Free Radical Reactions
Selectivity in Free Radical Reactions: Bromination vs. Chlorination
Last updated: May 28th, 2026 |
The Selectivity of Free-Radical Bromination vs Chlorination. A Detailed Answer
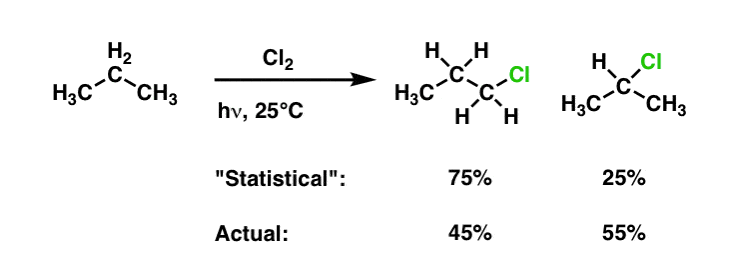
In last article on radicals (See Article – Selectivity in Free Radical Reactions) we saw this data that compares the chlorination of propane vs. the bromination of propane.
For chlorination, the reaction is selective for secondary C-H over primary C-H by a factor of 55/(45/3) = 3.6 to 1
For bromination, the reaction is selective for secondary C-H over primary C-H by a factor of 97/(3/3) = 97 to 1.
{kind=link}
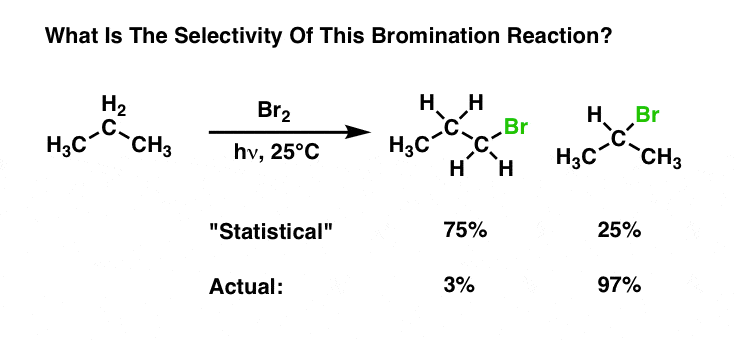
Wow!!! Is bromination ridiculously more selective than chlorination, or what?? (Again, if you want to know how this was calculated, go back to the last post.)
Today we’re going to try to answer, “why is bromine more selective than chlorine” ? You might think that going from 4:1 to 97:1 will involve a huge difference in energies. But as we’ll see, it’s more subtle than you might expect. A few kcal/mol can make a huge difference! (See article – 1 kcal/mol Is A Lot, Actually)
Table of Contents
- The Selectivity-Generating Step Is Breakage Of The C–H Bond By A Halogen Radical (Propagation Step #1)
- Background: Activation Energy And The Arrhenius Equation (Yes, This Is Relevant)
- Heat Increases The Average Velocity (And Energy) Of Molecules
- As A Reaction Mixture Is Heated, A Larger Proportion Of Molecules Will Have Sufficient Activation Energy (Ea) To React
- Selectivity Is Proportional To Differences In Activation Energy For The Key Step
- Small Differences In Activation Energies (~ 3 kcal/mol) Can Mean LARGE Differences In Selectivity (97:1)
- So WHY Is The Difference For Activation Energies Greater For Bromination Than For Chlorination?
- The Transition State For Chlorination Resembles The Reactants (An “Early” Transition State) Which Are Close Together In Energy. So Selectivity Is Low.
- The Transition State For Bromination Resembles The Products (A “Late” Transition State) Which Are Farther Apart In Energy. So Selectivity Is High.
