Reactions of Aromatic Molecules
Electrophilic Aromatic Substitutions (2): Nitration and Sulfonation
Last updated: May 30th, 2026 |
- Aromatic rings undergo nitration and sulfonation through the electrophilic aromatic substitution mechanism.
- Aromatic rings can undergo nitration when treated with nitric acid HNO3 in addition to the strong acid H2SO4.
- This leads to the formation of the nitronium ion NO2+ which is the active electrophile.
- Aromatic rings can undergo sulfonylation when treated with sulfur trioxide (SO3) in the presence of strong acid (H2SO4).
- The active electrophile is HSO3(+)
Table of Contents
- Recap: The Three Stages Of Electrophilic Aromatic Substitution Reactions
- Addition of NO2 (Nitration)
- Nitration of Benzene
- The Nitronium Ion (NO2+) Is The Key Electrophile In Aromatic Nitration
- Addition of SO3H (Sulfonation)
- Summary: Aromatic Sulfonation and Nitration
- Notes
- Bonus Topic: Reversibility of Sulfonation
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Recap: The Three Stages Of Electrophilic Aromatic Substitution Reactions
In the previous post on halogenation via electrophilic aromatic substitution, we saw that this electrophilic aromatic substitution reaction proceeded in three distinct stages:
- Activation. Since halogens (Cl2, Br2) don’t usually react with aromatic molecules at a reasonable rate, a Lewis acid catalyst (e.g. FeCl3) is added to “activate” the electrophile toward attack
- Attack of Electrophile By The Aromatic Ring. The activated electrophile is attacked by the aromatic ring, resulting in a carbocation intermediate (this is the rate determining step, and Step 1 in the generic mechanism of electrophilic aromatic substitution).
- Deprotonation. The carbocation intermediate is deprotonated by a weak base, restoring aromaticity (Step 2 in the generic mechanism of electrophilic aromatic substitution).
In today’s post we’ll cover the mechanism of two other important electrophilic aromatic substitution reactions that proceed through Brønsted acid catalysis – nitration and sulfonylation.
2. Addition of NO2 (Nitration)
Don’t Try This At Home
“Nitration” is the name we give to the process of attaching the nitro group (NO2) to a molecule.
If we skipped this reaction in previous chapters, it’s because nitrating functional groups other than aromatic rings leads to some pretty explosive products. For example, nitration of the tri-ol glycerol produces the infamous nitroglycerin, with three excellent leaving groups (ONO2 ; technically, these are “nitrate” groups, not nitro groups). The slightest disturbance to this liquid leads to a chain reaction resulting in the rapid generation of hot gas. [Note 1]
4 moles of liquid nitrogylcerin creates 35 moles of gas: 6 moles of N2, 12 moles of CO, 10 moles of H2O, and 7 moles of O2.
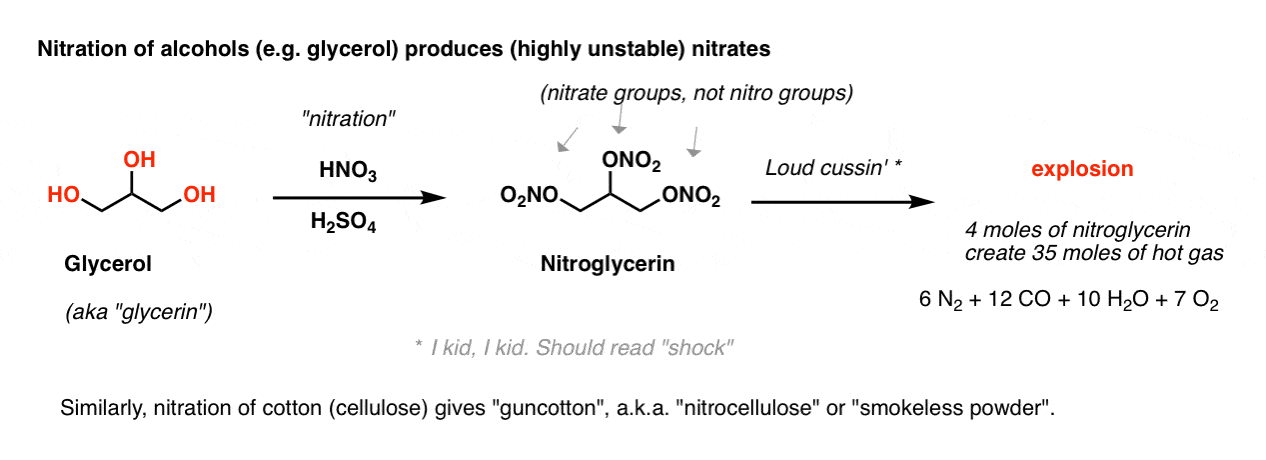
[Note to readers: don’t try to make nitroglycerin].
My dad and his neighborhood buddies all made black powder and other explosives when they were kids, but making “nitro” was considered the Holy Grail. One day, my dad got all the material ready for making nitroglycerin, but on the day when it came to actually making the stuff, he chickened out. Wise move, Dad. It’s pretty hard to to do surgery when you are missing a couple of fingers.
