Reactions of Aromatic Molecules
Aromatic Synthesis (3): Sulfonyl Blocking Groups
Last updated: May 29th, 2026 |
The Sulfonyl Blocking-Group Strategy For Synthesis of Aromatic Molecules
Or, how to just get the “ortho” product without any para- .
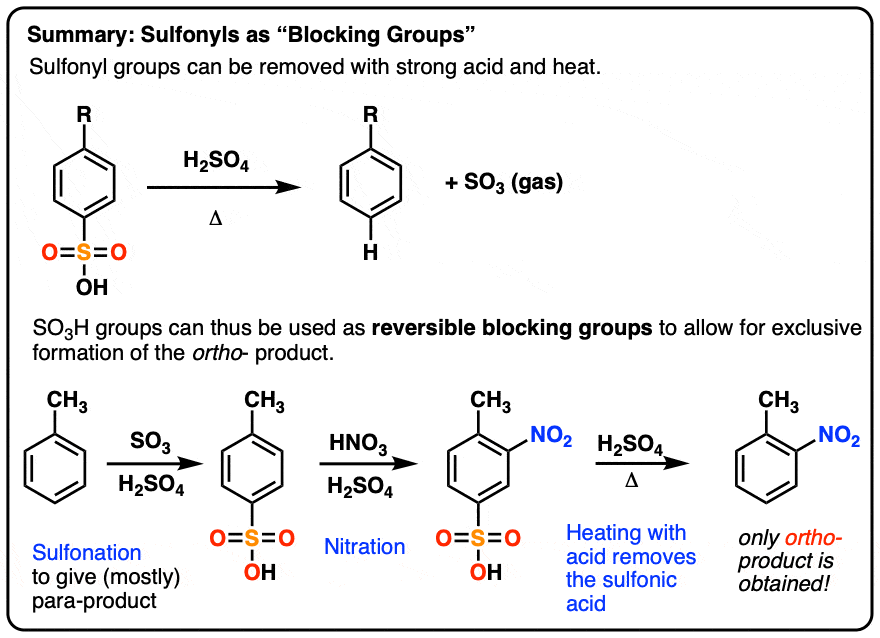
Table of Contents
- Why do para products tend to be favored over ortho- products?
- How Do You Get Just The ortho- Product?
- The Sulfonyl “Blocking Group” Strategy
- The Sulfonyl Blocking Group Strategy In Action
- Combining Sulfonyl Blocking Groups With Polarity Reversal
- Two Practice Problems
- Summary: Sulfonyl Blocking Groups
- Notes
- Quiz Yourself!
1. I Just Want The ORTHO- Product, Thank You
Question: Just how selective are ortho- para, directors for the ortho- and para- products, respectively?
There are two ortho positions and one para. All else being equal, we’d expect a ratio of about 2:1 favoring the ortho.
Is that what we get? No.
In reality, most electrophilic aromatic substitutions give a ratio of products slightly favoring para over ortho. A 60:40 ratio is typical.
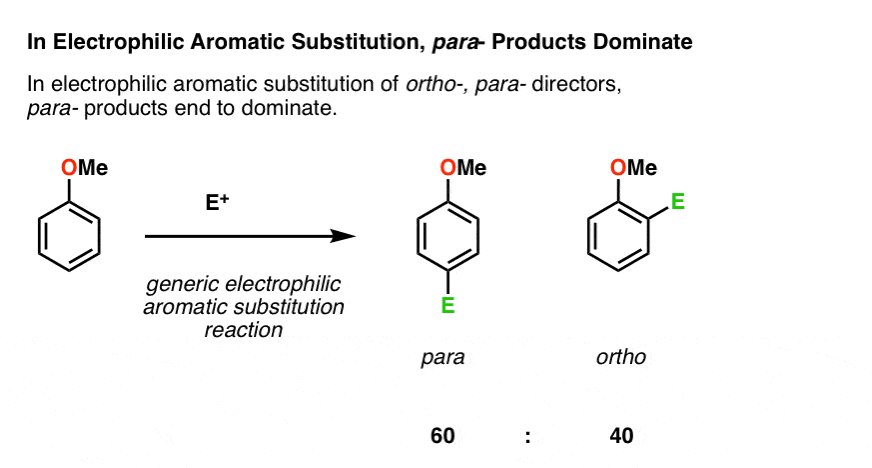
Why the preference for para?
Steric effects, mostly. The ortho- positions are adjacent to the substituent, which can block the path of the electrophile. The para- position is therefore more accessible for the electrophile to attack.
This 60:40 ratio is just a rough number, and depends on the particular substituent. To really drive products to the para-, use a really bulky group like t-butyl. This gives para products almost exclusively.
2. How Do You Get Just The ortho- Product?
This preference for the para– product can be annoying. What if we just want the ortho– product?
