Alkene Reactions
Rearrangements in Alkene Addition Reactions
Last updated: May 7th, 2026 |
Carbocation Rearrangements In Alkene Addition Reactions
- Alkene addition reactions that go through carbocation intermediates (such as addition of HCl, HBr, HI and H3O+) may occur with rearrangement of the carbocation intermediate if a more stable carbocation can be formed.
- Recall that carbocation stability increases with substitution, so tertiary carbocations are more stable than secondary carbocations, which are in turn more stable than primary carbocations.
- Rearrangements take the form of hydride shifts where a hydrogen atom and its pair of electrons migrates from an adjacent carbon, resulting in a more stable carbocation, or alkyl shifts where an alkyl group migrates with its pair of electrons.
- In certain cases, ring expansion reactions may also occur, where formation of a more stable carbocation is accompanied by release of ring strain from a small ring (e.g. cyclobutane)
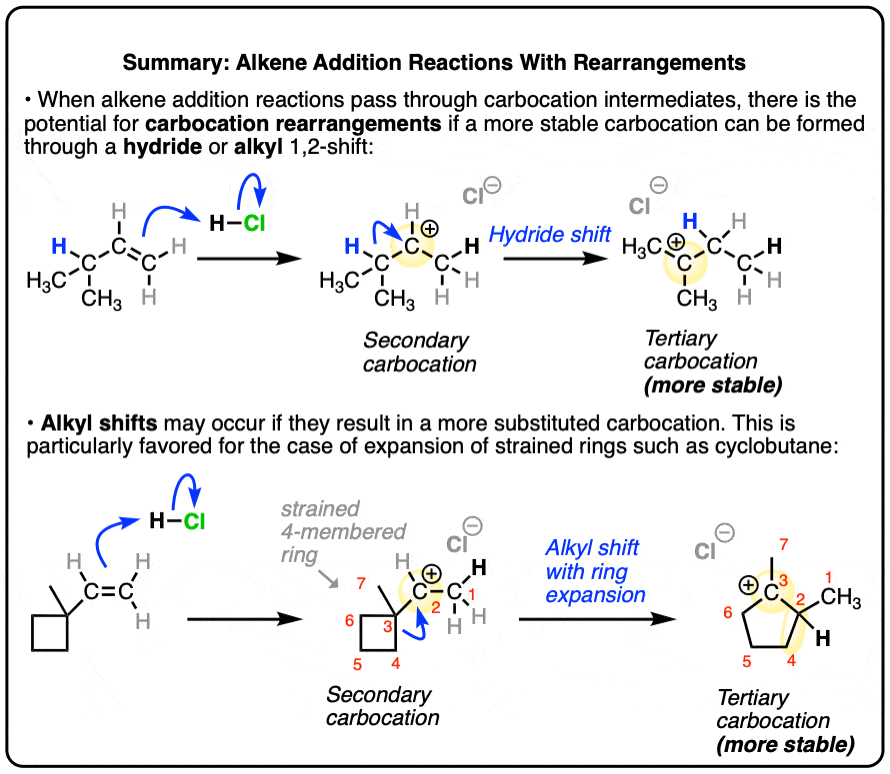
Table of Contents
- An Alkene Addition Reaction With A Twist
- Hydride Shifts In Alkene Additions, Step 1: Attack Of Acid By The Nucleophile
- The Key Rearrangement Step: Hydride Shift
- Step Three: Attack Of Nucleophile On The Carbocation
- Alkene Addition Reactions With Alkyl Shifts
- Alkene Addition Reactions With Ring Expansion
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. An Addition With A Twist
In exploring reactions that proceed along the carbocation pathway, every once in awhile you might see an example of an addition reaction that looks a little… strange. The alkene is gone, two new bonds have formed, but the positions of the new bonds is a little out of the ordinary. Like in this example!
