Organic Chemistry Tips and Tricks
Alkene Addition Pattern #3: The “Concerted” Pathway
Last updated: January 23rd, 2024 |
“Concerted” Mechanisms In Alkene Addition Reactions: Hydroboration, Hydrogenation, Epoxidation, Dihydroxylation, And Simmons-Smith Cyclopropanation.
In contrast to alkene addition reactions in the Carbocation Pathway and the 3-Membered Ring Pathway, we saw in the last two posts that hydroboration of alkenes is anomalous. The regioselectivity of the reaction is “anti-Markovnikov” and the stereochemistry of the addition is “syn“.
We also saw that the “syn” stereochemistry is due to the concerted nature of the mechanism proposed for this reaction.
So… are there any other reactions of alkenes that produce similar outcomes to those of hydroboration?
That is, products of syn addition as a result of a concerted mechanism?
There sure are!
Table of Contents
- Concerted Mechanisms In Alkene Addition Reactions
- Hydrogenation Of Alkenes With H2 And A Metal Catalyst Such As Pd-C
- Epoxidation Of Alkenes With Peroxyacids Such As meta-Chloroperoxybenzoic Acid
- Dihydroxylation Of Alkenes With Osmium Tetroxide (OsO4)
- Cyclopropanation Of Alkenes With The Simmons-Smith Reagent (Zinc-Copper Couple)
- Dichlorocyclopropanation Of Alkenes With Chloroform And Base (Giving Dichlorocarbene)
- Summary: The Concerted Pathway In Alkene Addition Reactions
- Notes
- (Advanced) References and Further Reading
1. Concerted Mechanisms In Alkene Addition Reactions
Just to review the previous post, here’s a drawing of the transition state for the hydroboration of alkenes showing the concerted mechanism that results in syn addition.
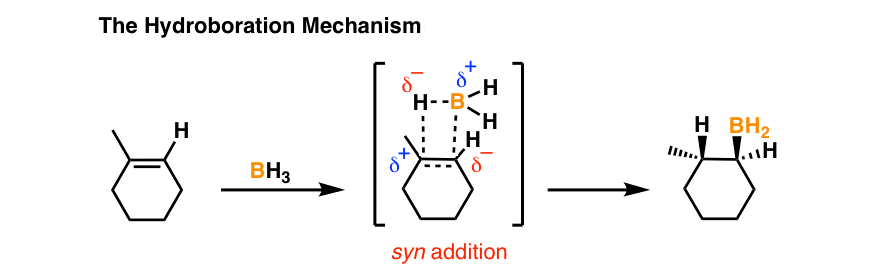
Several other reactions of alkenes that proceed through a concerted transition state are the following:
- Hydrogenation (Pd-C, H2)
- Dihydroxylation (OsO4)
- Epoxidation (RCO3H ; meta-chloroperoxybenzoic acid, m-CPBA is a common reagent in this family)
- Cyclopropanation (CH2I2, Zn-Cu)
- Dichlorocyclopropanation (CHCl3, KOH)
Although the exact mechanism of each reaction is not necessarily the same, each of these reactions does proceed through a concerted transition state and the stereochemistry of the addition is syn. One important thing to note here is that, unlike hydroboration, each of the reactions is adding identical atoms to each carbon of the alkene, so the issue of “regioselectivity” is moot.
The fact that the reaction products have these characteristics in common (if not the exact mechanism) still allows us to group them together as a loosely connected “family” – the “Concerted Pathway”, if you will.
Let’s go through them one by one:
2. Hydrogenation Of Alkenes With H2 And A Metal Catalyst Such As Pd-C
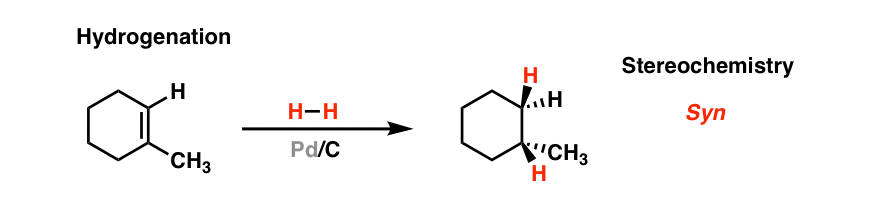
Treatment of alkenes with hydrogen gas and a “noble” metal catalyst such as palladium (Pd) or platinum (Pt) [nickel, rhodium, ruthenium and other metals also find use] results in the addition of two atoms of hydrogen to the same face of the alkene. Under these conditions, the alkene and hydrogen gas are both “adsorbed” on to the surface of the metal.
