Alcohols, Epoxides and Ethers
Protecting Groups For Alcohols
Last updated: June 2nd, 2026 |
Alcohol Protecting Groups
- There are many times when it’s useful to mask the reactivity of alcohols since their relatively high acidity interferes with strongly basic reagents like Grignard reagents.
- The most common protecting group for alcohols is silyl ethers.They are easily formed by treating alcohols with R3SiCl in the presence of base, and then easily removed with a source of fluoride ion F(-) since Si-F bonds are very strong.
- Silyl ethers are inert to Grignard reagents, strong bases, and oxidants (although will be removed with strong acid).
- Tetrahydropyranyl (THP) ethers are also useful protecting groups for alcohols.
- Ordinary ethers are generally not used as protecting groups since their removal requires harsh conditions
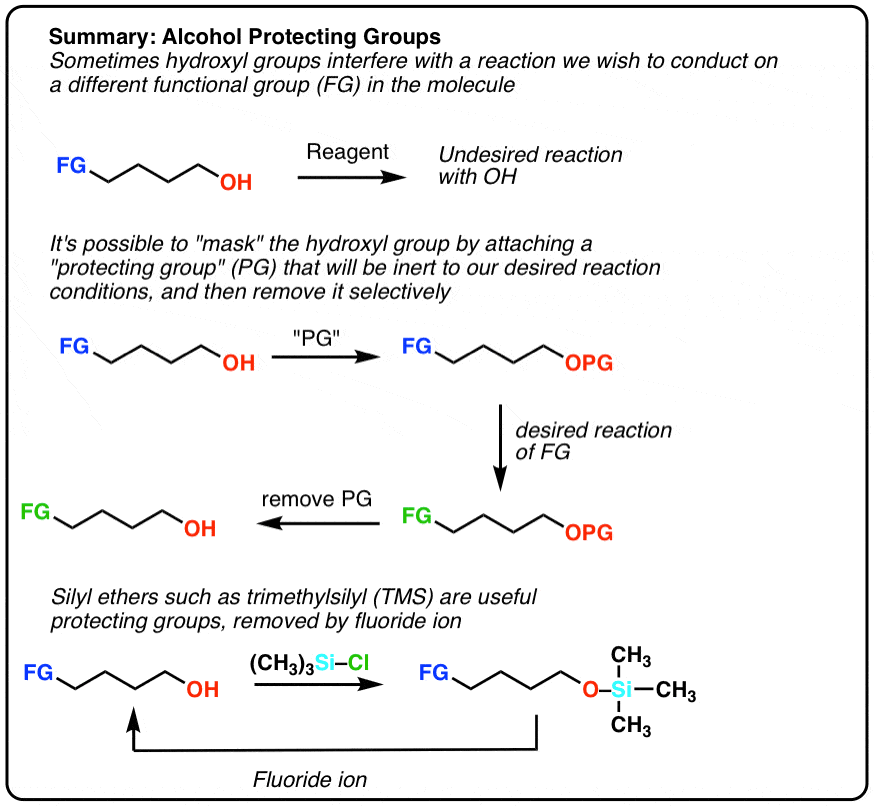
Table of Contents
- When Alcohols Get In The Way
- Protecting Groups Are Like “Painter’s Tape”
- What Would Be A Chemical Equivalent of “Painter’s Tape”?
- One Potential Solution: Ethers (Spoiler: They’re Not Great)
- A Better Way To Do It: Silyl Ethers
- A Successful Application of a Silyl Protecting Group Strategy
- Summary: Protecting Groups For Alcohols
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. When Alcohols Get In the Way
As we’ve seen in previous posts in this series, alcohols are very versatile functional groups that participate in a variety of reactions. They can be deprotonated with base (making them good nucleophiles in substitution reactions), protonated (making them good leaving groups in substitution and elimination reactions), oxidized to aldehydes or ketones, or transformed into better leaving groups (alkyl halides, or alkyl tosylates) allowing for a host of substitution and elimination reactions.
All this this versatility comes with a drawback, however. Sometimes alcohol functional groups can get in the way of other reactions we might like to do. Let me show you what I mean.
We’ve seen by now one of the most useful C-C bond forming reactions you learn in Org 1: nucleophilic substitution (SN2) of alkyl halides with acetylides (the conjugate base of acetylenes)
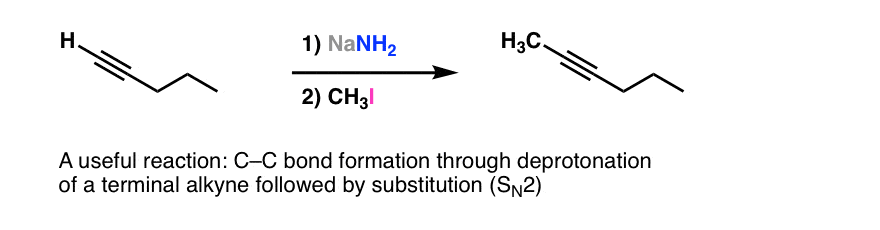
Since alkynes are like a blank canvas, this reaction can set up the introduction of many different types of functional groups through addition reactions.
Now let’s modify our substrate a bit. We’ll attach a hydroxyl group (OH) to the end of the molecule. Now let’s see what happens.
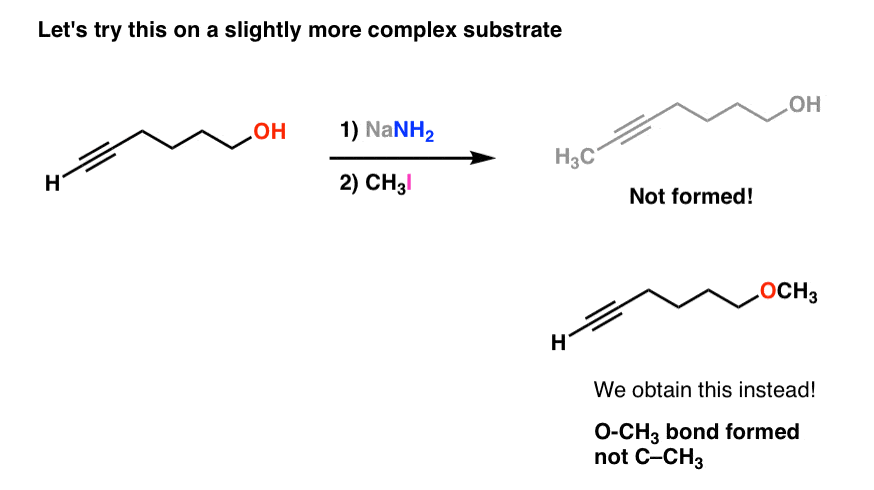
Look at what happened – we now formed a new O-CH3 bond instead of a C-C bond. What gives?