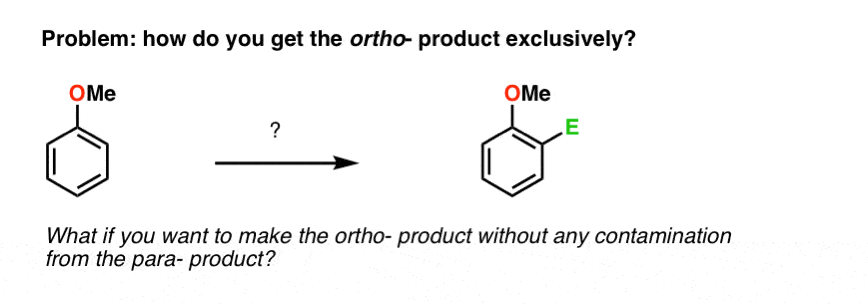
Sorry, not an option. At least: not yet. There aren’t any reactions we’ve learned that are selective for the ortho– product. So getting to the ortho- in one step, without ever having to separate it from the para-, just isn’t possible with the knowledge we have.
But, as often happens in organic chemistry, there IS a work-around. Here it is.
What if we take advantage of the natural preference for para- substitution, and install a group that can be reversibly added to an aromatic ring? This blocks the para position, which means that any subsequent reaction must go onto the ortho position.
Then we remove the blocking group, and voila! we have our ortho- substituted product exclusively.
3. The Sulfonyl “Blocking Group” Strategy
Have we seen any substituents that can be installed reversibly on benzene?
Yes. There are two: sulfonyl (SO3H) and t-butyl.
Here, we’ll mostly cover sulfonyl. (If you just can’t get enough of this topic – completely understandable! – I’ll cover t-butyl in the endnotes.)
Let’s review sulfonation:
- In the forward direction, treating an aromatic ring with heat, SO3 and acid, puts SO3H on the ring. [Note 1]
- To remove SO3H, we just heat the aromatic ring with strong acid (e.g. H2SO4), which eventually loses gaseous SO3.
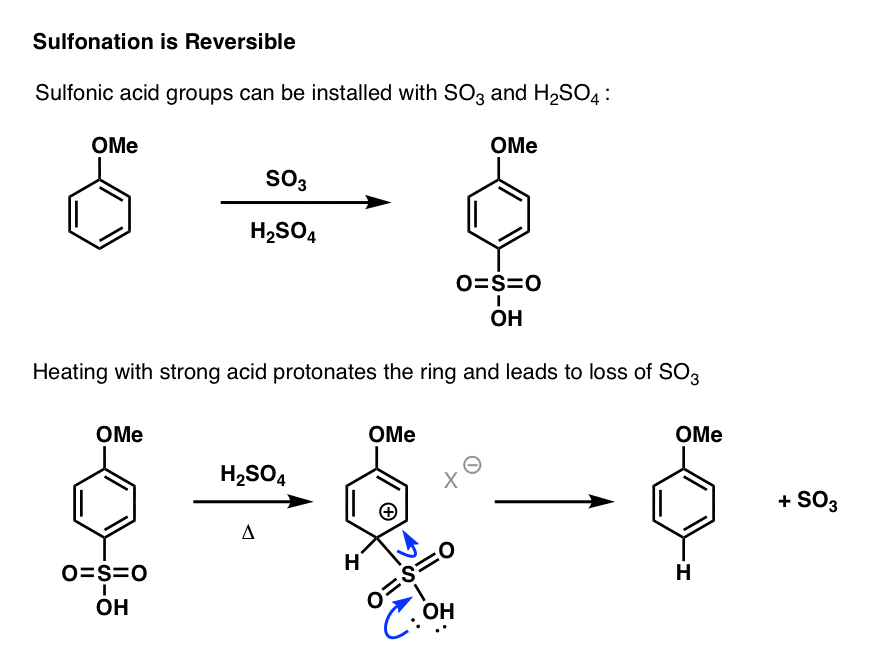
Here,. the aromatic ring is protonated at the carbon bearing the SO3H. [Note 2] In the re-aromatization event, SO3 is lost instead of H+. Once gaseous SO3 boils off, it’s not coming back.
4. Using SO3H As A Blocking Group
Let’s show a simple example of this blocking group strategy in action, beginning with methoxybenzene (“anisole”) toward the goal of synthesizing ortho-bromoanisole.
- Step 1 is to install the SO3H with SO3 and strong acid, which will go (mostly) to the para position.
- Step 2 is to install the desired substituent (bromine) on the ortho position.
- Step 3 is to remove SO3H with strong acid and heat, giving us our ortho- substituted product.