Similarly, nitration of cellulose (a polymer of the sugar glucose) produces “nitrocellulose“, which besides being used in smokeless powder, is a polymer used as film stock in the early days of the film industry. “Celluloid”, as it was called, is not a contact explosive, but it does have the unfortunate property of being prone to spontaneous combustion. (Hollywood later moved to cellulose acetate, a.k.a. “safety film”. )
3. Nitration of Benzene
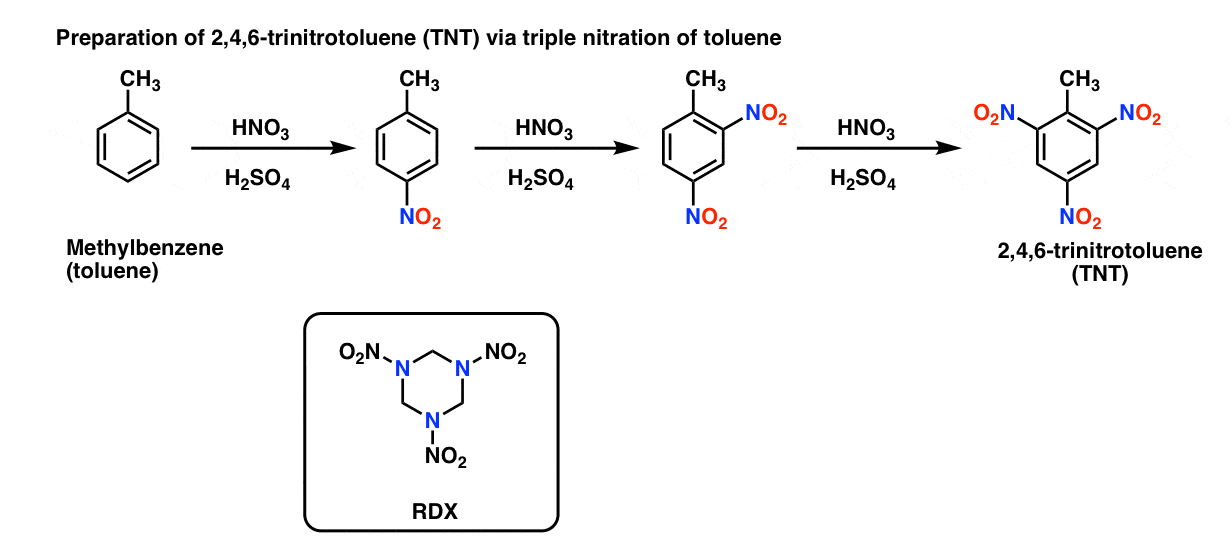
In contrast to nitration of alcohols, the nitration of benzene produces relatively stable nitro compounds that are much more difficult to detonate. For example, the high-explosive TNT (2,4,6-trinitrotoluene) is formed by triple nitration of toluene. Another explosive, RDX comes from nitration of trihydro-1,3,5-triazine. [Note 2]
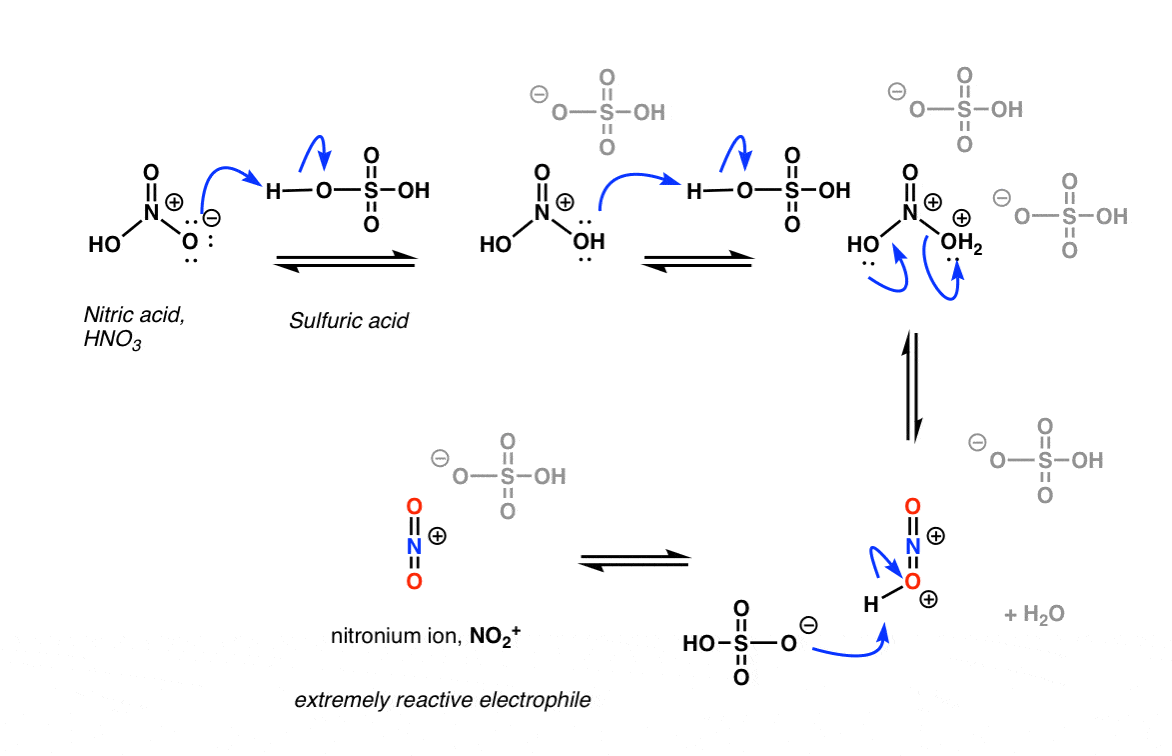
The key reagent for nitration is nitric acid, HNO3. By itself, nitric acid is a relatively slow-acting electrophile, especially in the presence of a poor nucleophile such as benzene. [Note – in the case of phenol and other aromatic rings with strongly activating groups, HNO3 by itself is sufficient for nitration].
Upon addition of acid, however – the usual choice being sulfuric acid, H2SO4 – a rapid reaction with benzene ensues.