In the transition state for this reaction, each of the two hydrogen atoms are delivered to the same face of the alkene. The rate of the reaction is surface area dependent: dispersing the metal on finely divided carbon (charcoal) drastically improves the reaction rate, hence the use of charcoal (finely divided carbon).[Note 1]
3. Epoxidation Of Alkenes With Peroxyacids Such As meta-Chloroperoxybenzoic Acid (m-CPBA)
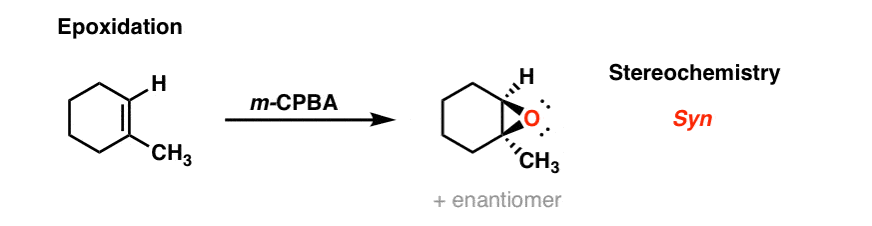
Treatment of an alkene with a peroxyacid such as m-CPBA results in formation of an epoxide (“oxirane”). This also occurs through a concerted transition state:
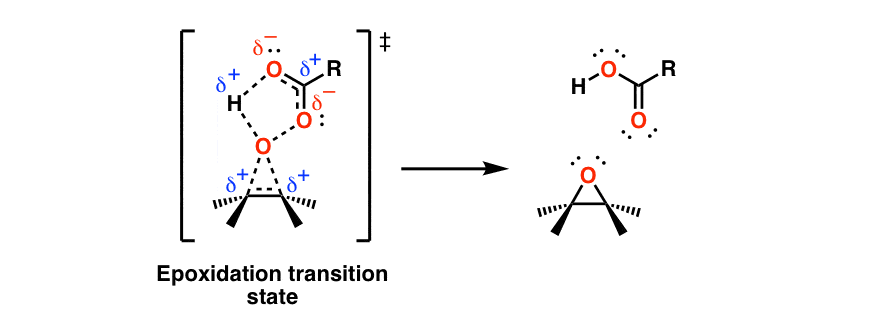
Note that as the (weak) O–O bond breaks, the proton from the peroxy acid is picked up by the (former) carbonyl oxygen.
4. Dihydroxylation Of Alkenes With Osmium Tetroxide (OsO4)
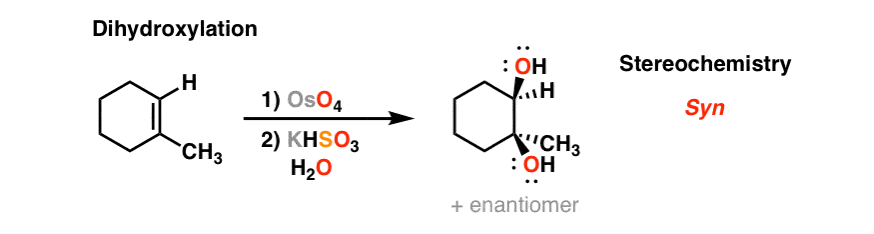
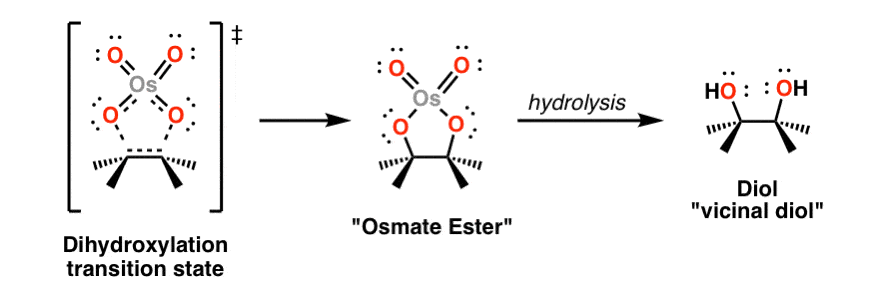
Osmium tetroxide, OsO4, (See post: Osmium Tetroxide OsO4) will add to alkenes in a concerted process to form two new C-O bonds. The stereochemistry is also syn.
An intermediate in this reaction is a cyclic compound containing osmium, called an osmate ester. The second step shown in grey (KHSO3, H2O) results in breakage of the O-Os bonds and formation of the alcohols. This is called “hydrolysis”. KHSO3 is a reducing agent and aids in the workup of the toxic osmium.