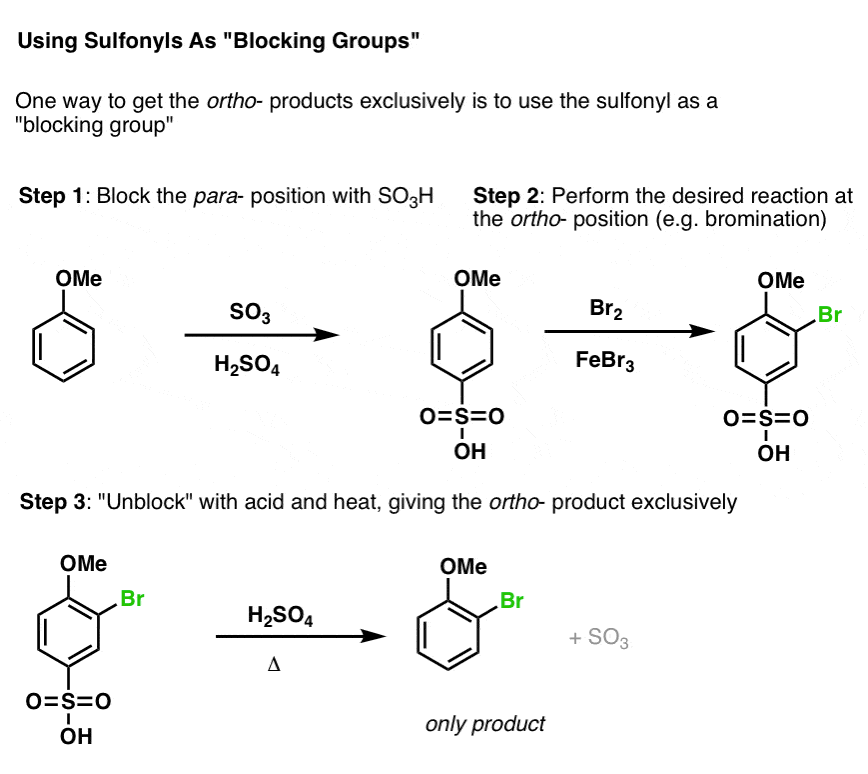
And there we go. After removal of the sulfonyl, we’re left with only ortho-bromoanisole.
Hooray!
5. Combining Blocking Sulfonyl Groups With “Polarity Reversal”
We can combine this blocking group strategy with the “polarity reversal” and “order of operations” strategies we learned earlier.
For example: how could we use this to make o-methyl aniline (aka o-toluidine)?
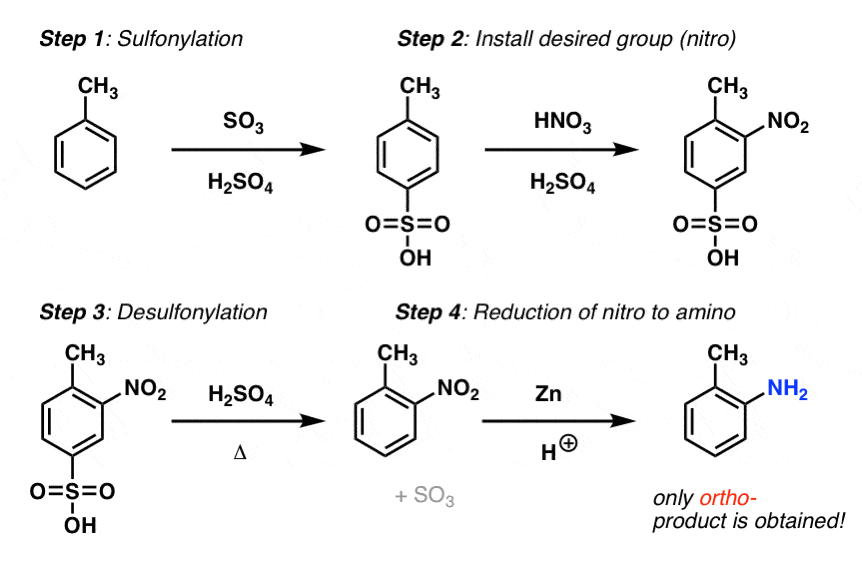
- We saw that we can’t form C-NH2 bonds directly through electrophilic aromatic substitution, but we can form C-NO2 and reduce to the NH2. This means we need to install NO2 on the ortho position.
- This results in the following order of operations: 1) sulfonylation, 2) nitration, 3) removal of SO3H using strong acid and heat, and 4) reduction of NO2 to NH2 using a reductant like zinc and acid (Zn/HCl).
- (it’s probably best to leave the reduction until the end; NH2, being basic, will interfere with the de-blocking step)
While this is one way to do it, it’s not the only way. There’s always an element of choose-your-own-adventure in synthesis.