The answer, of course, is that our strong base NaNH2 deprotonated the strongest acid [OH, pKa of 16 versus acetylide C-H, pKa of 25] and the resulting alkoxide [R–O– ] then attacked CH3-I, resulting in a substitution reaction with displacement of iodide ion [Note 1]
Here’s another example of the the principle at work. Here, we’d like to perform a substitution reaction of C-Br with C-C . So why does this reaction not lead to formation of a C-C bond?
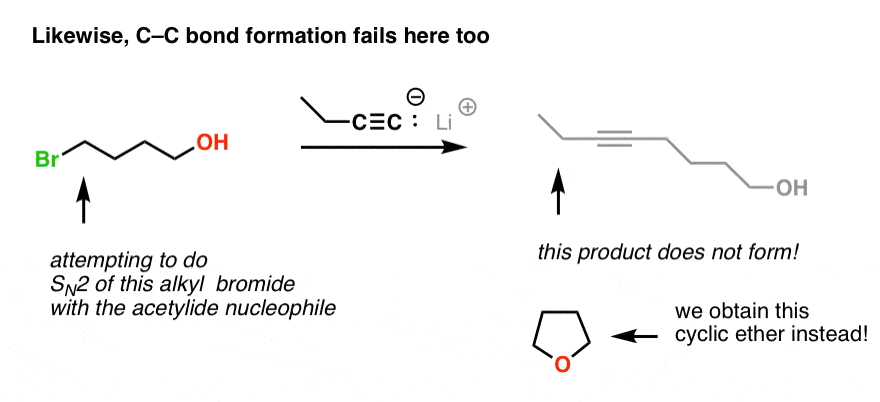
Same reason! Our acetylide ion is a strong base, and deprotonates the O-H group, which then participates in an SN2 reaction with the alkyl halide 4 bonds away (forming a five membered ring).
This is a textbook example of what we saw in our last post – an intramolecular SN2 reaction. [Why doesn’t it do substitution first? Acid-base reactions are fast, relative to substitution reactions].
So how could we have prevented this from occurring?
2. Protecting Groups Are Like Painter’s Tape
It’s reminiscent of a problem anyone who has painted a room would understand. Imagine you’re helping your cousin paint his room in a hideous shade of yellow-green so completely uncool to the untrained eye that only a hipster could appreciate it. Then you come to one of those annoying wall outlets. You could paint over it of course.

But now it’s useless if your cousin wants to plug in that 1965 Smith-Corona electric typewriter he found at a thrift store that he’s using to write his “novel”. Surely there’s a way to do this that doesn’t destroy our outlet. So what do you do?
Painter’s tape to the rescue!

Cover the outlet with painter’s tape, paint to your heart’s content, then remove the tape. THEN you can plug in the typewriter. Simple!
3. A Chemical Equivalent Of Painter’s Tape
Wouldn’t it be nice if we had a “chemical equivalent” of painter’s tape for alcohols. Something that could
- mask the reactivity of the OH group
- be inert to a large set of reaction conditions, and
- be easily and selectively removed to reveal the OH group once we’re done.
That would allow us to perform a synthesis of our desired molecule (second scheme above). Here I’m using “PG” to stand for “protective group”.
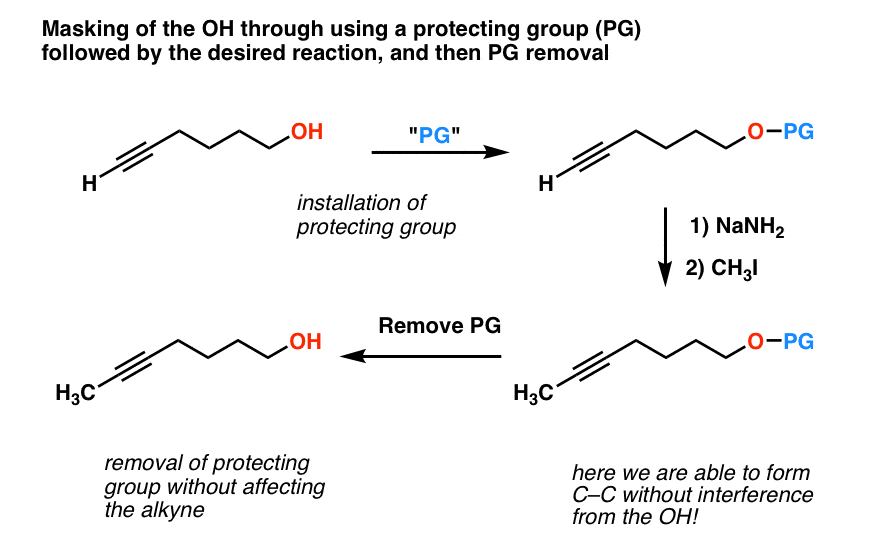
Well, you might have guessed by now that enterprising chemists have developed a solution for this problem. It’s very clever, in fact.
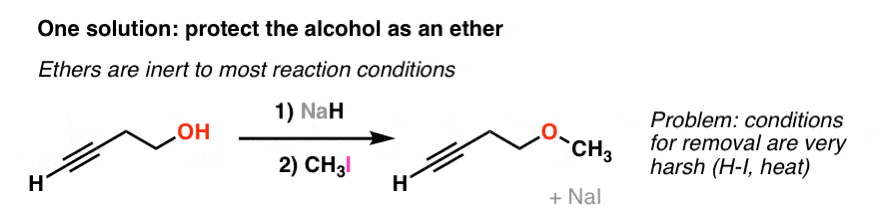
4. One Potential Solution: Ethers
As we’ve discussed earlier, ethers are quite possibly the most boring functional group you can encounter. The only important reaction of ethers you cover in Org 1 is how to cleave them with very strong acid (e.g. with hydroiodic acid, HI). That’s it. Other than that, ethers are inert to pretty much any other reaction condition you can name.
For the chemical equivalent of “painter’s tape”, boring is good! It means that we can “protect” a hydroxyl group as an ether without worrying about it being affected by reactions we might like to do on the rest of the molecule (like addition of an acetylide to an alkyl halide, for example).
There’s just one problem: ethers require very harsh conditions in order to break (hydroiodic acid, HI). That’s like destroying the village in order to save it: such conditions will likely torch whatever other functional groups are on your molecule.