The key to the process is protonation of OH on nitric acid, which converts it to H2O. Being a much better leaving group than HO(-), H2O is rapidly lost from nitric acid to give the highly reactive “nitronium ion”, NO2+.
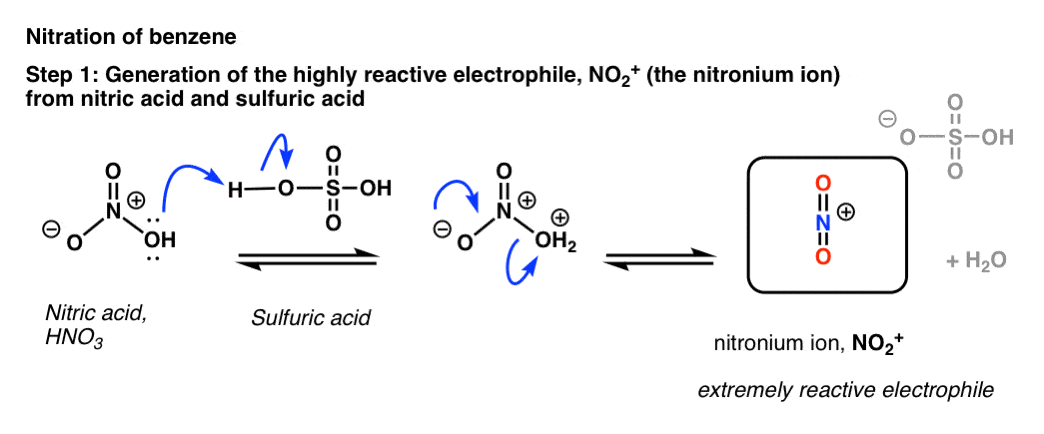
[note: the mechanism drawn above is a little oversimplified. Two equivalents of H2SO4 are usually best – protonate HNO3 twice (not once) followed by loss of water, and then deprotonation of HNO2+.
To see an image of the mechanism hover here for a pop-up image or open image link.
4. The Nitronium Ion (NO2+) Is The Key Electrophile In Aromatic Nitration
The nitronium ion is what really gets the job done here.
You might recall that this is just like what we saw with the halogens, where use of a Lewis acid in that case produced an activated electrophile that reacted much more quickly with benzene.
So what happens when the nitronium ion meets benzene?
In the first (and rate-determining) step of electrophilic aromatic substitution, the nitronium ion (NO2+) is attacked by a pair of pi electrons on the aromatic ring to give a carbocation intermediate, forming C–N and breaking C–C (pi) and N–O (pi).
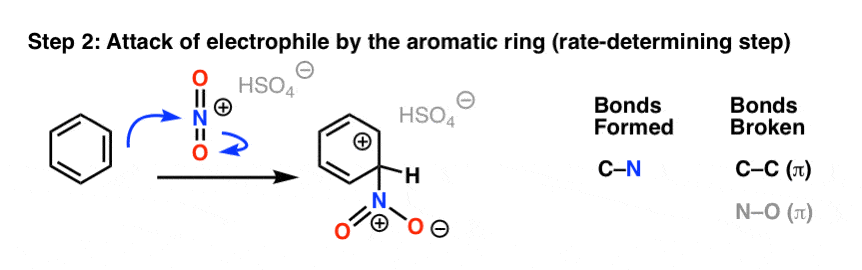
The second step of electrophilic aromatic substitution, which is relatively fast, is an acid-base reaction. A weak base (such as water, or the HSO4– ion left after protonation of HNO3) removes a proton from carbon bearing the nitro group, breaking C–H and re-forming C–C pi. Aromaticity is restored.
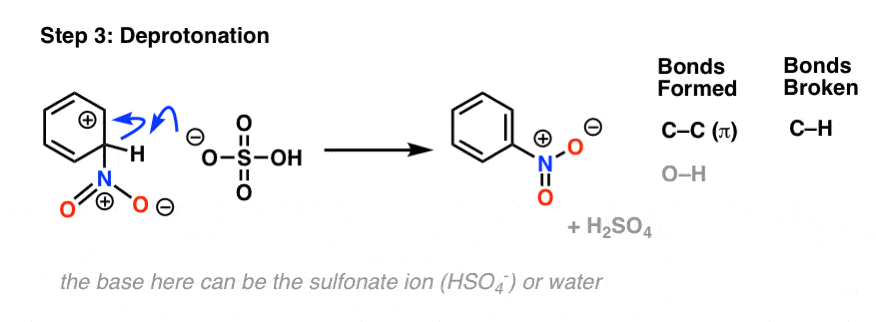
The last two steps we covered previously in the generic electrophilic aromatic substitution mechanism, and they are actually very similar between all electrophilic aromatic substitution reactions.
5. Addition of SO3H (Sulfonation)
The sulfonyl group, SO3H can also be added to an aromatic ring via electrophilic aromatic substitution.