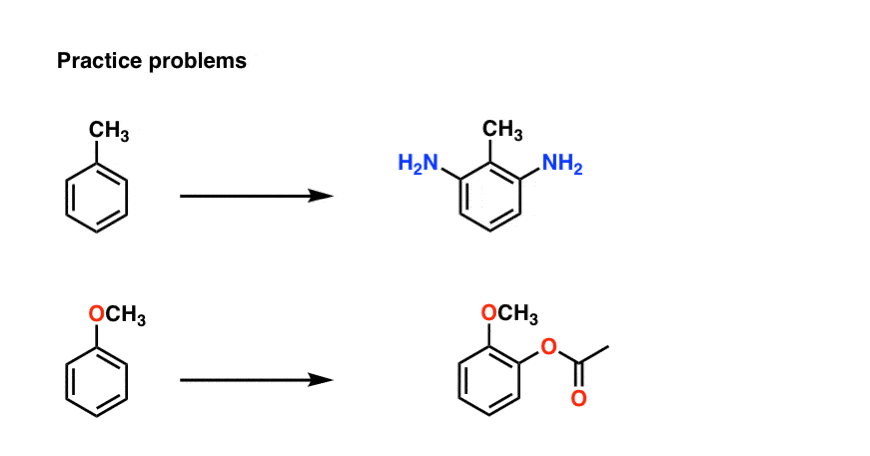
6. Two Practice Problems
Why not try some of your own? Here’s a few examples to practice with using this strategy:
7. Summary: Sulfonyl Blocking Groups
When you need the ortho- and only the ortho-, a blocking group strategy like this one is a useful trick to have in your toolbox.
Having covered some synthetic strategies, it’s likely worth our time to devote a whole post just to worked examples. That will come next!
Notes
Note 1. This will not occur with 100% selectivity for the para position; there will be some ortho- product as well, and it will need to be separated out at some point. So in one sense we are just switching the step at which we have to separate out the undesired product.
Note 2. In contrast to ortho-, meta-, or para-, the carbon attached to the substituent is referred to as the ipso– carbon.
t-Butyl As A Blocking Group
To be brutally frank, the sulfonyl strategy doesn’t get a ton of use in modern organic chemistry. One of the problems is that the resulting sulfonic acid groups are quite polar, and this can present its share of problems with isolation and purification. Ask anyone who’s done ion-exchange chromatography about how much they like concentrating their aqueous fractions. Yeah, no.
A different tack is to employ t-butyl groups as blocking groups. The t-butyl groups are nice and greasy – perfect for flash chromatography.
First, let’s review. How are t-butyl groups installed and removed again?
Installation is via Friedel-Crafts alkylation. We can use either t-BuCl with AlCl3 or 2-methylpropene with strong acid.
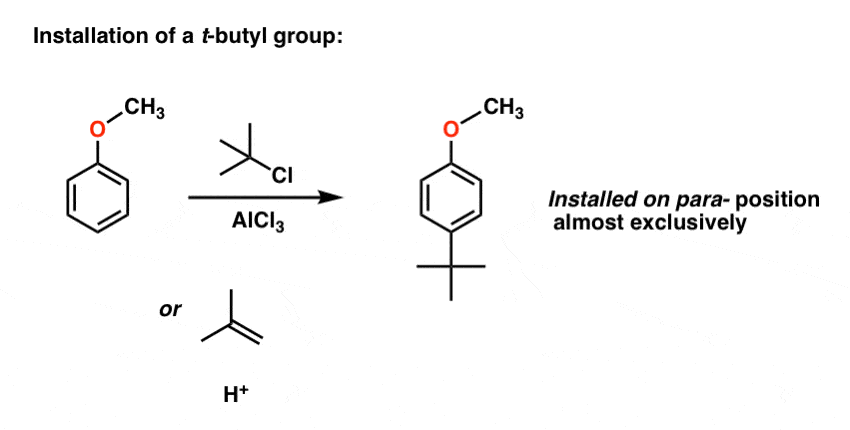
Removal of the t-butyl group is achieved by heating with an excess of aluminum chloride (AlCl3) using benzene as solvent. (This also happens to remove the methyl group from anisole as well). These are not exactly mild conditions, which limits the scope of the reaction somewhat a lot, but… onward.
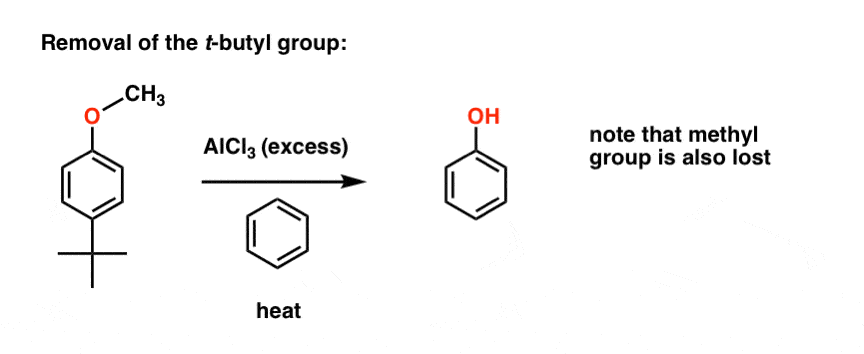
t-Butyl as a Blocking Group: In Action
Here’s an example of this blocking group being used toward the synthesis of a 2-hydroxy benzophenone derivative. Starting with anisole (methoxybenzene), the t-butyl group is added to the para position. Next, a Friedel-Crafts acylation results in exclusive formation of the ortho– product. Finally, removal of the t-butyl with AlCl3 and benzene results in the final product.