5. A Better Way To Do It: Silyl Ethers
Fortunately a very clever solution has been devised. Instead of making a typical ether (e.g. forming an O-C bond), we form a silyl ether (i.e. make an O-Si bond, not an O–C bond). It’s even easier to form than a “normal” ether, and shares the property of being inert to many types of reaction conditions. In most introductory courses the most common silyl ether used is trimethylsilyl (TMS) although there are others [Generally, the bulkier the groups around silicon, the harder it is to cleave the O–Si bond]
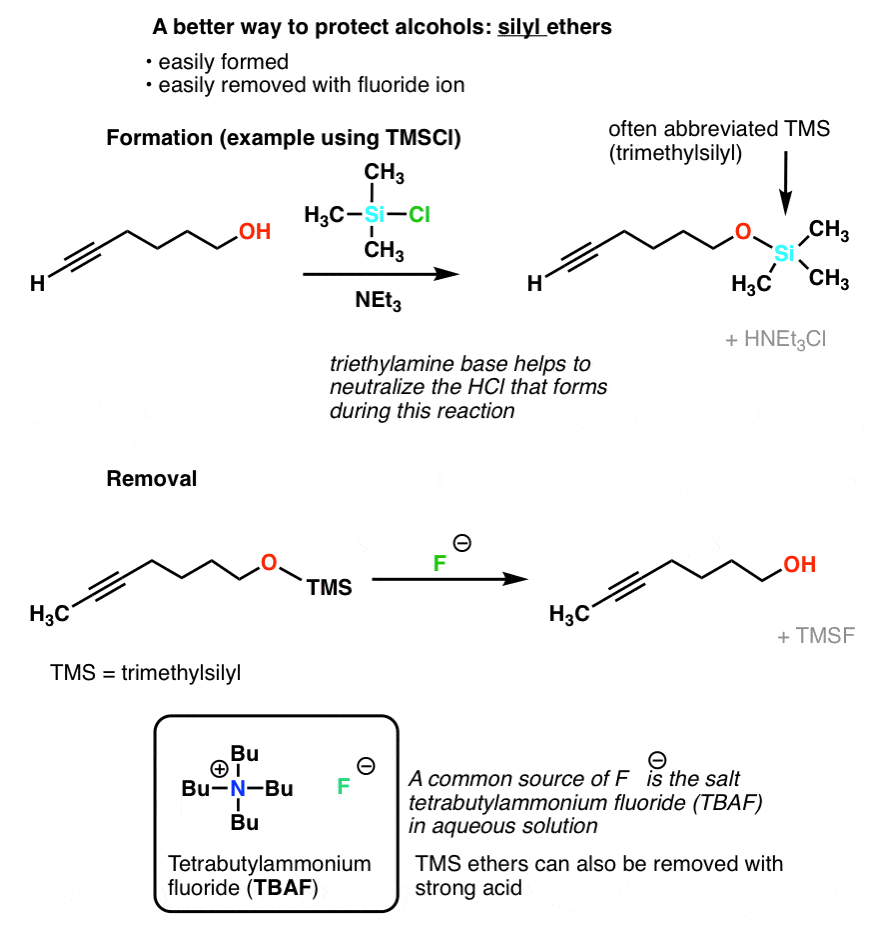
The main advantage of silyl ethers is that they’re easily cleavable. The Si-F bond is unusually strong – even stronger than Si-O. Addition of a source of fluoride ion (F-) will lead to cleavage of Si-O bonds without affecting the rest of the molecule. A typical source of fluoride ion is the salt tetrabutylammonium fluoride (TBAF).
Are there other protecting groups for alcohols? You betcha. For more information, see Note 2.
6. A Successful Application of A Silyl Ether Protective Group Strategy
So let’s go back to our second example. How could we get this sequence to work? Let’s “protect” the free alcohol as a silyl ether (TMS) and follow along.
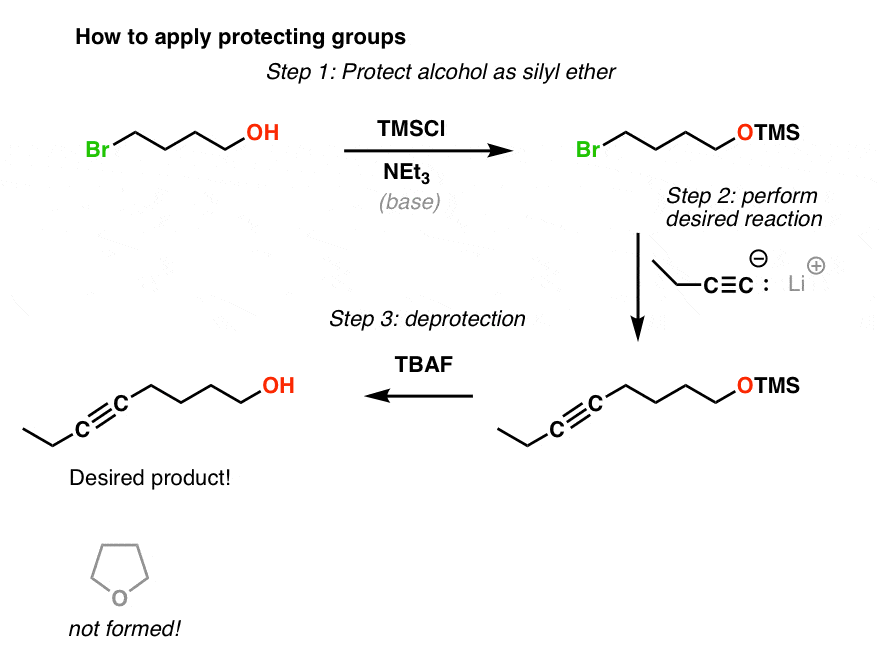
There you have it. All we needed to get our desired reaction to work was a way of masking the OH until we were done performing our surgery on the other half of the molecule.
7. Summary: Protecting Groups For Alcohols
This post barely scratches the surface of protecting groups for alcohols. Protecting groups are used for alcohols in a variety of different situations, far beyond the SN2 examples we covered here. For instance, when we talk about Grignard reagents, we’ll see that they can’t be formed in the presence of alcohols, so we have to protect them. Another example might be if you wanted to selectively oxidize one of two different alcohols in a molecule. We can add more posts on this topic as we go along.
You’ll notice that the vast majority of molecules you encounter in Org 1 and Org 2 have only one important functional group. It’s very rare that you’ll be given a substitution reaction, for example, that has two nucleophiles of comparable strength. Learning how to deal with molecules that have more than one key functional group is, in my opinion, where Org 2 ends and Org 3 begins.