- Summary: Selectivity For Free-Radical Chlorination vs Bromination
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
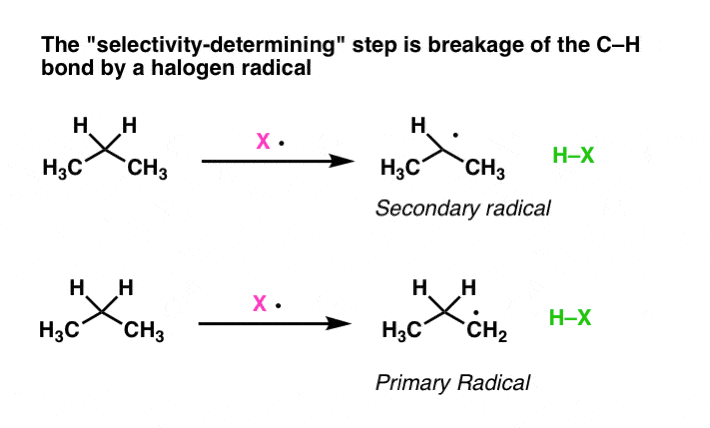
1. The Selectivity-Generating Step Is Breakage Of The C–H Bond By A Halogen Radical (Propagation Step #1)
The question that this post hopes to answer is “Why is bromine more “selective” for the secondary carbon than chlorine?”.
It’s kind of a long answer. This post goes through the data and makes the scientific argument. In the next post I’ll put forward a simple analogy that simplifies this idea for many students.
The first thing to note is formation of the different chloropropanes happens during the chain propagation step (that is, after initiation). So for our purposes here we are only going to analyze the two propagation steps and assume that initiation has already occurred. In other words, this step:
2. Background: Activation Energy And The Arrhenius Equation (Yes, This Is Relevant)
Before addressing the topic of selectivity directly, let’s first begin by talking about activation energy. You might recall that in order for a reaction to occur, the reactants must come into contact with each other with sufficient energy to overcome the repulsions between them (electron clouds). They also must collide in such a way that allows for transfer of electrons (aka “orbital overlap”)
In other words, the rate is equal to:
[concentration of molecules with sufficient energy] *[probability they collide in the “right way”]
This is dealt with by the Arrhenius equation:

Here, the “probability they collide in the right way” is dealt with by the pre-exponential factor A and is unique for each reaction. The exponential factor e-Ea/RT is what’s known as the “distribution function” and this is how we calculate the % of molecules in solution that have sufficient energy to react (heretofore known as the “activation energy”. )
3. Heat Increases The Average Velocity (And Energy) Of Molecules
Note the fact that this is temperature dependent. Why might that be? Well, as a collection of molecules is heated, the average speed of those molecules will increase. This is described by a related function known as the Boltzmann distribution, shown right here at three different temperatures [a=1, a=2, and a=5]. The y-axis shows the # of molecules (“distribution”) and the x-axis shows energy [thank you Wikipedia]
Heat Increases The Average Velocity Of Molecules
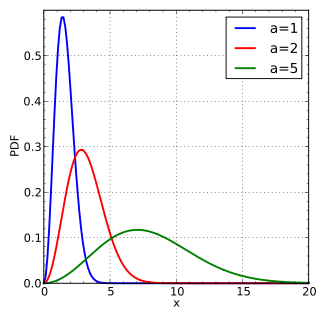
This shows the “distribution” (aka “number”) of molecules at specific energies. The hump in the middle is the “most probable” energy, and notice how it resembles a bell curve with tails at both ends. At the far right of the scale are the most energetic molecules. Note what happens when temperature is increased: on average, molecules have greater energy, and also the right-hand “tail” extends further out.
4. As A Reaction Mixture Is Heated, A Larger Proportion Of Molecules Will Have Sufficient Activation Energy (Ea) To React
Now imagine we have a reaction with activation energy Ea. At very cold temperatures, very few molecules have sufficient energy to react. But as the reaction mixture is heated, the proportion of those molecules increases. Ergo, the rate of the reaction increases with heat. Here is an example of how it works. Note how in this case below, the reaction doesn’t occur at 300K, but starts to occur at a reasonable rate at 330K!