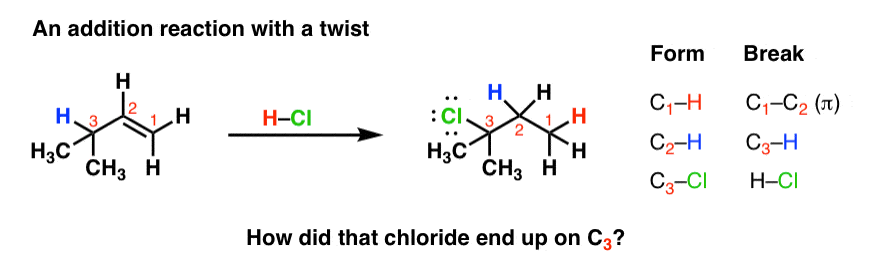
If you tally up the bonds that form and the bonds that break, we notice that there is an extra set of C-H bond forming/breaking events.
If you’ve seen the previous articles in the substitution and elimination series, this should look familiar. It’s a telltale sign that a rearrangement has taken place.
2. Hydride Shifts In Alkene Additions, Step 1: Attack Of Acid By The Nucleophile
The first step in this reaction we’ve seen before: attack of the alkene upon the electrophile (in this case, the H of H-Cl). The result is a carbocation.
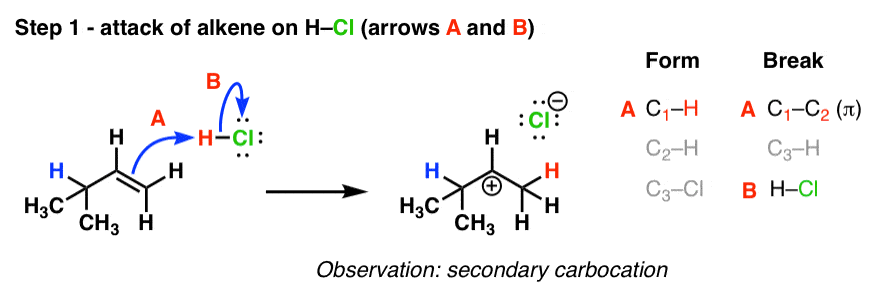
Note that the carbocation that’s been formed is a secondary carbocation, and it’s adjacent to a tertiary carbon.
3. The Key Rearrangement Step: Hydride Shift
In this next step, the lone pair in the C-H bond migrates from the tertiary carbon to the secondary, forming a new (tertiary) carbocation. The driving force for this reaction is formation of the more stable carbocation.
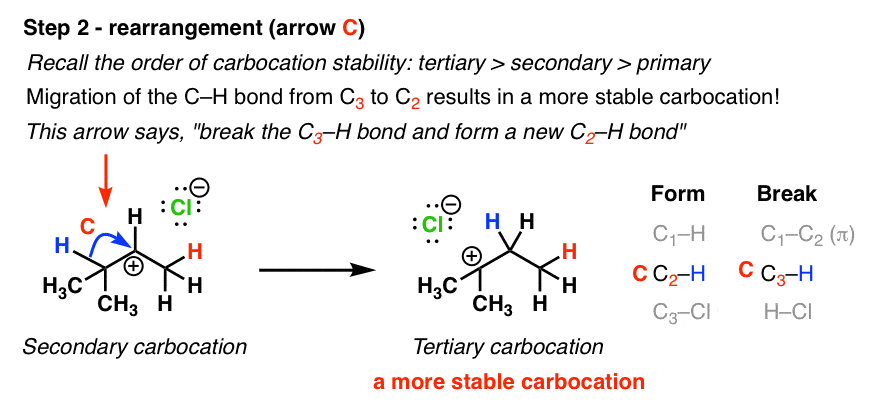
Note how it’s just one arrow we’re drawing here! The same arrow shows C-H bond breakage and C-H bond forming.