5. Cyclopropanation Of Alkenes With The Simmons-Smith Reagent (Zinc-Copper Couple)
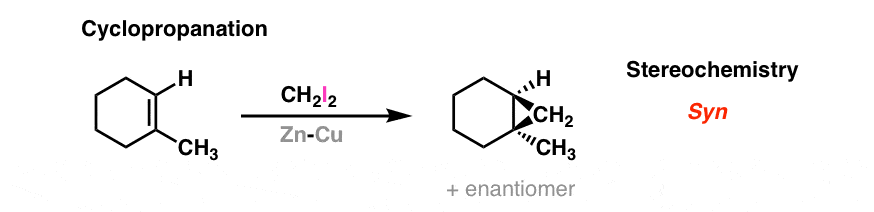
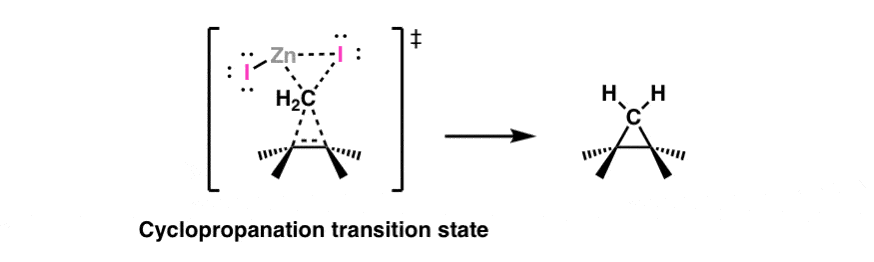
In a reaction sometimes known as the “Simmons-Smith reaction”, diiodomethane (CH2I2) and zinc-copper couple (“Zn-Cu’) form a “carbene” (actually, a carbenoid to be more precise). Alkenes add to this species to give cyclopropanes. The stereochemistry of the addition is syn. Here is the transition state generally drawn for this reaction:
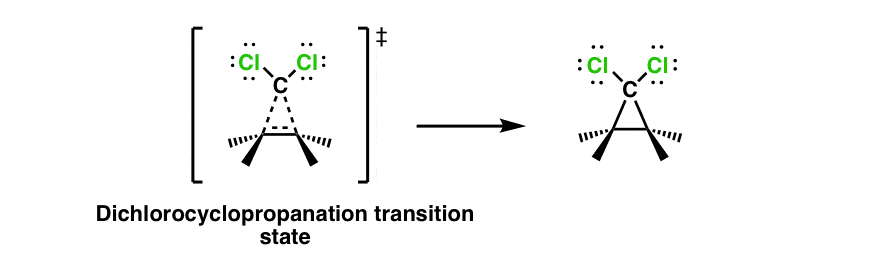
6. Dichlorocyclopropanation Of Alkenes With Chloroform And Base (Giving Dichlorocarbene)
When treated with strong base, chloroform (CHCl3) is deprotonated to give its conjugate base. Loss of chloride ion from this species results in Cl2C: , otherwise known as a “dichlorocarbene”. As in the reaction above, alkenes can add to this carbene to give a cyclopropane. The reaction proceeds through this transition state (empty p orbital and orbital lobe containing lone pair of electrons not shown)
7. Summary: The Concerted Pathway In Alkene Addition Reactions
To summarize, each of the reactions in this post proceed through a concerted transition state to give products of syn stereochemistry. The stereochemistry of the alkene is preserved in the stereochemistry of the product (that is, they are all stereospecific reactions). The regiochemistry is not relevant in any of these cases except for hydroboration, which is anti-Markovnikov.
So how do we go about drawing arrow-pushing mechanisms for these reactions? An excellent question! More on that dilemma in the next post.
NEXT POST: An Arrow Pushing Dilemma In Concerted Addition Reactions
Notes
Note 1 : this assumes the use of “heterogeneous” catalysts, such as Pd/C, Pt/C, etc which, rather than dissolve in solution, are suspended in it. There are also “homogeneous” catalysts for hydrogenation, such as Wilkinson’s catalyst. Since this reagent involves mechanisms of the d-block metals, this blog is not going to get into that. [Mike does, though!]