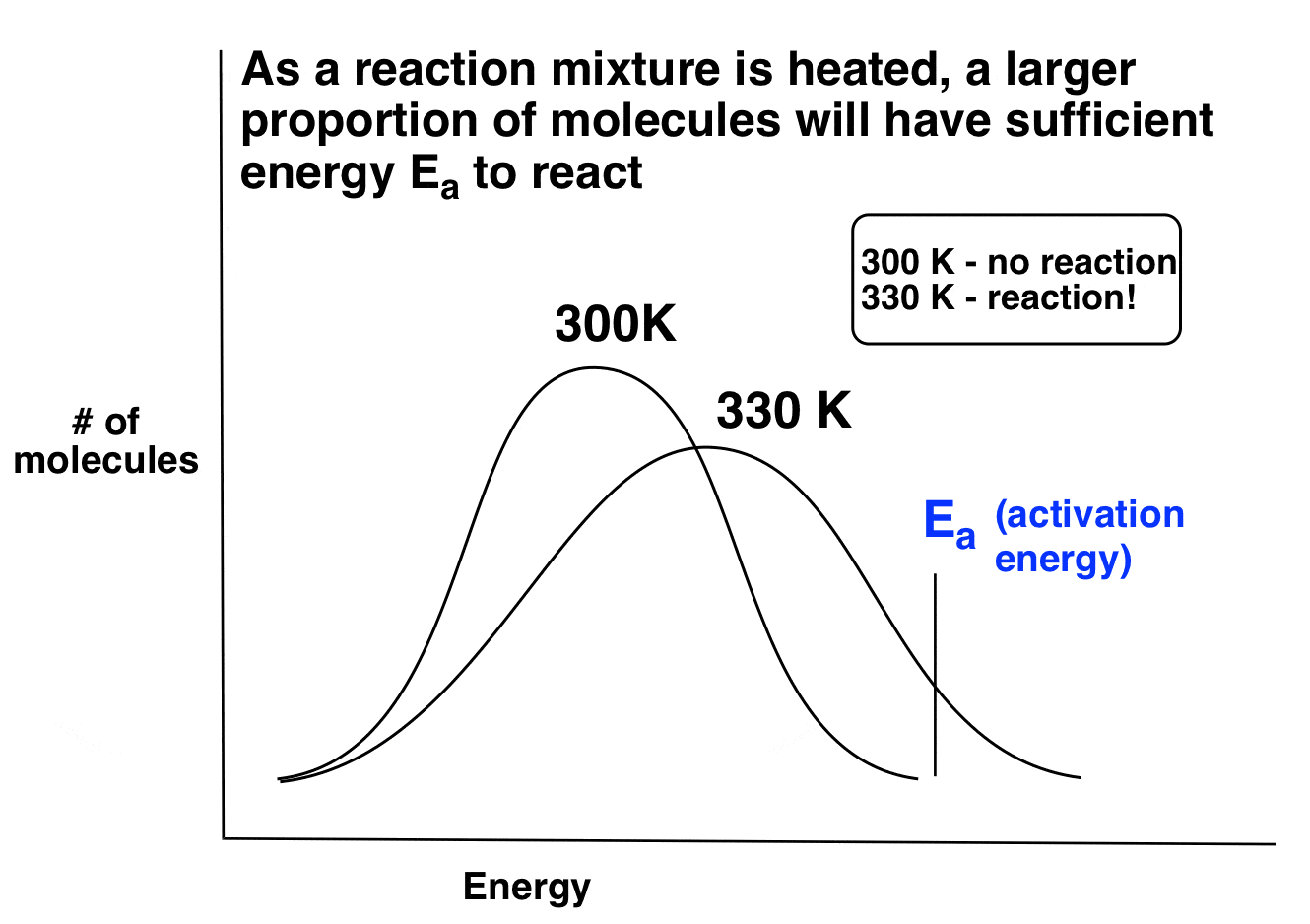
Okay, that covers changing the reaction temperature. But what does this have to do with the selectivity of halogenation?
5. Selectivity For Bromination vs Chlorination Is Proportional To Differences In Activation Energy For The Key Step
Here, we’re keeping reaction temperature constant, but the activation energy for each reaction is slightly different.
We have four reactions in total to think about (two different halogens and two different C-H bonds). The activation energy for each reaction has been experimentally measured [Ref 2] (and here, we’re talking about the activation of C-H bond breaking [i.e. propagation] not initiation. Here’s what they look like.

This is actually all the information we need to be able to make a rough estimate of selectivities.
For a reaction at 300K, we can calculate RT using the gas constant (1.987 cal/ K mol) and plug in the activation energy for each reaction. By dividing the two equations by each other, the pre-exponential factor A will roughly cancel out and we can obtain estimates for selectivities.