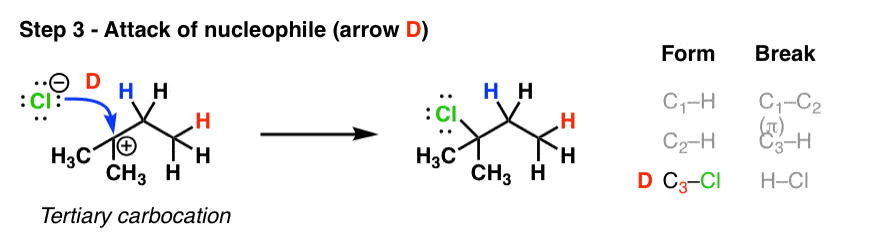
4. Step Three: Attack Of Nucleophile On The Carbocation
We’ve also seen the third step before. Attack of the nucleophile (chloride ion) upon the new carbocation gives us our new alkyl halide!
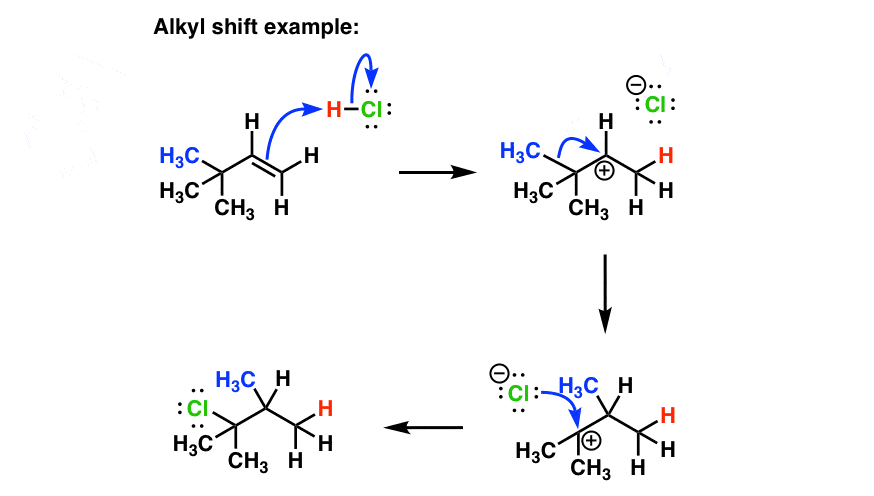
5. Alkene Addition Reactions With Alkyl Shifts
Rearrangements can also occur with alkyl shifts, as seen in the example below. Note again that the rearrangement step is represented by just one curved arrow!
6. Alkene Addition Reactions With Ring Expansion
Finally, one of the cases that students often find very difficult is in recognizing reactions that occur with rings (ring expansion or ring contraction). Although perhaps difficult to see, in fact it proceeds through exactly the same mechanism as in the cases above. Note again that we’re depicting the rearrangement reaction with a single curved arrow. [Hint – if you’re doing this on your own, it might help to draw the ugly version first].
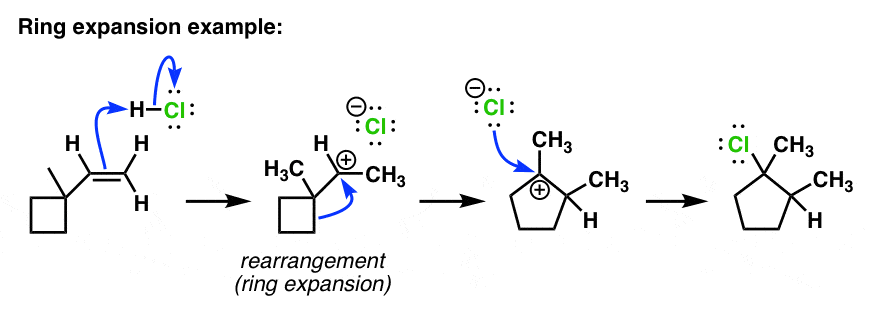
So why is it that the carbon from the ring migrates, and not the CH3 as before? A fair question. Migration of the CH3 would indeed produce a tertiary carbocation. However, migration of the CH2 from the ring not only produces a tertiary carbon but incrases the size of the ring from 4-membered to 5-membered, which relieves considerable ring strain present in the cyclobutane ring (worth about 26 kcal/mol).
