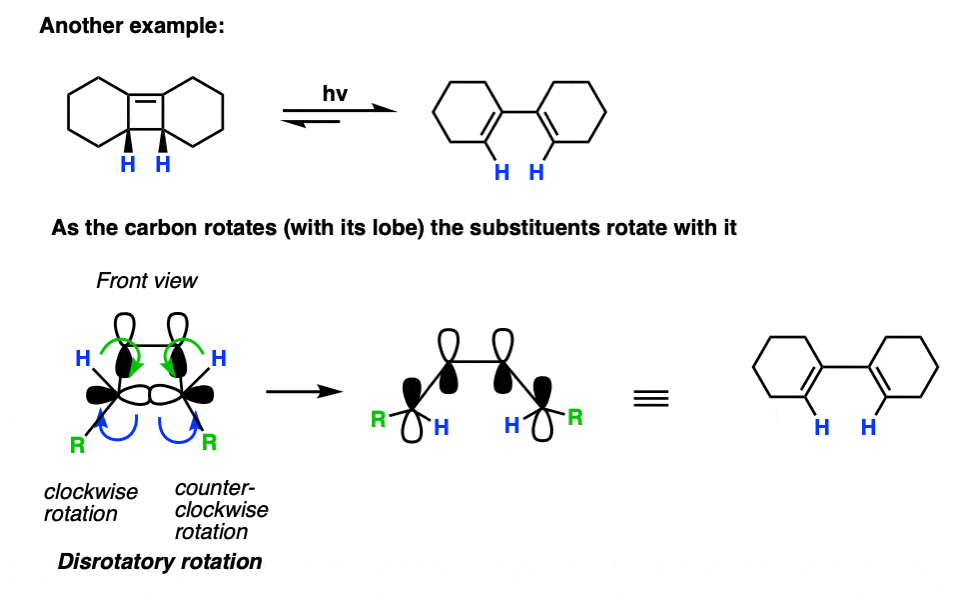
Note that in this case, only one disrotatory option is possible, since the alternative would result in a highly strained trans- double bond within a cyclohexene ring.
12. Summary: Thermal vs Photochemical Ring Opening and Closing
This has been a lot. Here’s what we’ve seen.
- Cyclobutene can undergo electrocyclic ring-opening to give 1,3-butadiene. Conversely, 1,3-butadiene can undergo electrocyclic ring-closing to give cyclobutene. These species are in equilibrium, and the reaction is concerted and goes through a common transition state.
- Since two pi bonds are involved these are classifed as “4π” electrocyclic reactions.
- These reactions are stereospecific.
- The products can be predicted by examining the orbitals of the HOMO on the terminal carbons of the pi system.
- Under thermal conditions (i.e. heat) the terminal carbons (and their substituents) rotate in a conrotatory fashion (both clockwise or both counterclockwise) during the electrocyclic reaction.
- Under photochemical conditions (i.e. UV light, hν) the terminal carbons (and their substituents) rotate in a disrotatory fashion (one clockwise and one counterclockwise) during the electrocyclic reaction.
This can all be summarized in this short table:

So what about larger pi systems? In the next post we’ll cover 6 pi electron systems (and 8 if we have time). The same concepts will apply!
Notes
Note 1. As so often happens in organic chemistry, experimental observations preceded the theoretical understanding of those observations. The details of this process were not completely worked out until the 1960’s with Woodward and Hoffmann’s work on the conservation of orbital symmetry [Ref]. (Despite how “obvious” the textbooks make these reactions appear to us now, organic chemistry is not a series of logical deductions, like math.)
Note 2. Woodward and Hoffmann definition of electrocyclic reactions:

Note 3. Definition of a stereospecific reaction, from IUPAC: “A reaction is termed stereospecific if starting materials differing only in their configuration are converted into stereoisomeric products.”
Note 4. For our purposes we treat, “both clockwise” and “both counterclockwise” as equivalent, but there are cases where one of the two modes is favored. This is called, “torqueselectivity”, (see ref.)
Note 5. Conrotatory and disrotatory woodward definition.