Notice that this also pops off the methyl ether – not an easy thing to do! Mild, these conditions are not.
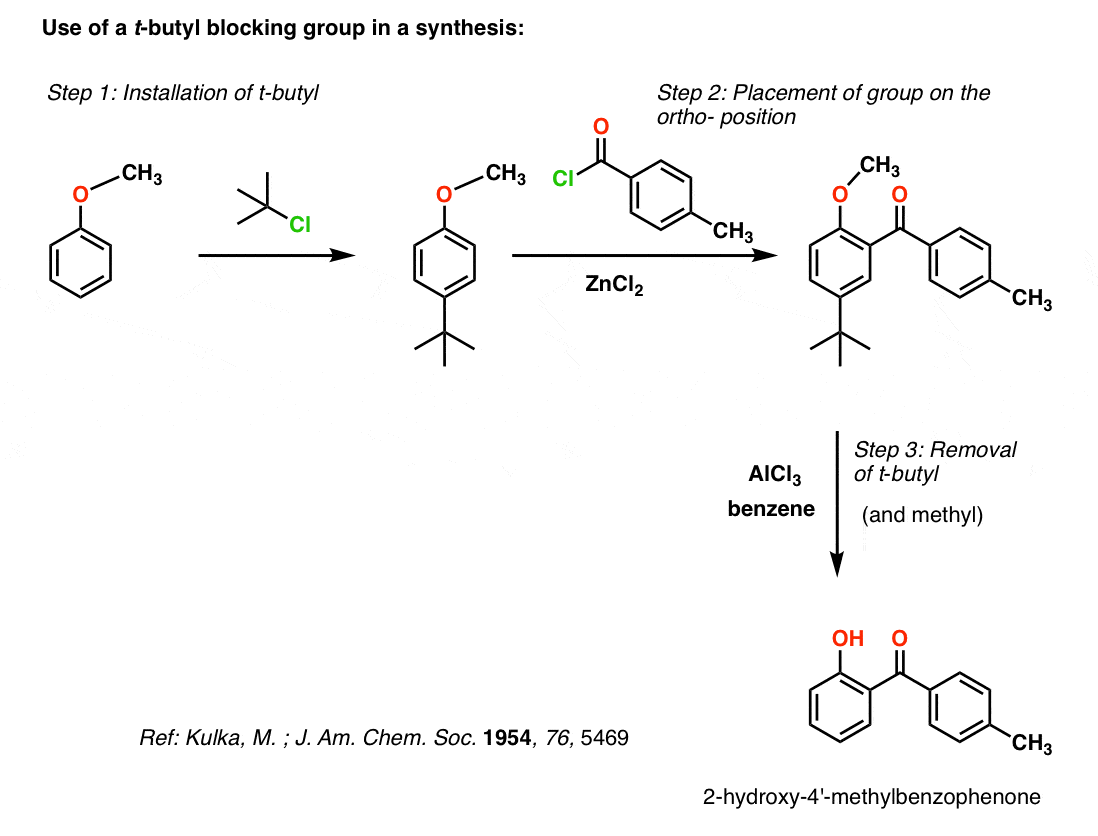
So how does the removal of the t-butyl group happen?
The reaction probably begins by protonating the ring with trace acid (e.g. HCl) present either in AlCl3 or from reaction of AlCl3 with trace water. Protonation of the ring at the para position can then set up re-aromatization not by loss of H+, but by loss of the t-butyl cation. The t-butyl cation is then quickly deprotonated to give 2-methylpropene (“isobutylene”) in an E1 reaction. To stop the isobutylene from Friedel-Crafting back to the para– position, benzene (or toluene) is used as solvent (or co-solvent), which eventually results in formation of t-butylbenzene.
This method does occasionally see use in synthesis. For example, in the synthesis of some resveratrol derivatives, Hou et. al. were trying to dimerize a stilbene derivative. To cut down on the number of potential products, they found it useful to block two positions of a phenol with t-butyl groups, which were later removed using AlCl3, nitromethane, and toluene. Reference here.