That about does it for the carbocation pathway of alkene addition reactions. In the next post we’ll go into the second (of three) major pathways for alkene addition mechanisms.
NEXT POST: Bromination of Alkenes – How Does It Work?
Notes
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
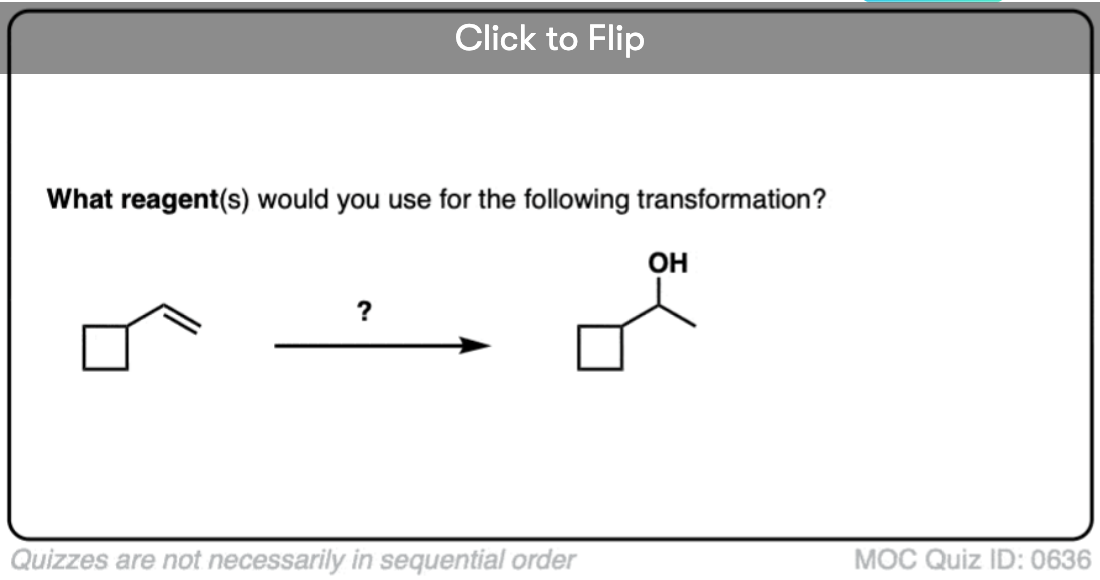
Become a MOC member to see the clickable quiz with answers on the back.
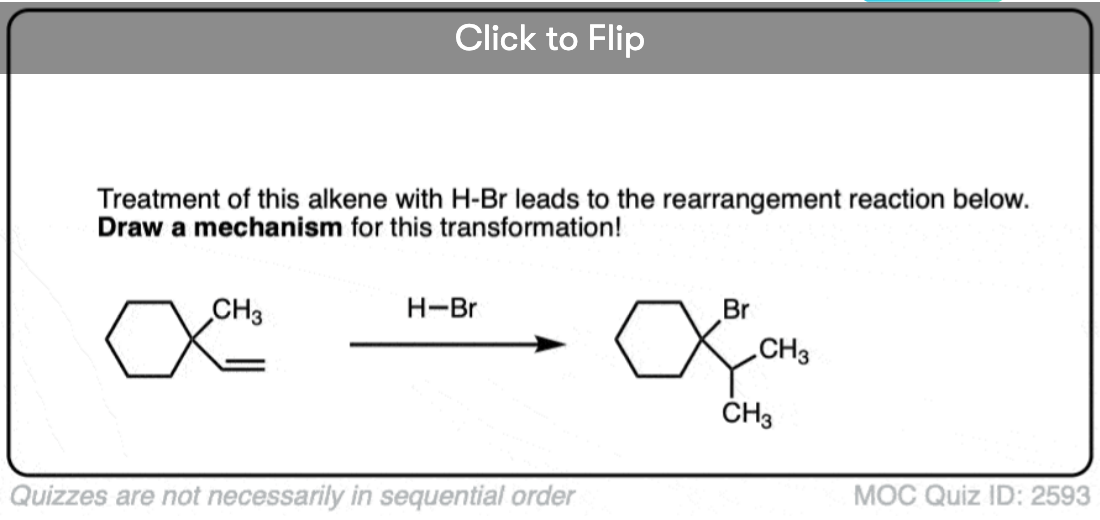
Become a MOC member to see the clickable quiz with answers on the back.