Note 6. You might wonder: what happens if photochemical ring opening occurred in a conrotatory fashion? Would that really be such a big deal? Well, since the orbital symmetry differs from π3, the electrons would not be part of the pi system (at least initially), and would instead have “di-radical” character. These “disallowed” pathways can occur sometimes, but usually have transition states that are higher in energy than the “allowed” pathway.
Quiz Yourself!
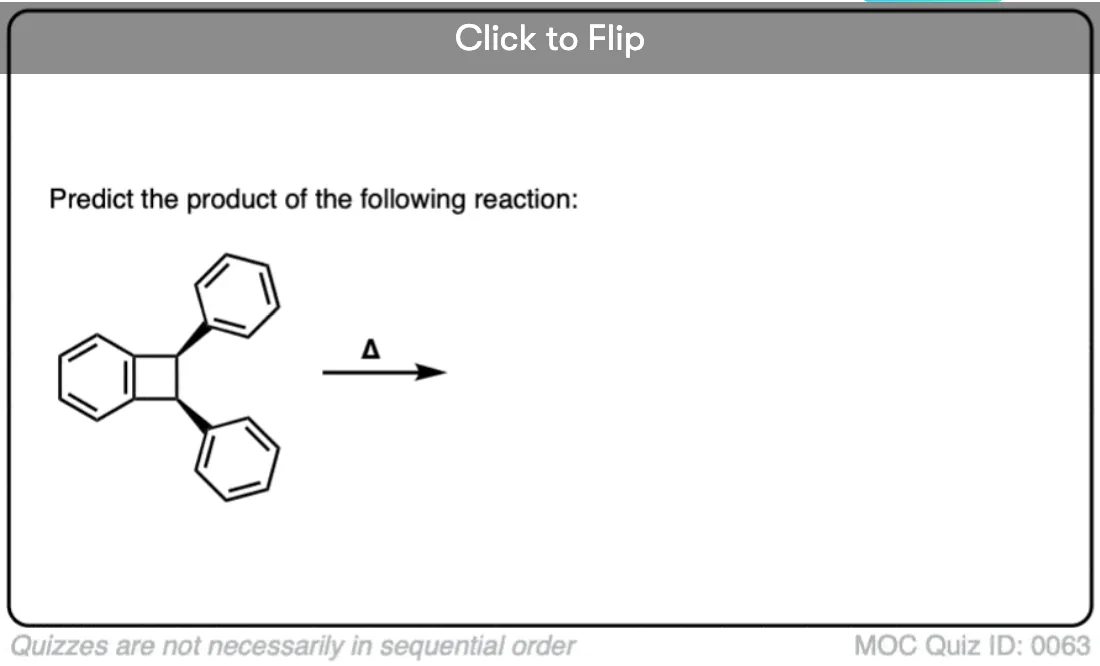
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
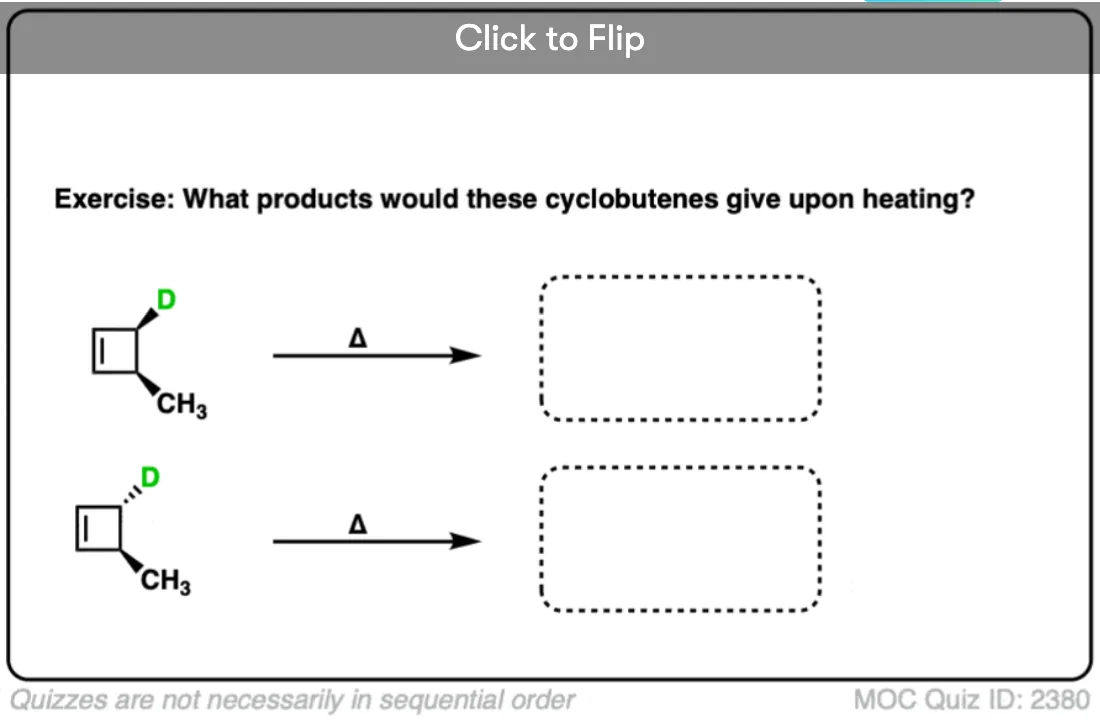
Become a MOC member to see the clickable quiz with answers on the back.