It’s at that point that you need to learn understand the relative reactivity of different functional groups, their compatibility with different reagents, and also how to plan a synthesis such that only one key functional group will participate in the reaction.
Next Post – Thiols And Thioethers
Notes
Note 1. Isn’t it possible that NaNH2 could have deprotonated a little bit of the alkyne? Sure! But remember that acid base reactions are equilibrium, and the alcohol is a far stronger acid (pKa ~16 ) than the alkyne (pka 25). Even if that acetylide formed, it would be quickly protonated by any spare alcohol R-OH swimming around, giving rise to the alkoxide. [back to article]
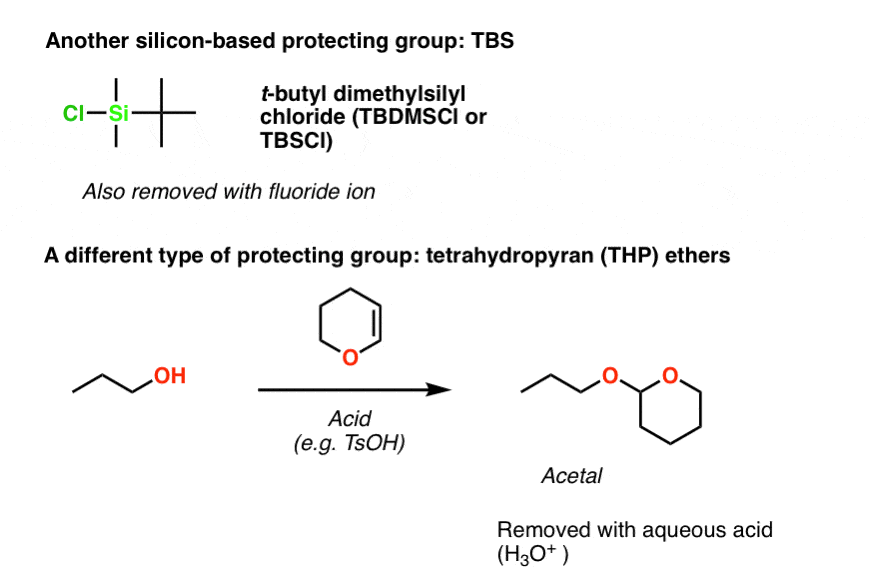
Note 2. Two more protecting groups that come up quite frequently are TBS (t-butyldimethylsilyl) and THP (tetrahydropyranyl) ethers. TBS is installed the exact same way TMS is – by using TBSCl in the presence of a base like NEt3.
THP ethers are slightly different. They are installed by adding dihydropyran (an “enol ether”) in the presence of strong acid. The enol ether is protonated at carbon by the strong acid, resulting in an oxonium ion, which is then attacked by the alcohol to give the ether. This is actually a special type of ether where two OR groups are attached to the same carbon. It’s a masked ketone, which we refer to as an acetal. We talk about acetals here in this blog post. [back to article]
For even more protecting groups for alcohols, see this handout by the group of Prof. Andrew Myers at Harvard. It’s phenomenal.
Quiz Yourself!
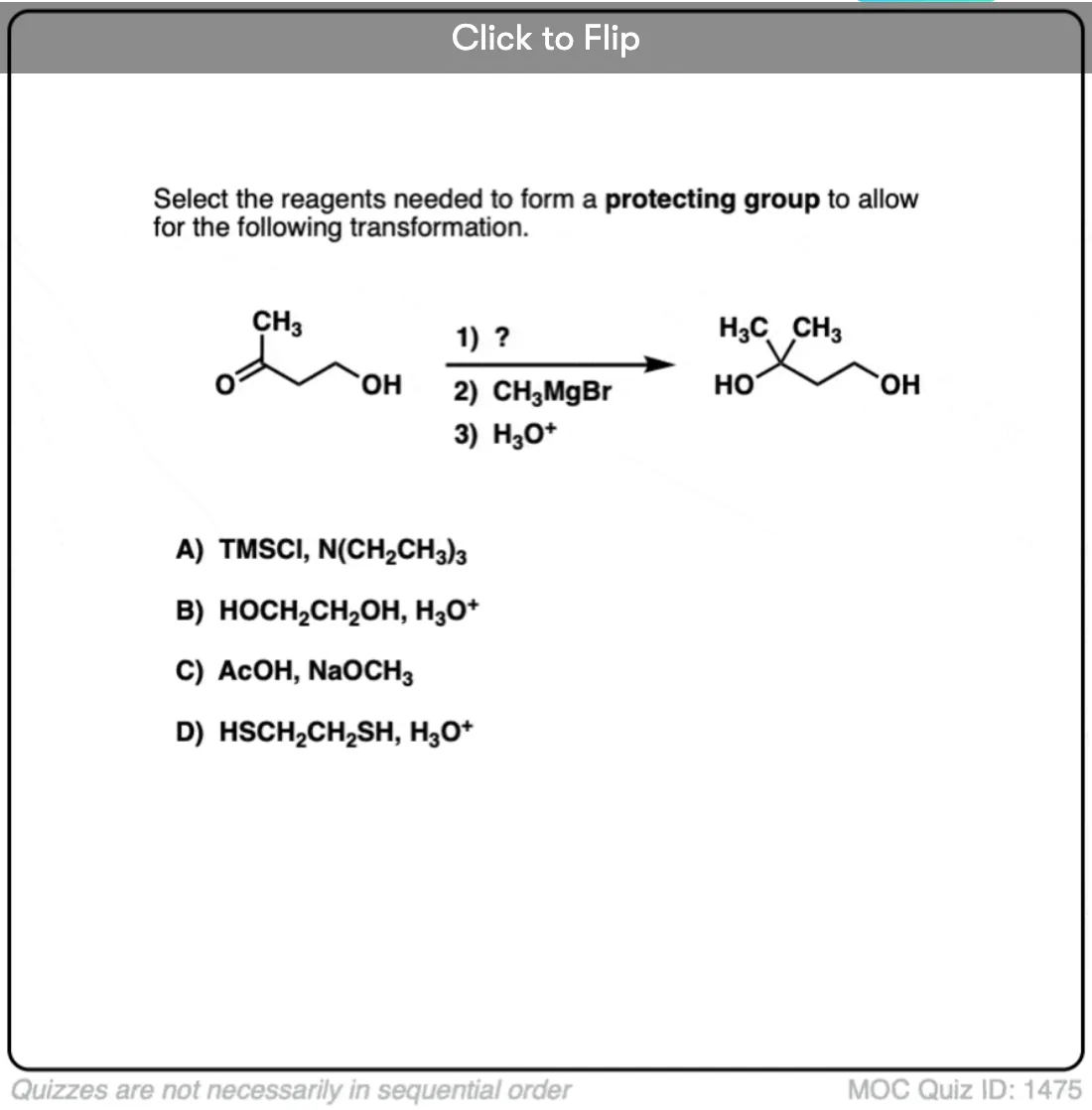
Become a MOC member to see the clickable quiz with answers on the back.