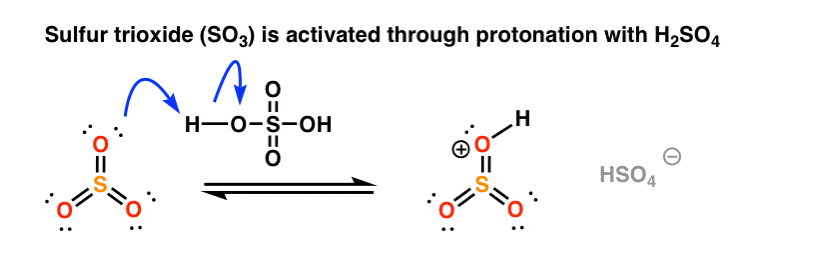
In this case the electrophilic reagent is sulfur trioxide, SO3 (a gas) which can be introduced by bubbling through the solvent. On its own, SO3 is not particularly reactive with aromatic rings, but as we’ve seen, addition of an acid can increase electrophilicity (and reaction rate) considerably. [See post: The power of acid catalysis]
Like nitric acid, sulfur trioxide is “activated” by the addition of a proton from sulfuric acid. [Note: this combination of SO3 and H2SO4 is called “fuming sulfuric acid“, or “oleum” if you want to go for some really old school nomenclature. ]
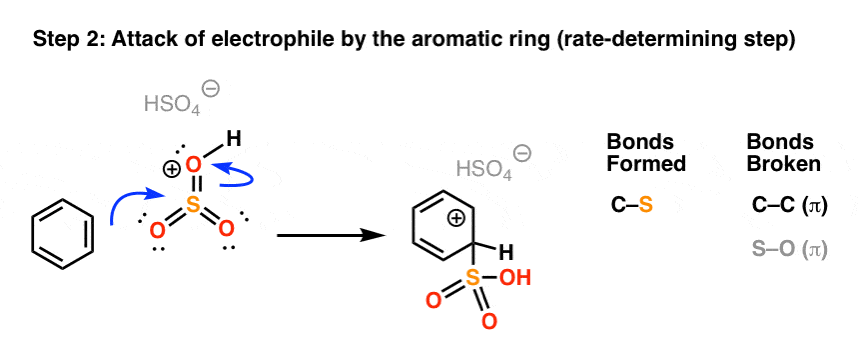
In the rate determining step, the highly electrophilic SO3H(+) is then attacked by the aromatic ring to give the carbocation intermediate, forming C-S and breaking C-C (pi).
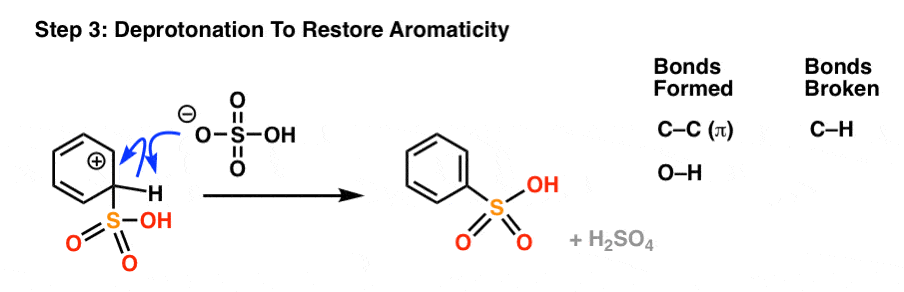
As with all electrophilic aromatic substitutions, the C-H bond is then deprotonated with a weak base to regenerate the C–C pi bond and restore aromaticity, providing the sulfonic acid product.
And… that’s really it for the key mechanisms of nitration and sulfonation.
6. Summary: Aromatic Sulfonation and Nitration
If you start comparing reactions, you should see that the key mechanism of electrophilic aromatic substitution doesn’t really change except for the identity of the electrophile.
What really give each reaction its unique flavor is how the electrophile is activated, either by Lewis acid catalysis (chlorination, bromination) or Brønsted acid catalysis (nitration, sulfonation).
In the next post, we’ll cover the fifth and sixth of the six key electrophilic aromatic substitution reactions: Friedel-Crafts alkylation and Friedel-Crafts acylation, and how they are used to form new carbon-carbon bonds on aromatic rings.
Next Post: Friedel-Crafts Alkylation and Acylation
Notes
Note 1. I am told that if nitroglycerin is neutralized with sodium bicarbonate, it is “reasonably” safe to work with, so long as you do not breathe it or get it on your skin. (reasonably safe still means, “don’t do it”). Upon prolonged standing, nitroglycerin develops a brown-red color, which is a sign that it is going “off”.
I don’t know, but I wonder if the detonation process of nitroglycerin starts with homolytic cleavage of the weak O-NO2 bond, to give the very nasty (and strongly colored) •NO2 radical. This can abstract a hydrogen atom from nitroglycerin, and then – shabang! – homolytic cleavage everywhere.
Nitrogen tetroxide (which is in equilibrium with 2 moles of •NO2) is a useful propellant in rocket engines and the like.
Note 2. Preparation of TNT, and structure of RDX.
Sulfonation Is Reversible
See: Aromatic Synthesis (3) – Sulfonyl Blocking Groups for a full post on this topic.
Aromatic sulfonyl groups have a very interesting property. If treated with a strong enough acid in the absence of SO3, the sulfonic acid group can be removed.
Let’s have a look at how this happens.
We’ve been so busy talking about different electrophiles (chlorine, bromine, nitronium, sulfonium…) that we neglected to mention that H+ can act as an electrophile too.
We’ve ignored this because the reaction is so easily reversible – protonation of an aromatic ring is expected to be quickly followed by deprotonation back to the (stable, aromatic) starting material. It’s a cul-de-sac.
However, in the particular case of sulfonyl groups (and one other group, which I’ll mention in the next post) protonation of the ring can have important consequences, IF done in the absence of SO3.