The bottom line here is that due to the nature of the Arrhenius equation, the greater the difference between activation energies, the larger the selectivity. The effects can be dramatic, even when going from a difference of 1kcal/mol (for chlorination) to 3 kcal/mol (for bromination of primary vs. secondary)
6. Small Differences In Activation Energies (~ 3 kcal/mol) Can Mean LARGE Differences In Selectivity (97:1)
Just a bit of math using the Arrhenius equation, a rough calculation of selectivities based on differences in activation energy.
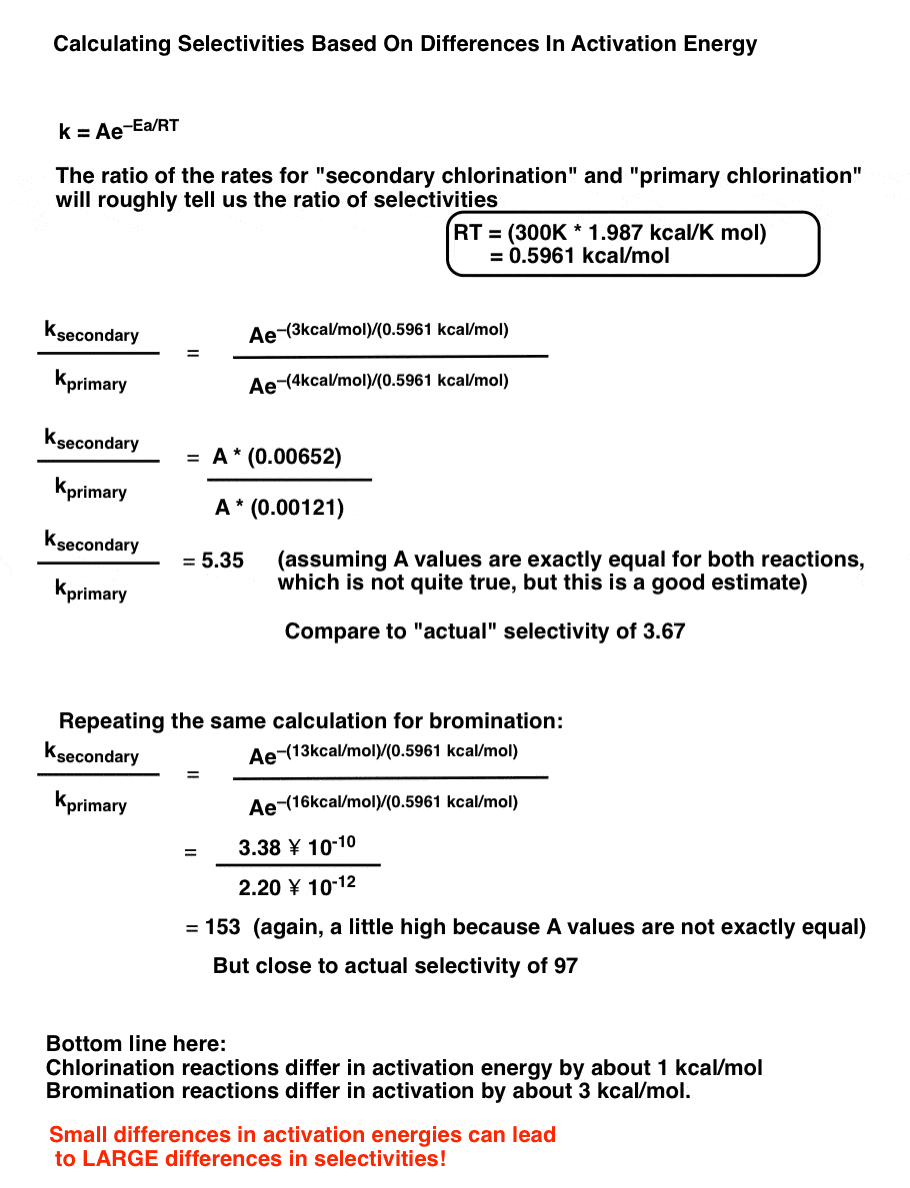
All this is fine – it’s based on experimental data for activation energies. But it just opens up another question.
WHY is the difference for activation energies greater for bromination (3 kcal/mol) than for chlorination (1 kcal/mol). Great question!
7. So WHY Is The Difference For Activation Energies Greater For Bromination Than For Chlorination?
Understanding this point begins with understanding the energy profile of these two different reactions (chlorination and bromination).