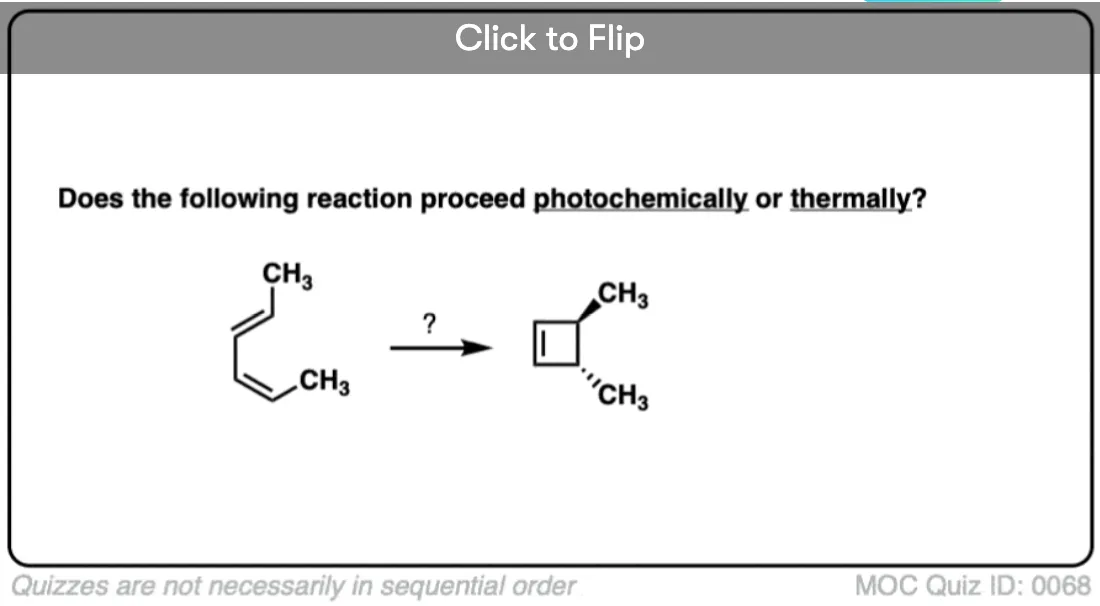
Become a MOC member to see the clickable quiz with answers on the back.