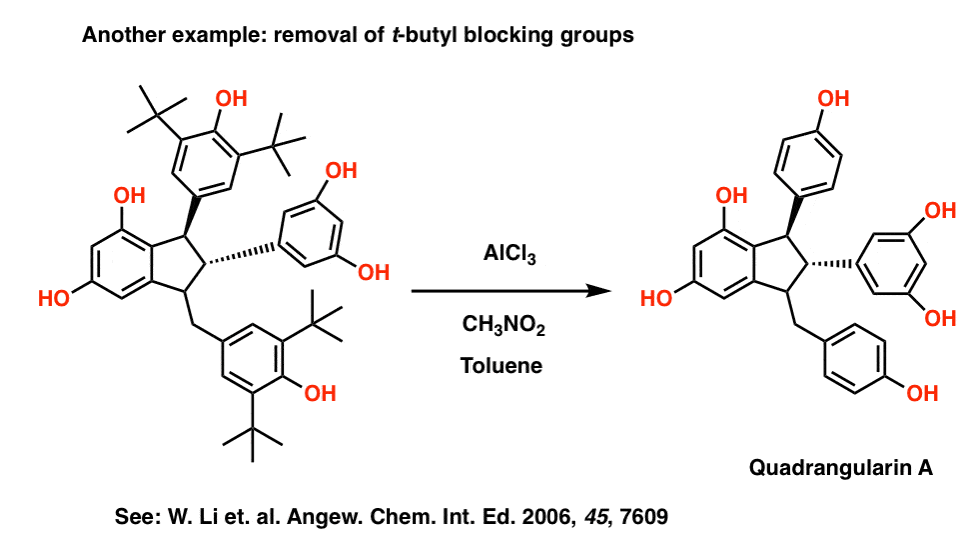
For more on the synthesis, check out Classics in Total Synthesis, volume 3 (Nicolaou and Chen) Chapter 20. And while you’re there, don’t miss chapter 23!
Quiz Yourself!
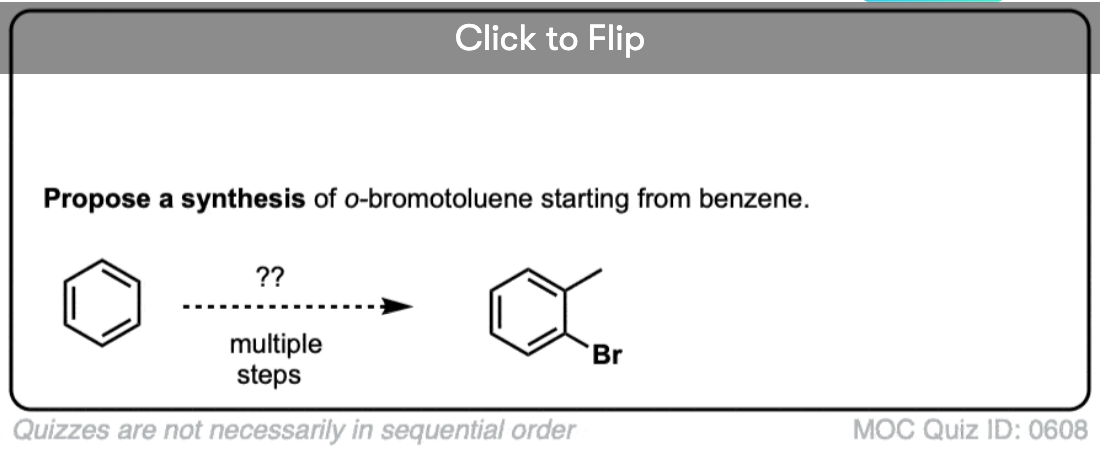
Become a MOC member to see the clickable quiz with answers on the back.
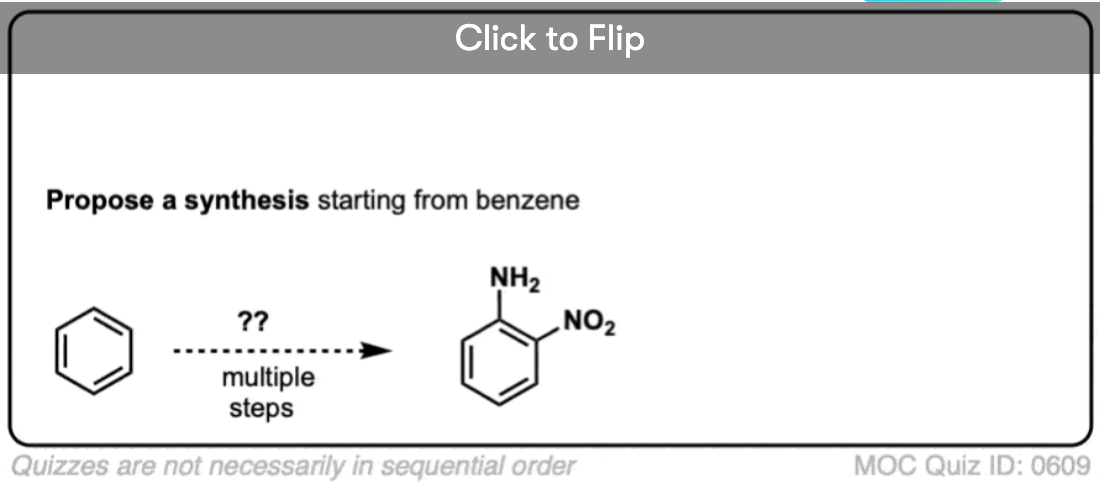
Become a MOC member to see the clickable quiz with answers on the back.