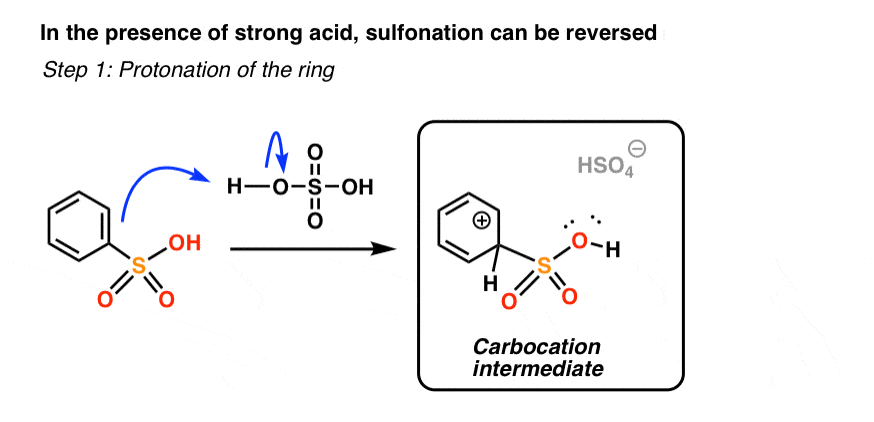
First, let’s see what “protonation of the ring” looks like.
If the ring is protonated at the exact same carbon as that bearing the sulfonyl group [you likely don’t need to know this, but this is called the “ipso” carbon], we generate a carbocation intermediate.
Does the product look familiar? It should – it’s the exact same carbocation intermediate that is generated when benzene attacks protonated SO3, in “step 2” of sulfonation.
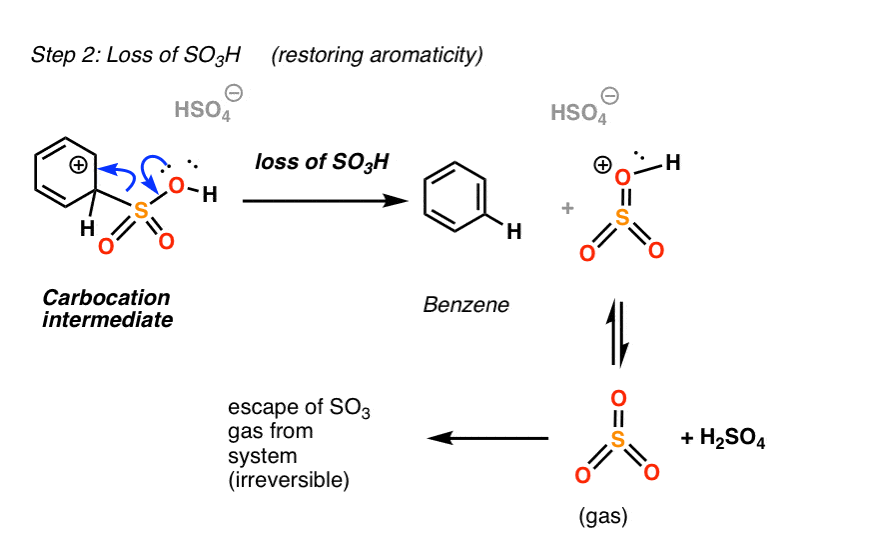
Most of the time, this intermediate will just be deprotonated to regenerate the aromatic sulfonic acid.
However, in the case of SO3H, there is another pathway to restore aromaticity that is not energetically available in the case of most other substituents. Instead of losing H, the intermediate can lose SO3H(+), which leads to the formation of benzene.
This is the reverse of electrophilic attack!
Now here’s where the “absence of SO3” part comes in. In the presence of a high concentration of SO3, benzene would just attack protonated SO3 again and re-form the sulfonic acid.
But if we just add acid in the absence of SO3, then this is less likely to occur. Furthermore, if we vent the reaction (say, by bubbling an inert gas like argon through the reaction mixture, which would eventually carry away any gaseous SO3 along with it), then gaseous SO3 will be slowly removed from the system. If you recall the principle of Le Châtelier, this means that our sulfonic acid is guaranteed to slowly revert back to benzene.
“OK, fine. It’s reversible. So what?” you may rightly ask.
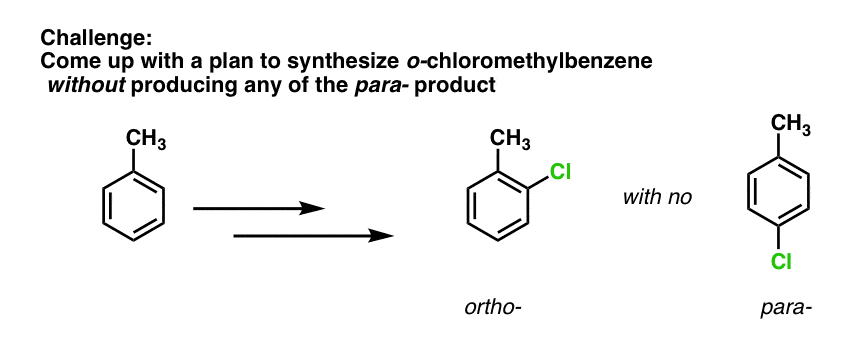
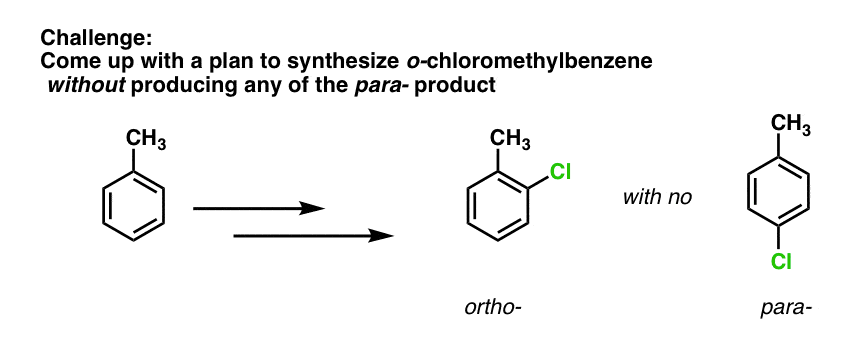
Well, this means that SO3H can be useful as a “blocking group” under some conditions. Again, we’ll have more to say on this when we get to synthetic strategy, but I’ll leave you with a challenge: how would you generate ortho-chloromethylbenzene from methylbenzene without any of the para- product?