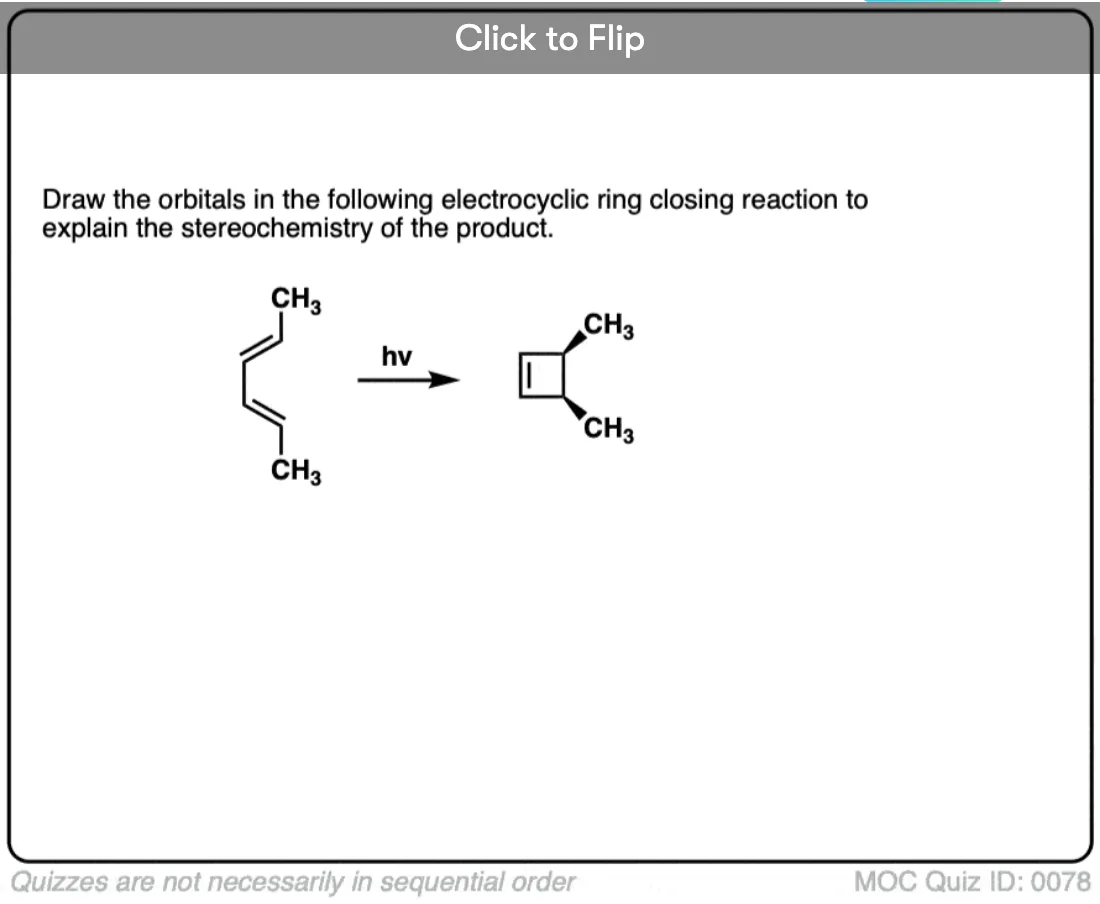
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
References and Further Reading
- Ueber einige Derivate des Cyclobutans.
Willstätter, R. and von Schmaedel, W.
Ber. Dtsch. Chem. Ges. 1905, 38: 1992-1999.
DOI:10.1002/cber.190503802130
Paper from 1905 where Willstätter and his co-worker form cyclobutene at elevated temperature, which isomerizes (through electrocyclic ring-opening) to butadiene. - The Preparation and Isomerization of cis- and trans- 3,4-dimethylcyclobutene
Rudolf E. K. Winter
Tetrahedron Letters, Volume 6, Issue 17, 1965, 1207-1212
DOI: 10.1016/S0040-4039(01)83997-6
Classic study on thermal 4pi electrocyclic ring-opening demonstrating that the reaction is stereospecific. - Photochemical Transformations of Conjugated Cycloheptadienes
O. L. Chapman, D. J. Pasto, G. W. Borden, and A. A. GriswoldO.L. Chapman, D. J. Pasto, G. W. Borden, and A. A. Griswold
Journal of the American Chemical Society 1962 84 (7), 1220-1224
DOI: 10.1021/ja00866a030
Some of the first examples of photochemical 4 pi electrocyclic ring closure of cyclic dienes to give cyclobutenes. - The Thermal Cyclization of 1,3-Dienes to Cyclobutenes
Kenneth M. Shumate, Peter N. Neuman, and G. J. Fonken
Journal of the American Chemical Society 1965 87 (17), 3996-3996
DOI: 10.1021/ja01095a047
Thermal ring closure of dienes to give cyclobutenes is usually disfavored due to ring strain. In this case the cyclic dienes are quite strained due to the presence of a trans double bond in the ring, which makes ring closure more favorable. - Energies of alternate electrocyclic pathways. Pyrolysis of cis-3,4-dimethylcyclobutene
John I. Brauman and William C. Archie Jr.