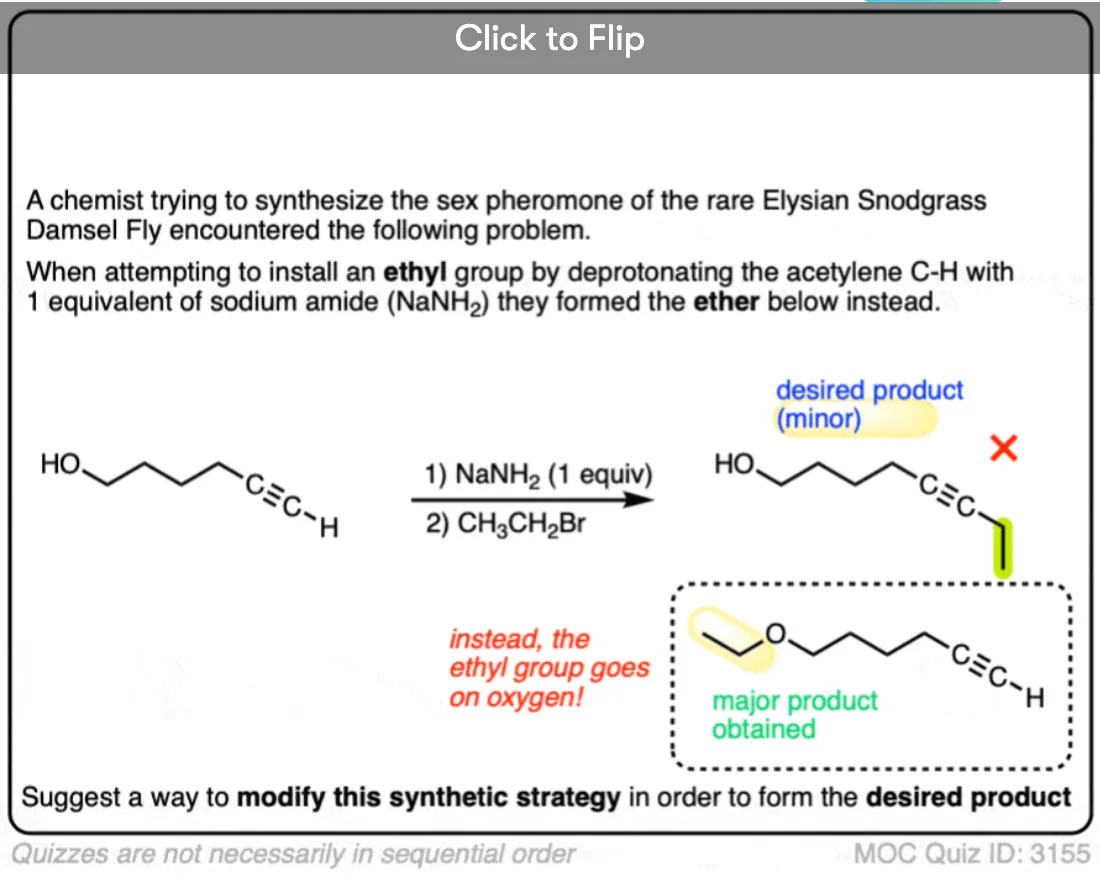
Become a MOC member to see the clickable quiz with answers on the back.
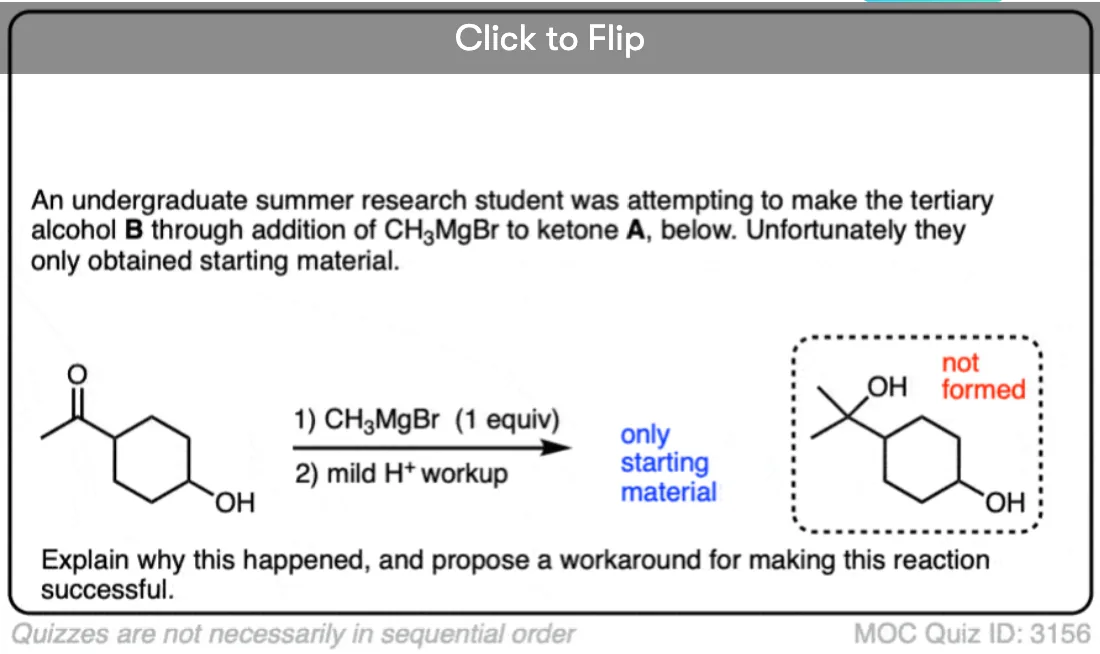
Become a MOC member to see the clickable quiz with answers on the back.