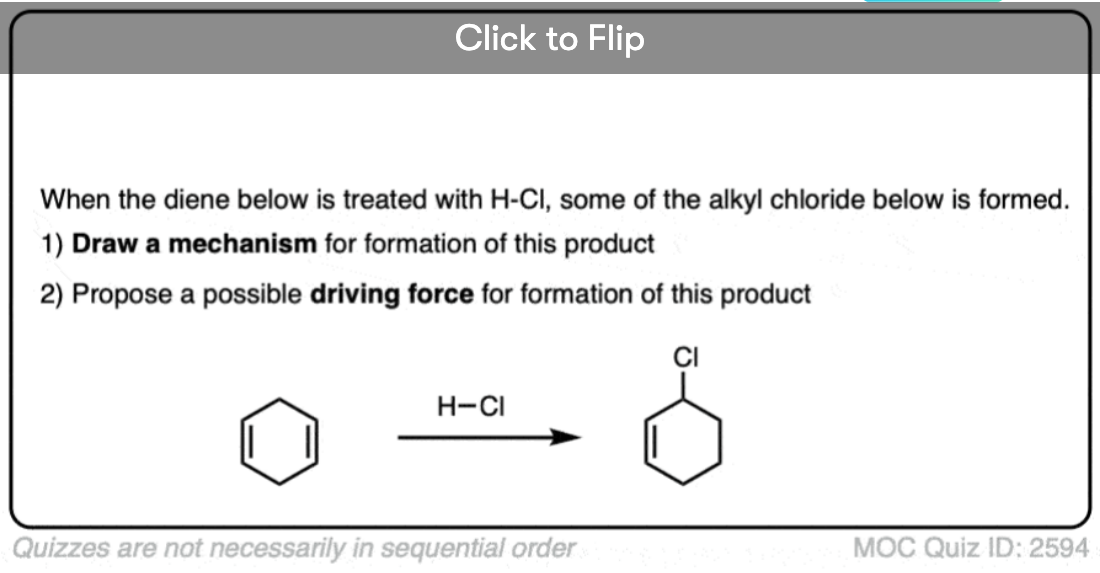
Become a MOC member to see the clickable quiz with answers on the back.
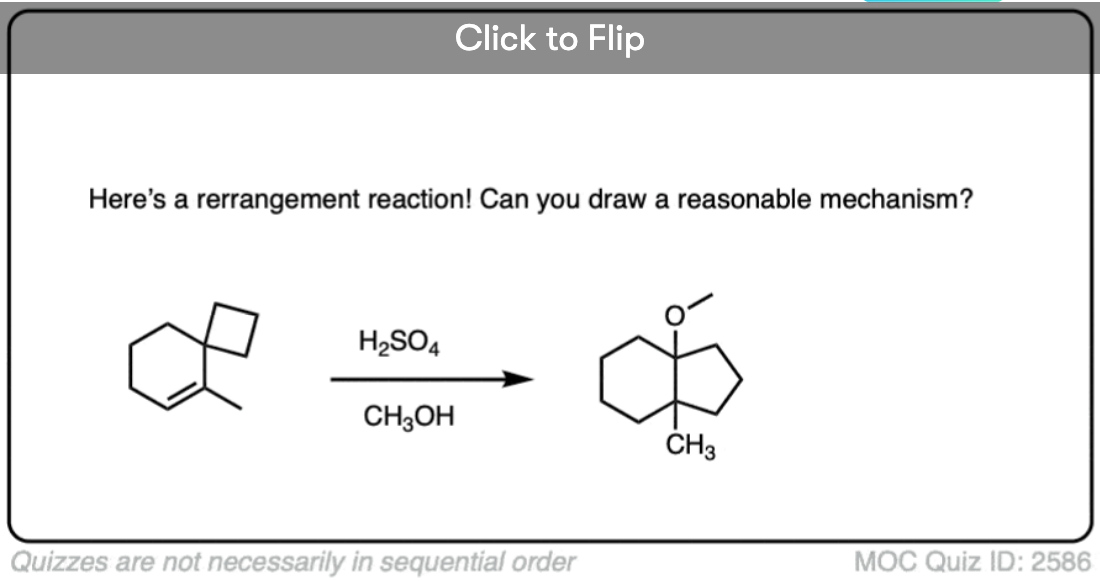
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- Ueber die Beziehung der Pinenhaloïdhydrate zu den Haloïdanhydriden des Borneols
Georg Wagner, W. Brickner
Ber. 1899, 32 (2), 2302-2325
DOI: 10.1002/cber.189903202168
The oldest examples of these rearrangements are in the pinene series. Wagner showed these rearrangements occur in conversions of pinene to bornyl compounds. - Über die Gleichgewichts‐Isomerie zwischen Bornylchlorid, Isobornylchlorid und Camphen‐chlorhydrat
Hans Meerwein and Konrad van Emster
Ber. 1922, 55 (8), 2500-2528
DOI: 10.1002/cber.19220550829
Prof. Hans Meerwein extended Wagner’s work to non-terpene series, and came up with the crucial insight that it proceeded through a carbonium ion – a very controversial insight at the time! Crucial experiment was finding rate was dependent on solvent polarity. Rate order was SO2 > MeNO2 > MeCN > PhNO2 > PhCN > PhOMe > PhBr > EtBr > PhCl > C6H6 > pet ether > ether. Furthermore, he found that certain acids considerably accelerated the rearrangement of camphene hydrochloride to isobornyl chloride. Alkyl migrations in carbocations are often called “Wagner-Meerwein” rearrangements after Georg Wagner and Prof. Hans Meerwein, who studied them more rigorously. - Mechanism of substitution at a saturated carbon atom. Part XXXII. The rôle of steric hindrance. (Section G) magnitude of steric effects, range of occurrence of steric and polar effects, and place of the Wagner Rearrangement in nucleophilic substitution and elimination
I. Dostrovsky, E. D. Hughes, and C. K. Ingold
J. Chem. Soc. 1946, 173-194
DOI: 10.1039/JR9460000173
Prof. Ingold formalized rules for carbocations:
1) It is necessary for rearrangement that initial bond breakage result in an atom with an incomplete octet
2) The system will only rearrange if the free energy change is in the right direction (i.e. the carbocation being rearranged to should be more stable, e.g. secondary -> tertiary).
This tied together SN1, E1, alkene addition with Wagner-Meerwein shifts in a unified framework. - Communications TO THE EDITOR
The Journal of Organic Chemistry 1962, 27 (5), 1926-1932
DOI: 10.1021/jo01052a098
Rigid steroid and diterpenoid systems show addition reactions where multiple hydride and alkyl shifts can occur. One example is dihydroipimaric acid. - The constitution and stereochemistry of euphol
H. R. Barton, J. F. McGhie, M. K. Pradhan, and S. A. Knight
J. Chem. Soc. 1955, 876-886
DOI: 10.1039/JR9550000876
Euphenol, which is similar to lanosterol, also undergoes addition via a carbocation intermediate which can undergo successive hydride and alkyl shifts. - The Structures of the Triterpenes Friedelin and Cerin
E. J. Corey and J. J. Ursprung
Journal of the American Chemical Society 1956, 78 (19), 5041-5051
DOI: 10.1021/ja01600a064
The path from Friedelin to Cerin is a crazy series of fun rearrangements! - Total synthesis of (±)-isocomene and related studies
Michael C. Pirrung
Journal of the American Chemical Society 1981, 103 (1), 82-87
DOI: 1021/ja00391a016
The rearrangement of vinyl cyclobutane opening to cyclopentane is accompanied by relief of ring strain, and this paper shows that can be applied fruitfully in sesquiterpene synthesis. - The Addition of Hydrogen Bromide to Simple Alkenes
Hilton M. Weiss
Journal of Chemical Education 1995, 72 (9), 848
DOI: 1021/ed072p848
A simple experiment suitable for undergraduate organic chemistry laboratory courses that demonstrates that it is possible for the intermediate carbocation to rearrange and give different products.