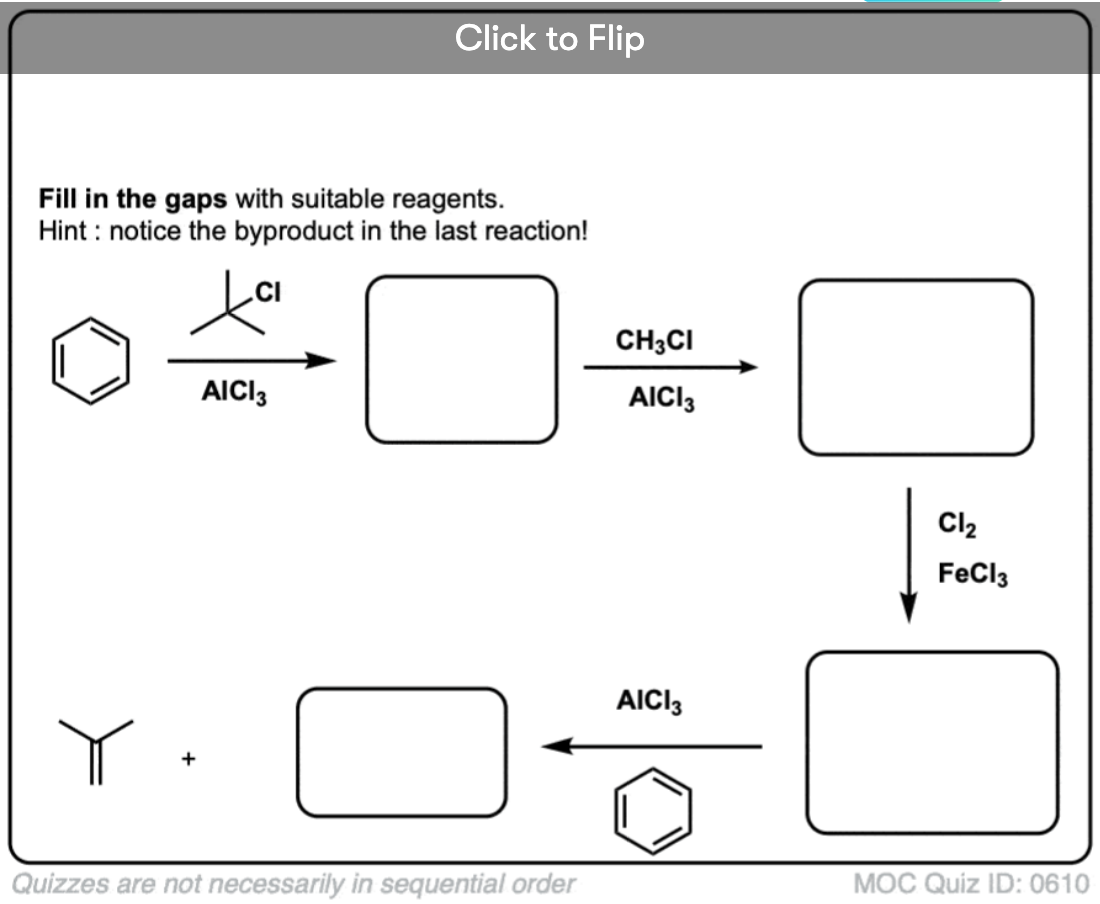
Become a MOC member to see the clickable quiz with answers on the back.
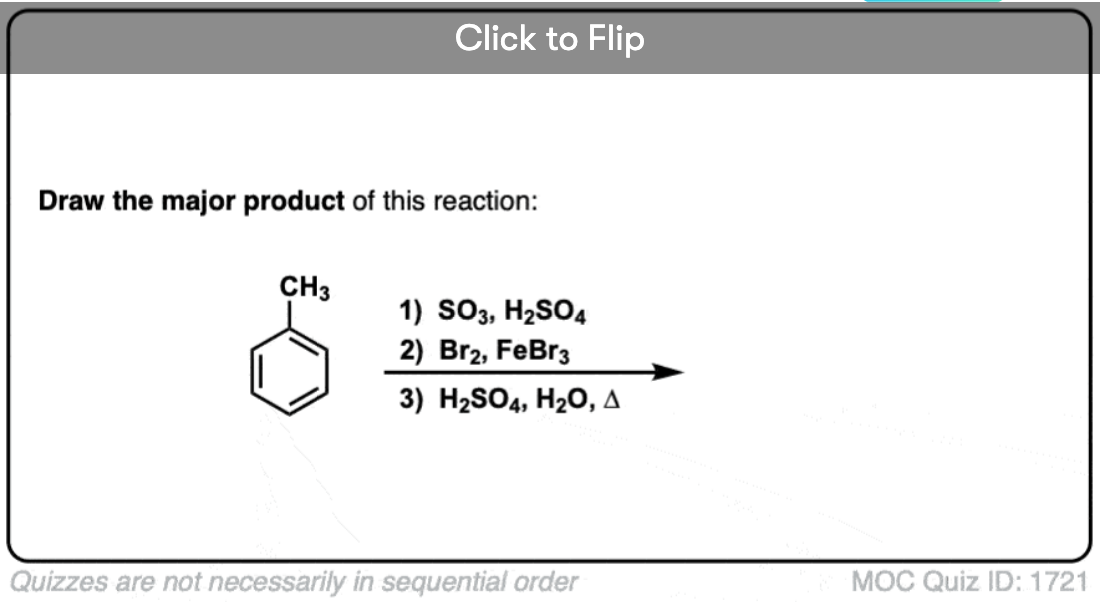
Become a MOC member to see the clickable quiz with answers on the back.