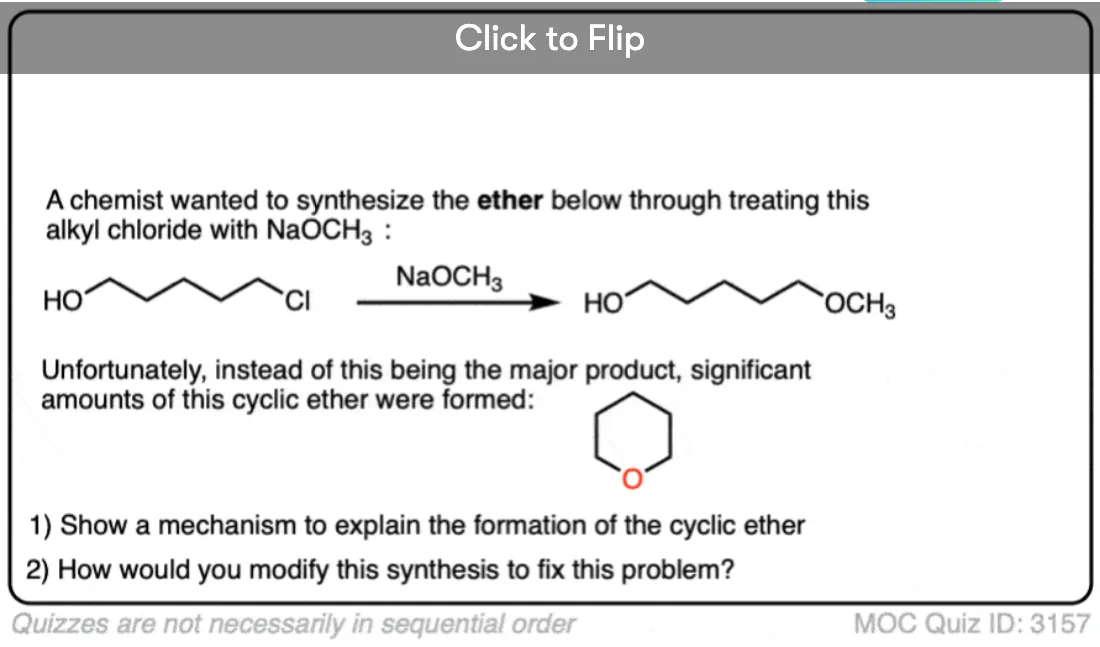
Become a MOC member to see the clickable quiz with answers on the back.
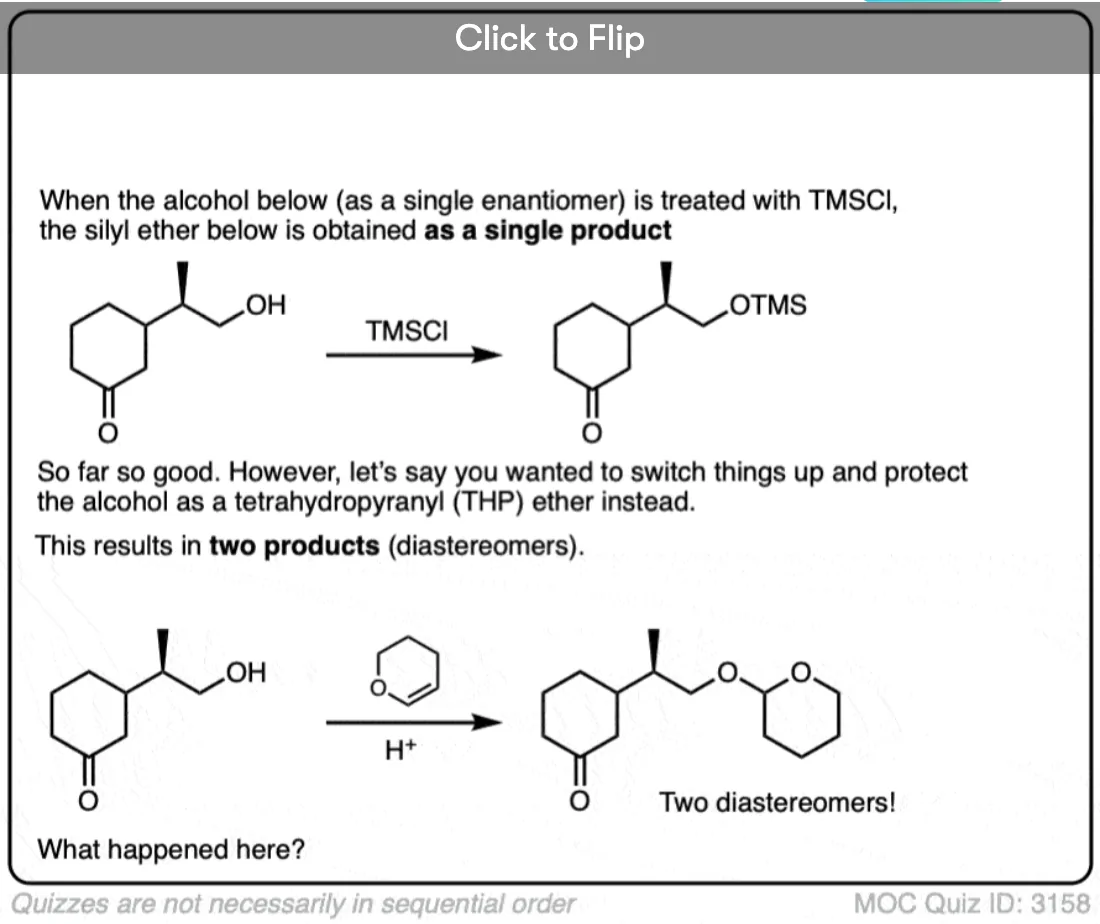
Become a MOC member to see the clickable quiz with answers on the back.