We’ll do the math in a second, but the key difference is that in chlorination, the key propagation step is exothermic and in bromination, the key propagation step is endothermic. This is because chlorination forms a strong H-Cl bond (103 kcal/mol) and bromination forms a much weaker H-Br bond (87 kcal/mol).
The two reaction coordinates roughly look like this:
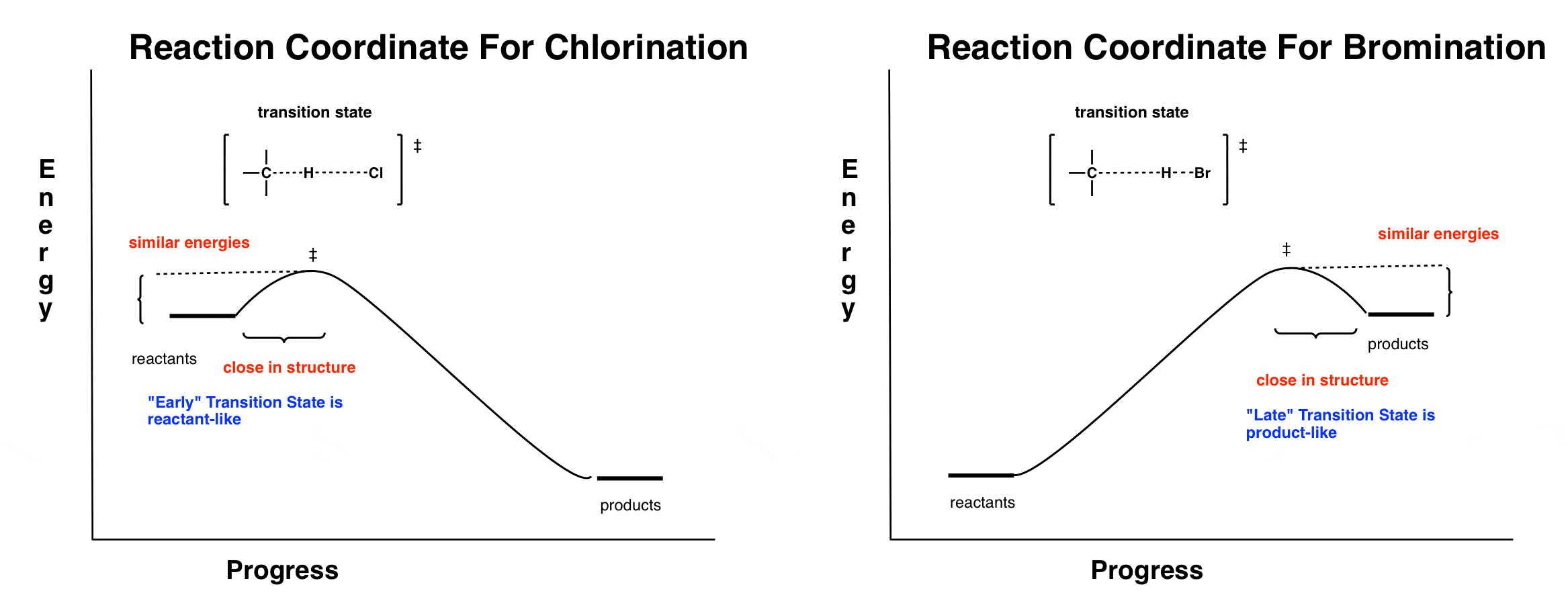
Look closely where the transition state is for each reaction.
In chlorination, the reaction is exothermic, and the transition state resembles the reactants. According to Hammond’s postulate, we could say that this transition state is “early”.
In bromination, the reaction is endothermic, and the transition state resembles the products. According to Hammond’s postulate we say that this transition state is “late”.
8. The Transition State For Chlorination Resembles The Reactants (An “Early” Transition State) Which Are Close Together In Energy. So Selectivity Is Low.
What this means is that for chlorination, the difference in activation energies between the two radical pathways (i.e. “secondary” and “primary”) will most closely resemble the reactants (which are identical in energy). So there will be a very small difference in activation energies between the two.