Journal of the American Chemical Society 1972 94 (12), 4262-4265
DOI: 10.1021/ja00767a038
Classic study on the stereochemistry of electrocyclic ring opening. - Direct Investigation of the Photochemical Ring-Opening Dynamics of Cyclobutene with Resonance Raman Intensities
Mary K. Lawless, Steven D. Wickham, and Richard A. Mathies
Journal of the American Chemical Society 1994 116 (4), 1593-1594
DOI: 10.1021/ja00083a064
Observing the photochemical ring opening of cyclcobutene to 1,3-butadiene is difficult since the high energies of the photons lead to multiple rearrangement products. This study uses Raman spectroscopy data to support disrotatory ring opening. - Substituent Effects on Rates and Stereoselectivities of Conrotatory Electrocyclic Reactions of Cyclobutenes. A Theoretical Study
Satomi Niwayama,E. Adam Kallel,David C. Spellmeyer,Chimin Sheu, and, and K. N. Houk*
The Journal of Organic Chemistry 1996 61 (8), 2813-2825
DOI: 10.1021/jo950884i
In the post above, we treated the two conrotatory cases of “both clockwise” or “both counterclockwise” as equal. This paper examines “torqueselectivity”, where one of the two possible rotation modes is favored over the other. - Stereochemistry of Electrocyclic Reactions
R. B. Woodward and Roald Hoffmann
Journal of the American Chemical Society 1965 87 (2), 395-397
DOI: 10.1021/ja01080a054
In this classic paper Woodward and Hoffmann define the terms “electrocyclic reaction”, “conrotatory”, and “disrotatory”. - Strain Energy of Small Ring Hydrocarbons. Influence of C−H Bond Dissociation Energies
Robert D. Bach* and and Olga DmitrenkoJournal of the American Chemical Society 2004 126 (13), 4444-4452
DOI: 10.1021/ja036309a
The strain energy of cyclobutene is 29.8 kcal/mol, compared to 26.3 kcal/mol for cyclobutane. - Orbital Symmetry and the Photochemistry of Cyclcobutene
William J. Leigh
Canadian Journal of Chemistry, 1993, 71, 147-155.
DOI: 10.1139/v93-021#.Xm5OsJNKh24
This paper has examples of photochemical ring-opening of several cyclobutenes. As sometimes happens, reality is messier than theory.
Very good Explanation. Easily Understand. 24-10-2025, Friday
Thank You Professor
Terpenes, Sesquiterpenes, Sesquiterpenoids in Marijuana & Metabolism within the Human Vision Centers?
Recently, the MO details of the photocyclic conrotary ring-opening of 7-dehydrocholesterol to pre-vitamin D3 (in skin), prior to the thermal 1,7 hydride shift, piqued my curiosity.
Beaudry et al. in Chemical Reviews, 2005, Vol. 105, No. 12 (attached below) write:
The biosyntheses of vitamin D3 (76a) and vitamin
D 2 (76b) are arguably the best-known examples of
electrocyclic reactions in Nature (Scheme 18). The
transformations of 7-dehydrocholesterol (74a) and
ergosterol (74b) into vitamin D 3 (76a) and vitamin
D 2 (76b), respectively, have been extensively re-
viewed, and they are therefore only briefly sum-
marized here.35 Photochemical conrotatory ring open-
ing of 74a leads to pre-vitamin D3 (75a).
However, they cite only structures and not the frontier MO analysis desired. I am unable to find the “extensive reviews” they are talking about. There is the book section “Photochemistry of vitamin D and its isomers and of simple trienes” apparently at https://onlinelibrary.wiley.com/doi/book/10.1002/9780470133415#page=311, but it’s from a 1979 book that I can’t seem to find database access for. I realize the solution must be “too trivial to publish” in research articles but I wonder why I can’t find the MO solution in most modern textbooks if it is the “best-studied” reaction. If the MO solution is so trivial, why don’t we encounter it earlier in our biochemical education?
Such nice explanation thank you so much
Very nice..I can understand easily thank you.🙏
Very understandable and clear.Also thorough and addressed all aspects without leaving anything to doubt