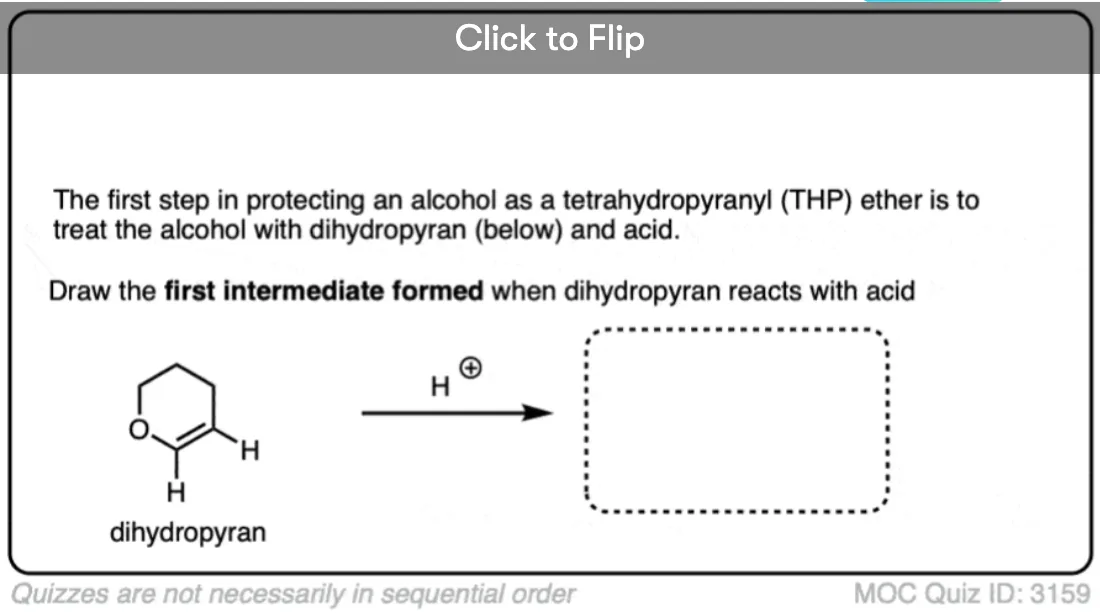
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
Protection as silyl ethers:
- Protection of hydroxyl groups as tert-butyldimethylsilyl derivatives
E. J. Corey and A. Venkateswarlu
Journal of the American Chemical Society 1972 94 (17), 6190-6191
DOI: 10.1021/ja00772a043
The original paper by Nobel Laureate Prof. E. J. Corey (Harvard) describing convenient conditions (TBS-Cl, DMF, imidazole) for the protection of alcohols as TBS-ethers. - GENERATION OF NONRACEMIC 2-(t-BUTYLDIMETHYLSILYLOXY)-3-BUTYNYLLITHIUM FROM (S)-ETHYL LACTATE: (S)-4-(t-BUTYLDIMETHYLSILYLOXY)-2-PENTYN-1-OL
James A. Marshall, Mathew M. Yanik, Nicholas D. Adams, Keith C. Ellis, and Harry R. Chobanian
Org. Synth. 2005, 81, 157
DOI: 10.15227/orgsyn.081.0157
The first step in this synthesis from Organic Syntheses (a source of reliable and reproducible synthetic organic experimental procedures) is O-silylation with TBSCl according to Corey’s procedure. - Protection of Hydroxy Groups by Silylation: Use in Peptide Synthesis and as Lipophilicity Modifiers for Peptides
John S. Davies, Clement L. Higginbotham, E. John Tremeer, Charles Brown, and Richard C. Treadgold
J. Chem. Soc., Perkin Trans. 1, 1992, 3043-3048
DOI:1039/p19920003043
This paper describes a kinetic study of the susceptibility to hydrolysis of various silyl ethers under acidic and basic conditions, and has a convenient table on the first page describing the results. - Symmetrical alkoxysilyl ethers. A new class of alcohol-protecting groups. Preparation of tert-butoxydiphenylsilyl ethers
John W. Gillard, Rejean Fortin, Howard E. Morton, Christiane Yoakim, Claude A. Quesnelle, Sylvain Daignault, and Yvan Guindon
The Journal of Organic Chemistry 1988 53 (11), 2602-2608
DOI: 1021/jo00246a038
This paper describes the use of -SiPh2OtBu as a protecting group for alcohols which is easier to remove with F– compared to conventional -TBDPS ethers. - Selective Deprotection of Silyl Ethers
Todd D. Nelson, R. David Crouch
Synthesis 1996; 1996(9): 1031-1069
DOI:1055/s-1996-4350
This review covers the selective removal of silyl ethers in the presence of similar or different silyl ether groups in the same molecule. In organic synthesis, deprotection strategies are just as important as protection strategies!Ethers can also be used for alcohol protection. Two common ether-based protecting groups are THP- (tetrahydropyranyl-) and MOM- (methoxymethyl-). - Pyridinium p-toluenesulfonate. A mild and efficient catalyst for the tetrahydropyranylation of alcohols
Masaaki Miyashita, Akira Yoshikoshi, and Paul A. Grieco
The Journal of Organic Chemistry 1977, 42 (23), 3772-3774
DOI: 1021/jo00443a038
While p-toluenesulfonic acid is generally used as the catalyst for tetrahydropyranylation of alcohols, PPTS (the pyridine salt of p-toluenesulfonic acid) can be used as an even milder catalyst for this reaction. This is of value when trying to protect an alcohol with several other delicate functional groups in the molecule. - Total synthesis of (S)-12-hydroxy-5,8,14-cis,-10-trans-eicosatetraenoic acid (Samuelsson’s HETE)
E. J. Corey, Haruki Niwa, and Jochen Knolle
Journal of the American Chemical Society 1978, 100 (6), 1942-1943
DOI: 10.1021/ja00474a058
This paper demonstrates a synthetic strategy using alcohol protecting groups. One of the -OH in a substrate groups is protected as a THP ether (conditions not mentioned), and later deprotected, using p-TSA in methanol, 1h at RT, 94% yield. - Diastereo‐ and Enantioselective Total Synthesis of Stigmatellin A
Dieter Enders Prof. Dr. Gunter Geibel Dr. Simon Osborne Dr.
Eur. J. 2000, 16 (8), 1302-1309
DOI: 10.1002/(SICI)1521-3765(20000417)6:8<1302::AID-CHEM1302>3.0.CO;2-J
9 -> 3 involves a MOM protection. Interestingly, this is selective for one of the hydroxyl groups in the molecule, since, as the paper says, “the second hydroxyl group is blocked by a hydrogen bond to the carbonyl function in [the] ortho-position”. This shows that the way we commonly draw structures does not always reflect the preferred conformation!