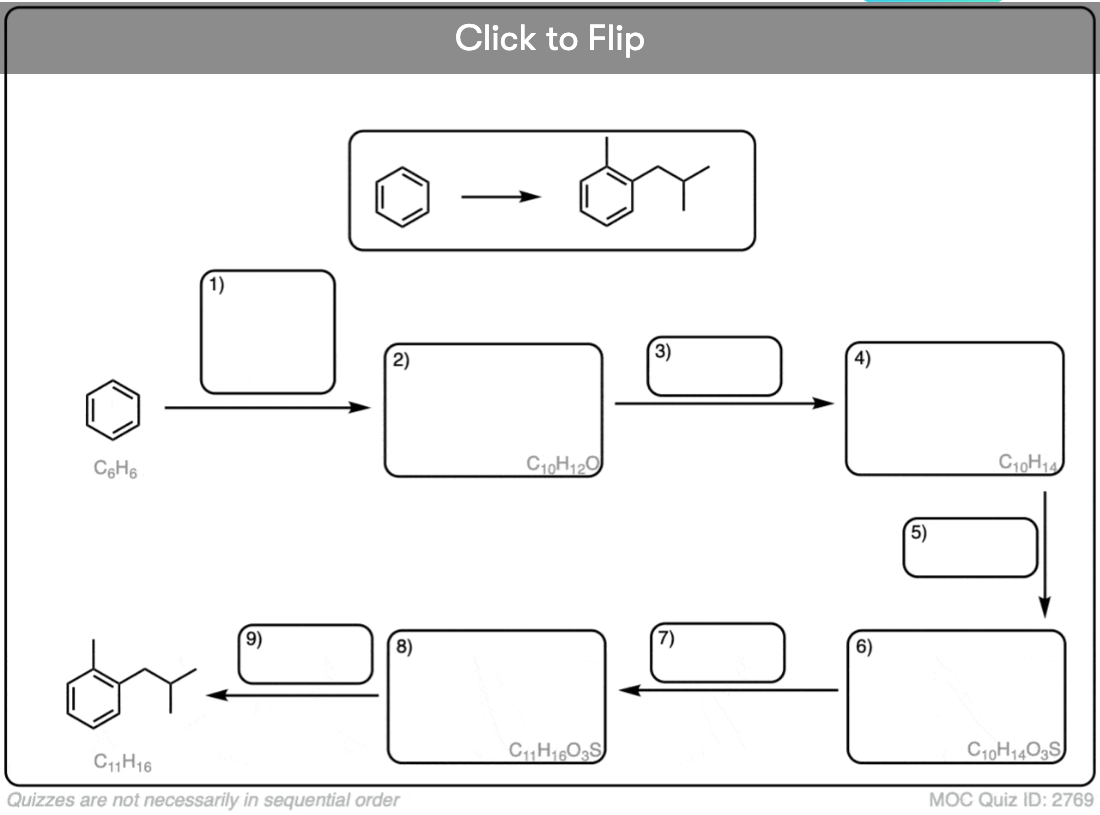
Become a MOC member to see the clickable quiz with answers on the back.
When you convert anisole to phenole you put 2 arrow between them but I think only arrow will be present bacause when anisole react with HCl then 1st protonation occur on oxygen then Cl- attack on sigma star orbit of C-O bond and formation of phenol and methyl chloride.Then why you put 2 arrow plss say
only one*
The purpose of the two arrows is just to show that the reaction eventually converts to phenol, without going into details on the mechanism. If I had to draw it out, as you suggest, the first arrow would be protonation of the anisole oxygen, and the second would be attack at the sigma star with Cl- .
Another nifty example of dealkylation is the formation of meta products following long reaction times/higher temps during poly-alkylation: https://en.wikipedia.org/wiki/Friedel%E2%80%93Crafts_reaction#Friedel%E2%80%93Crafts_dealkylation . The wikipedia page does say “needs citation” but I think I recall reading it in March’s advanced organic.
I have yet to meet a student I tutor who has actually needed to know this fun fact. So I fear to breath a word of it, because drawing a meta product for alkylation would get zero points on 99.9% of exams!