(Advanced) References and Further Reading
Simmons-Smith Reaction:
- Cyclopropanes from Unsaturated Compounds, Methylene Iodide, and Zinc-Copper Couple
Simmons, H. E.; Cairns, T. L.; Vladuchick, S. A.; Hoiness, S. A. Reactions 1973, 20, 1-133
DOI: 10.1002/0471264180.or020.01
This chapter in Organic Reactions has everything you need to know about the Simmons-Smith reaction, including mechanistic studies, experimental procedures, and substrate scope. - NORCARANE
D. Smith and H. E. Simmons
Org. Synth. 1961, 41, 72
DOI: 10.15227/orgsyn.041.0072
A detailed procedure for the Simmons-Smith reaction in Organic Syntheses, including the preparation of the Zn-Cu couple.Dichlorocyclopropanation with CCl2: - The Addition of Dichlorocarbene to Olefins
William von E. Doering and A. Kentaro Hoffmann
Journal of the American Chemical Society 1954 76 (23), 6162-6165
DOI: 10.1021/ja01652a087
The original paper describing the synthesis of dichlorocyclopropanes from alkenes with chloroform and KOtBu, by the legendary William von Eggers Doering. - 1,6-Methano[10]Annulene
E. Vogel, W. Klug, and A. Bruer
Org. Synth. 1974 54, 11
DOI: 10.15227/orgsyn.054.0011
The second step in this procedure is the synthesis of a dichlorocyclopropane, by addition of dichlorocarbene to an alkene.Dihydroxylation of alkenes with KMnO4: - Improved Preparation of 9(10),10(9)-Ketohydroxystearic Acids by Oxidation of Oleic Acid with Potassium Permanganate in Neutral Solution
Joseph E. Coleman, C. Ricciuti, and Daniel Swern
Journal of the American Chemical Society 1956, 78 (20), 5342-5345
DOI: 1021/ja01601a050 - The Mechanisms of Permanganate Oxidation. IV. Hydroxylation of Olefins and Related Reactions
Kenneth B. Wiberg and Klaus A. Saegebarth
Journal of the American Chemical Society 1957, 79 (11), 2822-2824
DOI: 1021/ja01568a042
Mechanistic studies of dihydroxylation of olefins with KMnO4. The first paper is by the same Swern of Swern oxidation fame! The disadvantage with KMnO4 is that it can overoxidize the substrate, giving ketols. Carrying out the oxidation in basic solution can minimize this. - dl-GLYCERALDEHYDE ETHYL ACETAL
J. Witzemann, Wm. Lloyd Evans, Henry Hass, and E. F. Schroeder
Org. Synth. 1931, 11, 52
DOI: 10.15227/orgsyn.011.0052
Typical procedure for dihydroxylation of an alkene with KMnO4. This procedure highlights one of the difficulties with using KMnO4 for oxidation; even though it is cheap, using it in stoichiometric quantities results in the formation of an equivalent amount of MnO2, which can be difficult to separate from the desired product.Dihydroxylation of alkenes with OsO4: - Osmium Tetraoxide Cis Hydroxylation of Unsaturated Substrates
Schroder, M.
Chem. Rev. 1980 80, 187
DOI: 10.1021/cr60324a003
This review covers the mechanism of dihydroxylation of olefins by OsO4 (including both catalytic and stoichiometric variants), illustrates various Os complexes, and describes the substrate scope of the reaction. - CATALYTIC OSMIUM TETROXIDE OXIDATION OF OLEFINS: cis-1,2-CYCLOHEXANEDIOL
VanRheenen, D. Y. Cha, and W. M. Hartley
Organic Syntheses, Coll. Vol. 6, p.342 (1988); Vol. 58, p.43 (1978).
DOI: 10.1021/orgsyn.058.0043
A reliable procedure for the Upjohn dihydroxylation in Organic Syntheses. Note that the name comes from the Upjohn company in Kalamazoo, MI (later acquired by Pfizer), where it was first developed.Epoxidation of alkenes with mCPBA: - Epoxidations with m-Chloroperbenzoic Acid
Nelson N. Schwartz and John H. Blumbergs
The Journal of Organic Chemistry 1964 29 (7), 1976-1979
DOI: 1021/jo01030a078
This paper describes mechanistic studies of m-CPBA oxidation that demonstrate that ionic intermediates are not involved in the reaction, and that the rate is insensitive to solvent polarity. - Experimental Geometry of the Epoxidation Transition State
Daniel A. Singleton, Steven R. Merrigan, Jian Liu, and K. N. Houk
Journal of the American Chemical Society 1997 119 (14), 3385-3386
DOI:1021/ja963656u
Combined experimental and theoretical studies of the epoxidation transition state, showing that both C-O bond forming events are nearly synchronous. - The mechanism of epoxidation of olefins by peracids
V. G. Dryuk
Tetrahedron Volume 32, Issue 23, 1976, Pages 2855-2866
DOI: 10.1016/0040-4020(76)80137-8
An account of the author’s work on kinetic studies of the epoxidation of olefins with peracids in order to determine the exact mechanism.
Great website. I have been a regular reader for some time now. Extremely useful and lucid content. Loved it Sir!
Are you sure that the hydrogenation of alkenes and alkynes is concerted?
The first step, addition of hydrogen and palladium to multiple bonds is concerted (hydropalladation). And the second step (reductive elimination) preserves the stereochemistry at the carbon. Since hydropalladation is usually beyond the scope of introductory organic chemistry, it is not too much of a stretch to say that hydrogenation is concerted.