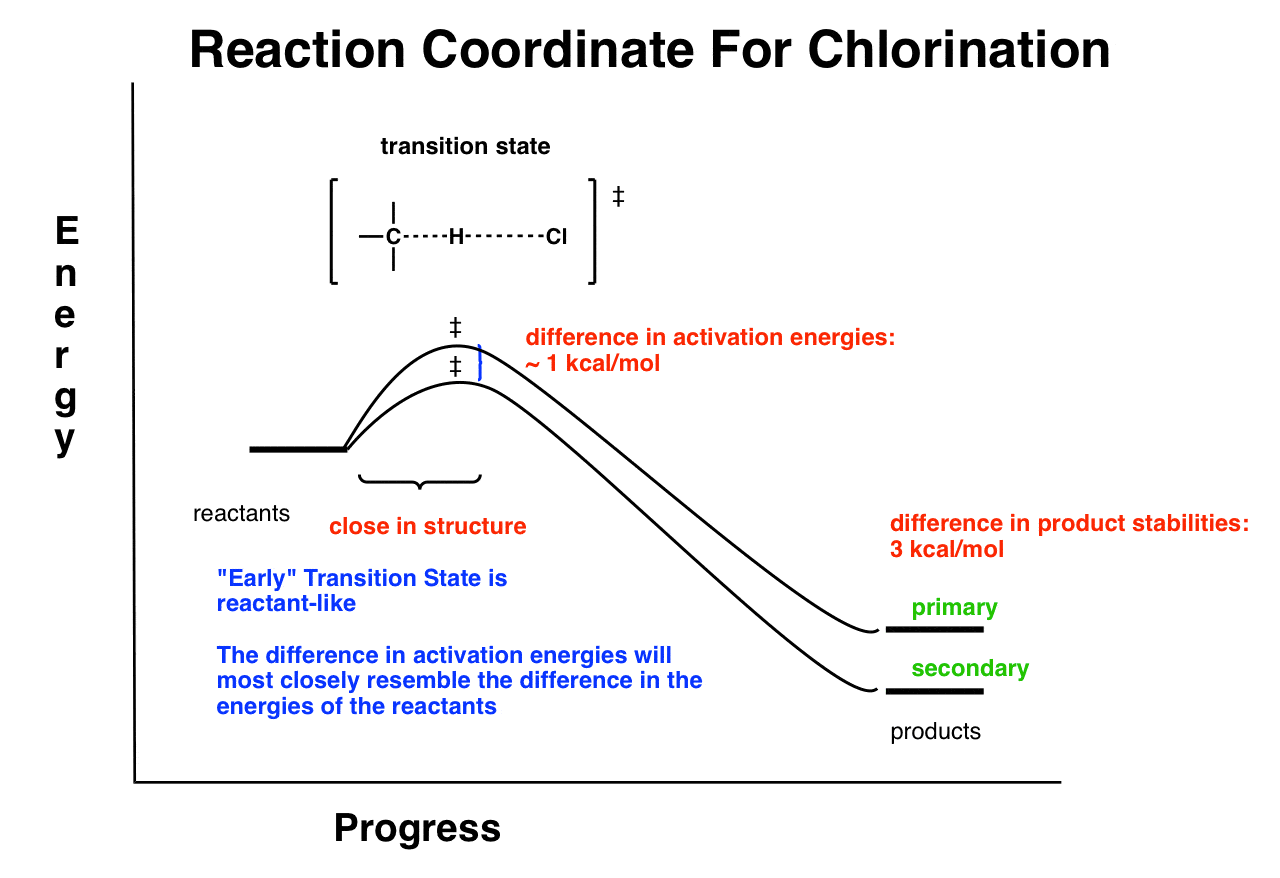
The difference in activation energies is small (1 kcal/mol) because of the early transition state. Note that even though there is a fairly large difference in energy between the products (3kcal/mol) it doesn’t affect the activation energies.
9. The Transition State For Bromination Resembles The Products (A “Late” Transition State) Which Are Farther Apart In Energy. So Selectivity Is High.
We can do the same analysis for bromination. Here, it’s a “late” transition state, so the difference in activation energies between primary and secondary will closely resemble the differences in energy between the two. So we would expect the activation energy difference to more closely resemble the difference between the energies of the products. And that is the case!