{kind=link}
{kind=link}
Quiz Yourself!

Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
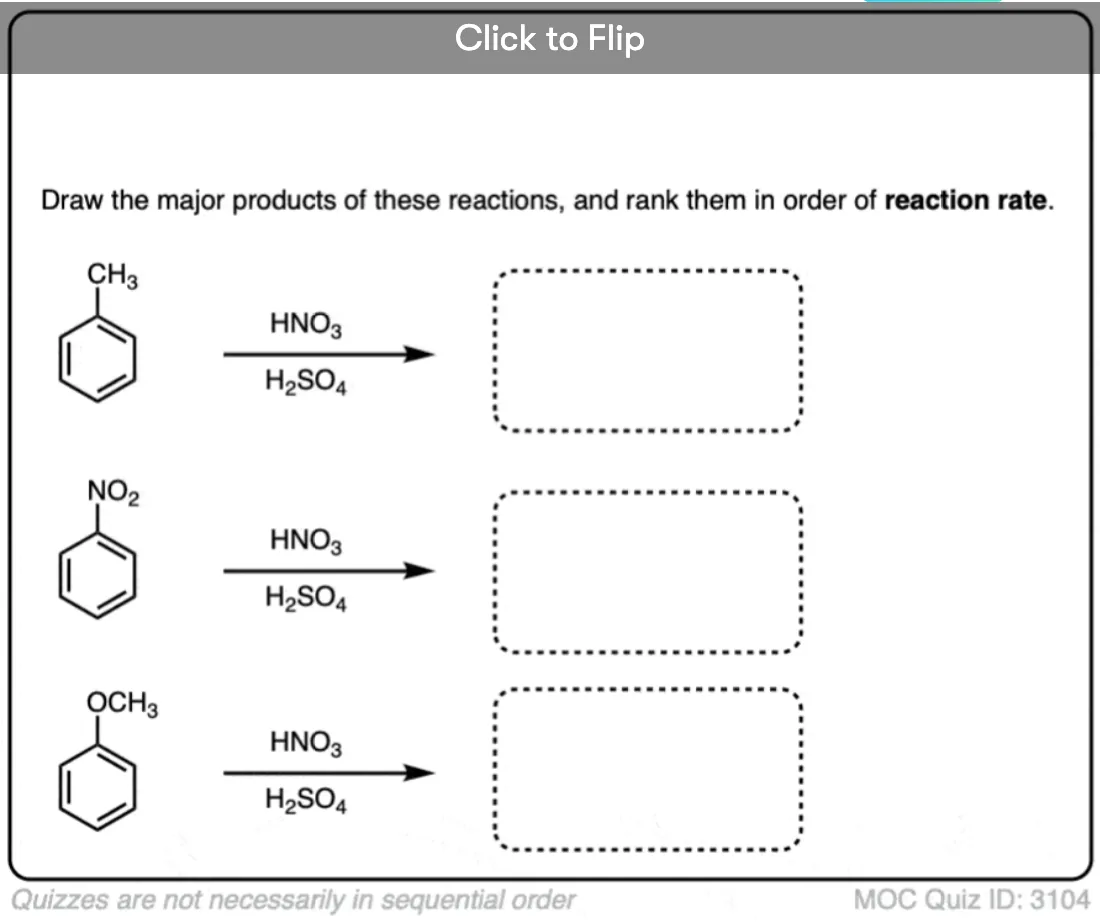
Become a MOC member to see the clickable quiz with answers on the back.
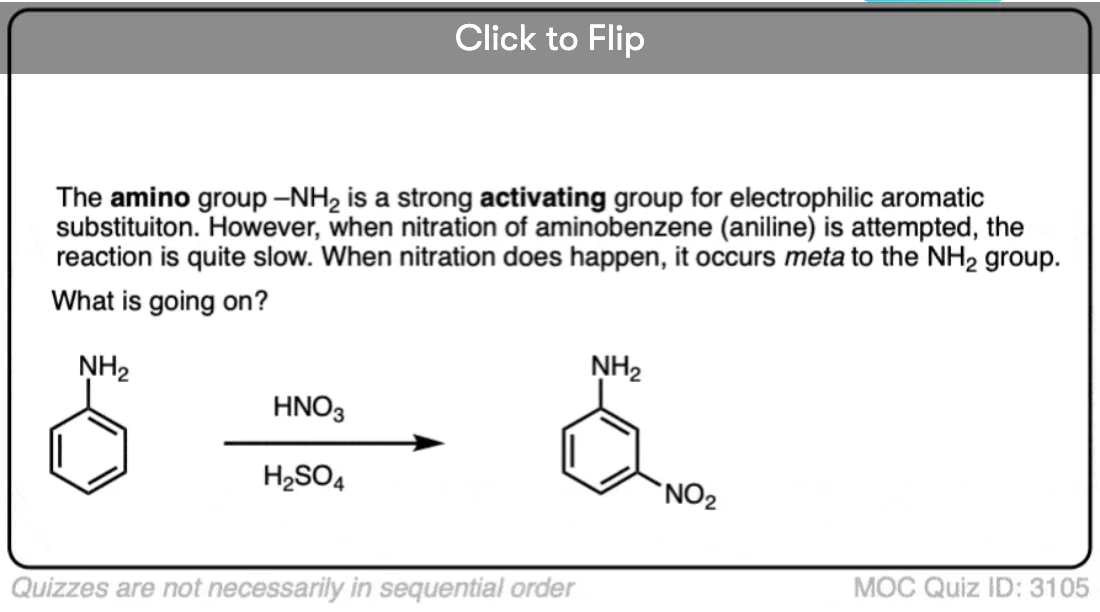
Become a MOC member to see the clickable quiz with answers on the back.