Protecting with the MOM- group is commonly done using MOM-Cl (methoxymethyl chloride). However, due to this compound’s extreme toxicity (it is a powerful alkylating agent and can alkylate proteins and nucleic acids in our bodies), alternative methods for protection are sought, and the following two papers by Nobel Laureate Prof. G. A. Olah give alternative methods for MOM- protection of alcohols without using MOM-Cl. - Iodotrimethylsilane-Catalyzed Preparation of Methoxymethyl Ethers of Primary and Secondary Alcohols with Dimethoxymethane
George A. Olah, Altaf Husain, Subhash C. Narang
Synthesis 1983, 11, 896-897
DOI: 10.1055/s-1983-30554 - Synthesis of terrein, a metabolite of Aspergillus terreus
Joseph Auerbach and Steven M. Weinreb
Chem. Soc. Chem. Comm. 1974, 298-299
DOI: 10.1039/C39740000298
This paper demonstrates the use of MOM- protection in total synthesis. Two -OH groups are initially protected as MOM ethers by deprotecting to the alkoxides with NaH, followed by MOM-Cl in DMF at RT. At the end of the synthesis, the protecting groups were removed using “a trace of concentrated HCl in MeOH at 62° for 15 min”.
is it possible in a diol to protect only one oh with THP?
Generally no. You can often selectively protect a primary alcohol with a trityl group (triphenylmethyl). There are other situations where you can selectively protect one of the alcohols but unless you are in graduate school it’s not worth getting into them.
I will try that. Although, the aldehyde is solid and not liquid.
Dissolve it up in THF or whatever solvent you’re using to make the enolate as a dilute solution (e.g. 0.2 M)
A million thanks. You are such a rare gem.
I have tried several times using NaOH, KOH, MgI2, as well as acid catalysis using HCl, but all to no avail (the aldehyde was not deprotonating to form chalcone). I am thinking of using NaH this time around and that’s why am concerned about protecting the hydroxy group.
Why not just pre-form the enolate with LDA in THF and then add your aldehyde dropwise at low temp? The OH could end up protonating your enolate, but it’s pretty sterically hindered.
I’m using 3,5-di-tert-butyl-4-hydroxybenzaldehyde. I want to condense it with Acetophenone to form (E)-3-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-phenylprop-2-en-1-one
I’m using 3,5-di-tert-butyl-4-hydroxybenzaldehyde. I want to condense it with Acetophenone to form (E)-3-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-phenylprop-2-en-1-one.
In that case, it would be hard to protect since it’s sandwiched between those t-butyl groups.
Have you tried running the reaction anyways? Do you pre-form the enolate of acetophenone and then add the aldehyde? Curious if the OH would give you problems.
Otherwise this is basically a Claisen-Schmidt reaction, of which about a billion examples exist, under conditions where the enolate forms reversibly and a slight excess of base, I would imagine you could get this to go.
I want to do an aldol condensation with hydroxybenzaldehyde using a strong base. How can I protect OH group in the presence of aldehyde ?
Which hydroxybenzaldehyde are you starting with?
The usual procedure would be to start with a methoxybenzaldehyde and then try to remove the CH3 after your aldol with something like TMSI.
One milder way to do it would be to treat the hydroxybenzadehyde with KH and BnBr to make the O-benzylated benzaldehyde and then after your aldol, treat with Pd-C, H2. Or paramethoxybenzyl (PMB) and deprotect with DDQ.
How would I deprotect OMe in presence of CH2OH?
Deprotecting OMe is never fun. It depends on how stable the rest of your molecule is. I would employ basic conditions (RS- , polar aprotic solvent) since acidic conditions will likely ionize the CH2OH. Without knowing what the rest of the molecule looks like, it’s hard to say.
I want to protect aliphatic OH in the presence of aromatic OH, give me the suggestion.
Without knowing what your molecule looks like it’s hard to say. It would be easy to selectively protect the aromatic OH because it’s considerably more acidic, and once you form the alkoxide it will be far more nucleophilic. One idea would be to just protect both groups, and then selectively deprotect the aromatic OH afterwards.
please tell the alcohol protection with uv active group
Usually 2-nitrobenzyl. See https://en.wikipedia.org/wiki/Photolabile_protecting_group
How would I protect an NH2 group during and EAS reaction and why would I need to do so?
Suppose you have a primary and a secondary alcohol in a molecule. Would TMS prefer one over the other? Why?
In theory primary alcohols are less sterically hindered than secondary alcohols. You would think that TMS might be selective for primary. It is, but to a very minor extent. In order to get very good selectivity for primary over secondary, you have to use a very bulky protecting group like TBDPSCl or TIPSCl or trityl chloride. But like I said you have to make the protecting group much bigger than TMS.
Hello,
I had a few questions, as i’m learning organic synthesis,
i understood that the silylmethylether can be attacted by R- if there is not a good leaving group such as (Br, Cl, OTs), is that correct because i didn’t read something about that here?
Thank you very much!
I’m not sure what “attracted by R- means”. Could you elaborate?
TBDMS-Cl is superior to TMS-Cl for selective protection of an alcohol group, why
Much bulkier and therefore more sensitive to steric factors. However I’d go even further and use TBDPS.
which protecting group can be used to protect OH group in presence of carboxyl functional group(not to be protected)?
Silyl groups are fine, benzyl groups are fine. Lots of options.
I am trying to protect aryl OH with trimethyl silyl chloride using HMDS base. In the reaction it is showing different spot on TLC. But when we do column chromatography. It only gives starting back. Does silyl protection breaks during column chromatography?
What is the solution? Please help me.
TMS groups are extremely labile, especially on a phenol hydroxyl group. If you’re going to go with a silyl group, then pick TBS or something heavier.
How does the addition/ removal of a protecting group affect stereochemistry? For example a hydroxyl OH group on a wedge or dash would it remain the same or be changed?
Won’t be affected. It’s like deprotonation, you’re only affecting the O-PG bond. To invert the stereochem you’d have to invert the C-O bond.
Why can’t tertiary alcohols be protected by tetrahydropyranyl
ether?
I wouldn’t say this a 100% restriction – there are likely exceptions – but if you’re being taught this, here’s at least two reasons.
1. THP protection involves using strong acid, and strong acid might protonate the OH, resulting in loss of water to form a (stable) tertiary carbocation.
2. Tertiary alcohols are quite sterically hindered, and the rate of attack of the tertiary alcohol at the electrophile produced by protonation of DHP might be slow.
Hope this helps! James
Thanks for the suggestion I will look into it in more details.
I am looking for ways to protect a hydroxyl group on the bone of a molecule that has a carboxyl group at the end that needs to remain active. Is it possible to mask an OH on the bone while to acid at the end of chain remains ” active ” ?
Richard
try to chlorinate the carboxyl group, then protect the hydroxyl group, and then add water to the molecule, producing your desired product. use a protecting agent that doesn’t react with aqueous hcl, which will be produced from the reaction of water with the cocl group on the end of your molecule.
Thanks for the amazingly detailed post! Love the example with the painter’s tape.