What about the solvents in the reaction. Does a CCl4 medium affect the rearrangement in any way?
The reaction generates carbocations, which are much less stable in non-polar solvents like CCl4. The best solvent for a carbocation rearrangement is a highly polar, ionizing solvent.
What about the stereochemistry of the ring expansion? How is the CH2 attack? Backside or frontside?! I mean, after the expansion, the CH3 (next to the Carbon with positive charge, out of the ring) will stay inside or outside the plane of the paper?
The group that is migrating remains on the same face. This is OK because the carbocation has an empty p orbital and in theory can be attacked from either face.
can we convert propene to propane -2-ol by using HCl ( markonikoff rule) and then by adding KOH
While HCl will give you 2-chloropropane, KOH will perform elimination (E2) to give you back propene.
But why not just add H3O+ (H2SO4 / H2O) and cut out the middleman? Or oxymercuration?
What about cyclopropane. will rearrangements take place to form cyclobutane.
Cyclopropane is a very, very interesting case. Carbocations on an alkyl group adjacent to a cyclopropane are particularly stable. The result is not ring expansion, but a non-classical carbocation called the cyclopropylcarbinyl cation: https://en.wikipedia.org/wiki/Carbocation#Specific_carbocations
Can a type of cyclopentene undergo a carbocation rearrangement to form a cyclohexane?
when 1 methyl 2 cyclohexene reacted with HBr than what will be the major product?
What if HCl is to be added across 1-Chloroethene ? Does the resulting carbocation stability and the position of the positive charge depend upon the fact that “for halogens inductive>mesomeric effect” ?
The chloride would stabilize the carbocation through resonance (donation of lone pair) . So protonation would occur adjacent to the carbon attached to chlorine, and the resulting product is a dihalide.
https://www.masterorganicchemistry.com/2013/05/24/alkyne-reaction-patterns-the-carbocation-pathway/
Thanks for the link . What about 2-Chloro-2-Butene ? Which carbon will become the carbocation ? Here , would the C3 carbon be more stable as it doesnt have much effect from the -Cl ‘s inductive effect ?
Actually the carbon attached to Cl will become the carbocation. Cl has a pair of electrons which can stabilize adjacent carbocations via pi donation. See this post https://www.masterorganicchemistry.com/2018/03/05/why-are-halogens-ortho-para-directors/
In case of addition of HBr to 3-methylcyclohexene..what will be the major product? I think it will be 1-bromo 1-methylcyclohexane but my teacher thinks otherwise, saying there will be no rearrangement. What do you think will happen? and why?
Hi – addition of HBr to 3-methylcyclohexene, proposing a rearrangement is very reasonable, as it would mean that a secondary carbocation would be rearranging to a tertiary carbocation.
Some profs are not very consistent when it comes to these things.
James
So if you had HCl addition to just vinylcyclobutane (no methyl group as in the example above)… would you give the product as 1-chloro-2-methylcyclobutane or 1-chloro-1-methylcyclobutane?
Do you mean cyclopentane instead of cyclobutane? If so, I’d think the 1-chloro-1-methylcyclopentane product would eventually result. Curious as to what experiment has to say.
yes, I meant cyclopentane as the product :(
I am also curious as to what experiment would say… hmm….
Are these rearrangements termed 1,2 hydride shifts and 1,2 alkyl shifts?
Yes they are – or Wagner-Meerwein shifts.
Love your post, thankyou
Glad you found it useful Kathy!
In the penultimate scheme, the 3rd structure, there’s a CH2 instead of a methyl CH3.
Great post, anyway.
Fixed. As always, thank you