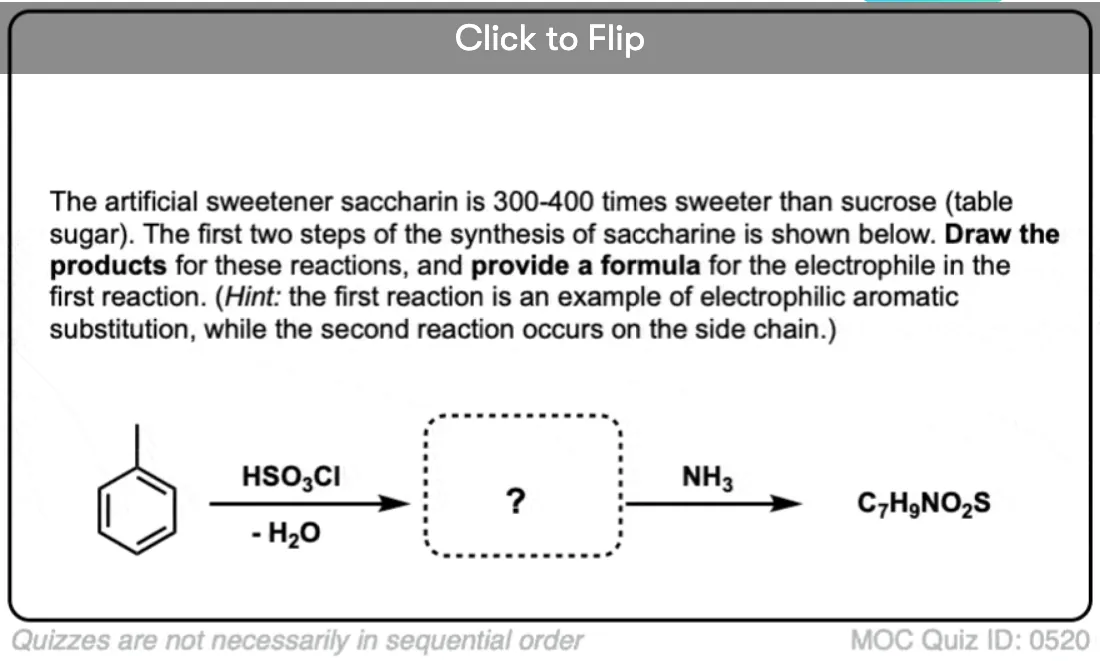
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
For more detailed references on these reactions, consult the sections in the reaction guide. The references here are highlights.
Nitration:
- Kinetics and Mechanism of Aromatic Nitration
J. GILLESPIE, E. D. HUGHES, C. K. INGOLD, D. J. MILLEN & R. I. REED
Nature volume 163, pages 599–600 (1949)
DOI: 10.1038/163599b0
Prof. Christopher Ingold carried out a lot of early mechanistic studies on electrophilic aromatic nitration, and laid the groundwork for our current understanding of the reaction. He first proposed that the nitric acid-sulfuric acid mixture (known as “mixed acid”) used in nitration generated the nitronium ion, [NO2]+, and that this was the reactive species in electrophilic nitration.The Nobel Laureate late Prof. George A. Olah also published a lot of work studying the mechanism of nitration, resulting in at least 50+ papers and a book on the topic. Along the way, he isolated the nitronium ion as a stable salt (which is now commercially available) and demonstrated that it underwent the same reactions as mixed acid system. - Aromatic Substitution. VII. Friedel-Crafts Type Nitration of Aromatics
Stephen J. Kuhn and George A. Olah
Journal of the American Chemical Society 1961, 83 (22), 4564-4571
DOI: 1021/ja01483a016 - Aromatic substitution. XXVIII. Mechanism of electrophilic aromatic substitutions
George A. Olah
Acc. Chem. Res., 1971, 4 (7), 240-248
DOI: 10.1021/ar50043a002 - Unified Mechanistic Concept of Electrophilic Aromatic Nitration: Convergence of Computational Results and Experimental Data
Pierre M. Esteves, José Walkimar de M. Carneiro, Sheila P. Cardoso, André H. Barbosa, Kenneth K. Laali, Golam Rasul, G. K. Surya Prakash, and George A. Olah
Journal of the American Chemical Society 2003, 125 (16), 4836-4849
DOI: 10.1021/ja021307w
This is the grand-daddy paper on nitration, summarizing a lifetime’s worth of work on the subject. If you don’t have time to read any of the other papers here, read this.Sulfonation: - Aromatic sulfonation with sulfur trioxide: mechanism and kinetic model
Samuel L. C. Moors, Xavier Deraet, Guy Van Assche, Paul Geerlings and Frank De Profta
Sci., 2017, 8, 680
DOI: 10.1039/c6sc03500k
A very recent paper on a classic reaction. This paper is a computational analysis of electrophilic aromatic sulfonation, attempting to clarify the kinetics and mechanism of the reaction. - Electrophilic Aromatic Sulfonation with SO3: Concerted or Classic SEAr Mechanism?
Gergana Koleva, Boris Galabov, Jing Kong, Henry F. Schaefer, III, and Paul von R. Schleyer
Journal of the American Chemical Society 2011, 133 (47), 19094-19101
DOI: 1021/ja201866h
The late Prof. P. v. R. Schleyer was a giant in computational Physical Organic chemistry. This is a computational study of aromatic sulfonation that attempts to clarify the mechanism. Computational modeling shows that a trimolecular reaction with 2 SO3 molecules is actually energetically favored.Prof. Hans Cerfontain (U. Amsterdam) did a lot of work on electrophilic aromatic sulfonation, publishing over 100 papers on this topic. The following two papers are just a selection: - Aromatic sulphonation. Part 92. Sulphonation of the three methylphenols and the six dimethylphenols in concentrated aqueous sulphuric acid; and the lsomerization of some of the resulting sulphonic acids and of m-xylene-2-and o-xylene-3-sulphonic acid
Hans J. A. Lambrechts, Zwaan R. H. Schaasberg-Nienhuis and Hans Cerfontain
Chem. Soc., Perkin Trans. 2, 1985, 669-675
DOI: 10.1039/P29850000669 - Kinetics of the desulfonation of benzenesulfonic acid and the toluenesulfonic acids in aqueous sulfuric acid
C. M. Wanders, H. Cerfontain
Rec. Trev. Chim. Pays-Bas 1967, 86 (11), 1199-1216
DOI: 10.1002/recl.19670861106
The desulfonation of arylsulfonic acids is a synthetically useful reaction, and this paper examines the kinetics of the reaction.The following two procedures from Organic Syntheses highlight the utility of desulfonation, allowing access to ortho-substituted aromatics. Whether such a route would actually be conducted or not depends on the target compound. With the improved chromatographic separation techniques available today, one might just separate the isomers from a crude mixture. - o-BROMOPHENOL
Ralph C. Huston and Murel M. Ballard
Org. Synth. 1934, 14, 14
DOI: 10.15227/orgsyn.014.0014 - 2,6-DINITROANILINE
Harry P. Schultz
Org. Synth. 1951, 31, 45
DOI: 10.15227/orgsyn.031.0045
‘,’Electrophilic Aromatic Substitutions (2) – Nitration and Sulfonation
what is the answer to the challenge? how to get only o isomer without any para isomer?
Use sulfonation to put an SO3H group on the para position, then perform your EAS, and finally, remove the SO3H
Good work.It’s helpful to undergraduate students.
Does Rate of reaction also depend on the speed with which C-H bond is broken, What if we use C6D6 instead of C6H6 in Nitration?
The rate of reaction of C6D6 versus C6H6 has been studied but the influence of D is less than 1% on the reaction rate.
There are no conventional deuterium isotope effects in EAS since the rate-determining step of EAS reactions is breakage of the C-C pi bond and not the second step (re-aromatization)
Which one to pick between options – SO3 and HSO3+ ?
My textbook says “the electrophilic reagent used in sulfonation of benzene is SO3”, but going by the exact mechanism, isn’t the electrophile SO3H?
Yes, protonated SO3 is the electrophile. Acids (and Lewis acids) tend to make electrophiles even more electrophilic.
HEY! WHAT WILL BE THE ANSWER TO THE CHALLENGE? WHAT ABOUT NITRATING IT FIRST NO2+ WILL NOY OCCUPY O POSITION DUE TO STERIC CROWDING HENCE OCCUPIES PARA . NOW ADD CL2 IN PRESENCE OF ALCL3 THAT5 WILL SURELY GOT TO ORTHO DUE TO REINFORCING
The answer to the challenge is to sulfonate first, which will go on the para position. Then perform chlorination which will go ortho. Then heat with acid to remove the para sulfonyl group.
I really appreciate for what you are doing. your posts really deepens the insight of this topic. Regarding this post, I just need the link of your next post where you have talked about one more case of Ipso Substitution other with SO3H.
Hey James, your nitration mechanism pop-up has transparent background, so, it’s virtually invisible when clicked on. Also, SO3 exists in gas (minor) and liquid or solid forms (major forms depending on the traces of water), so “bubbling” it through the solution is almost never the case. I always did this reaction with solid polymeric SO3 that I’d weigh out in the glove box and add to the flask, then a couple of drops of sulfuric acid, and we’re in business :) Oleum is a nasty stuff. Hate it with passion!
Thank you for the tip!
Re: SO3, my usual go-to would be SO3-pyridine complex, but I didn’t want to over-complicate things. I really appreciate you adding the comment, however!
I believe aqua regia is HCl and HNO3. https://en.wikipedia.org/wiki/Aqua_regia
You are 100% correct. Thank you for the correction.
Please help…. I am very much confused over this stuff?
There are some tutor recommendations (see in the tabs above).
What would happen if we try to introduce Cl- in p- methyl amino benzene? What possible difference may be there on trying to nitrate or sulfonate the same? ( other than the possibility of oxidation of aryl ring, that is…..)
Chlorination would be fine, but you wouldn’t need a Lewis acid. Unprotected anilines are highly activated and react rapidly w/ Cl2 in absence of Lewis acid.
On the other hand any procedure which requires acid will protonate the amine, resulting in an ammonium group. Nitration / sulfonylation will go meta to the amino group, since under the reaction conditions the amine is protonated to ammonium, and this is an EWG.
What might be the major product in case of chlorination? Actually I had thought that Cl would go ortho to amino group, but one of our Chemistry faculties at school insists that Cl should go ortho to CH3 group, ie meta to NH2 in order to avoid steric repulsion with amino group… which would, in his opinion, decrease the resonance energy of the benzene ring, (by sending NH2 out of the ring, ie.) what he describes as ORTHO EFFECT. You understand I am somewhat stuck on this and don’t know what to do…. Please help!
This is incorrect. The repulsion difference between the amino and nitro group is marginal, so the electronic effects are predominant here and amino-group is a way stronger activating EDG than a methyl, so the substitution will go ortho to amine.