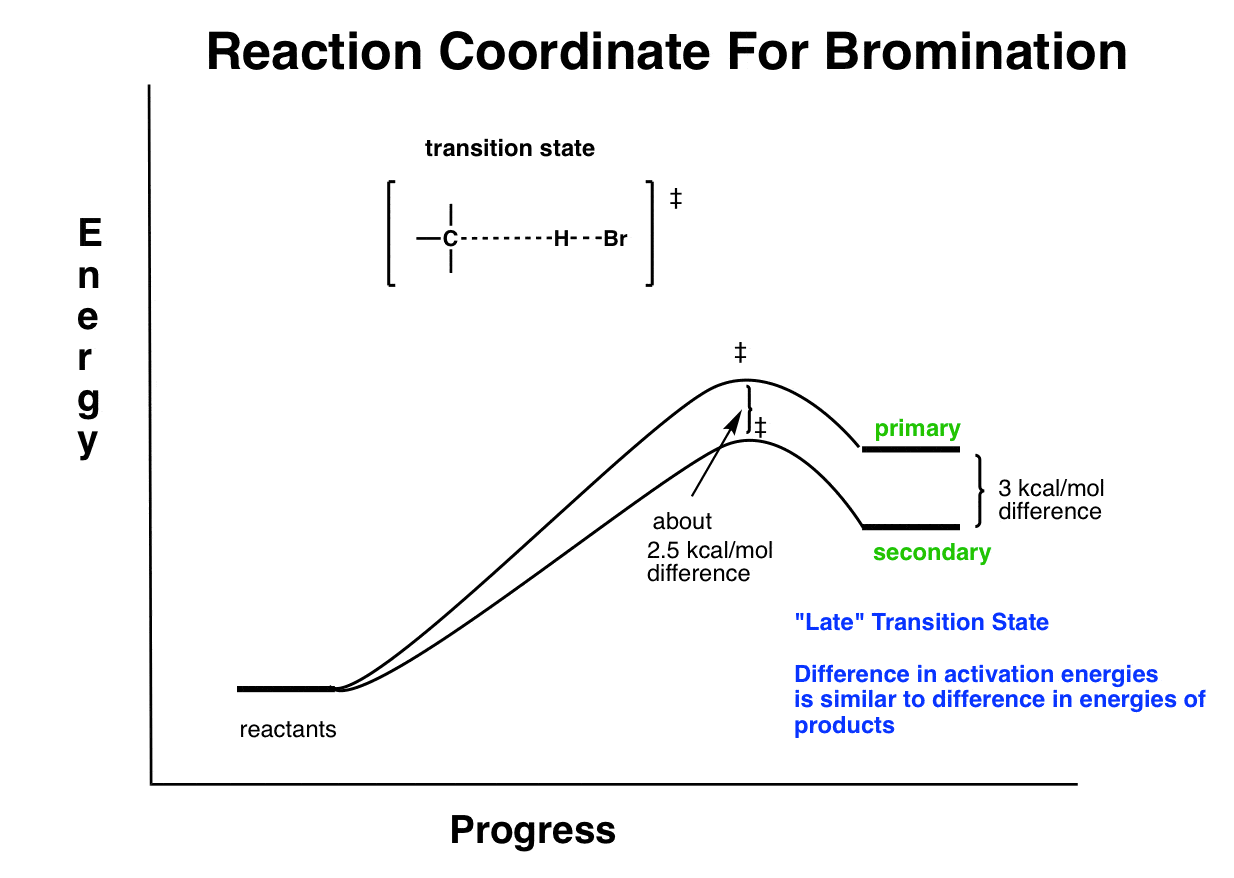
So the bottom line for today’s post is a few things:
10. Summary: Selectivity For Free-Radical Chlorination vs Bromination
1) going from an activation energy difference of 1kcal/mol to about 3 kcal/mol can mean the difference between a reaction with a selectivity of 3.5:1 and a reaction with a selectivity of 97:1. Wow!!
2) We compared a reaction with an “early” transition state and a “late” transition state and saw that the reaction with the “late” transition state was more selective. This is to be expected when we’re starting with identical reactants and there are significant differences in the energies of the products.
In the next post I’ll use a simple analogy to drive home the point in a way that has helped students to understand this point on a more intuitive level.
Next Post: Halogenation At Tiffany’s
Notes
Note 1. Calculations for the energies of each reaction, just using available tables of bond energies.
Chlorination:
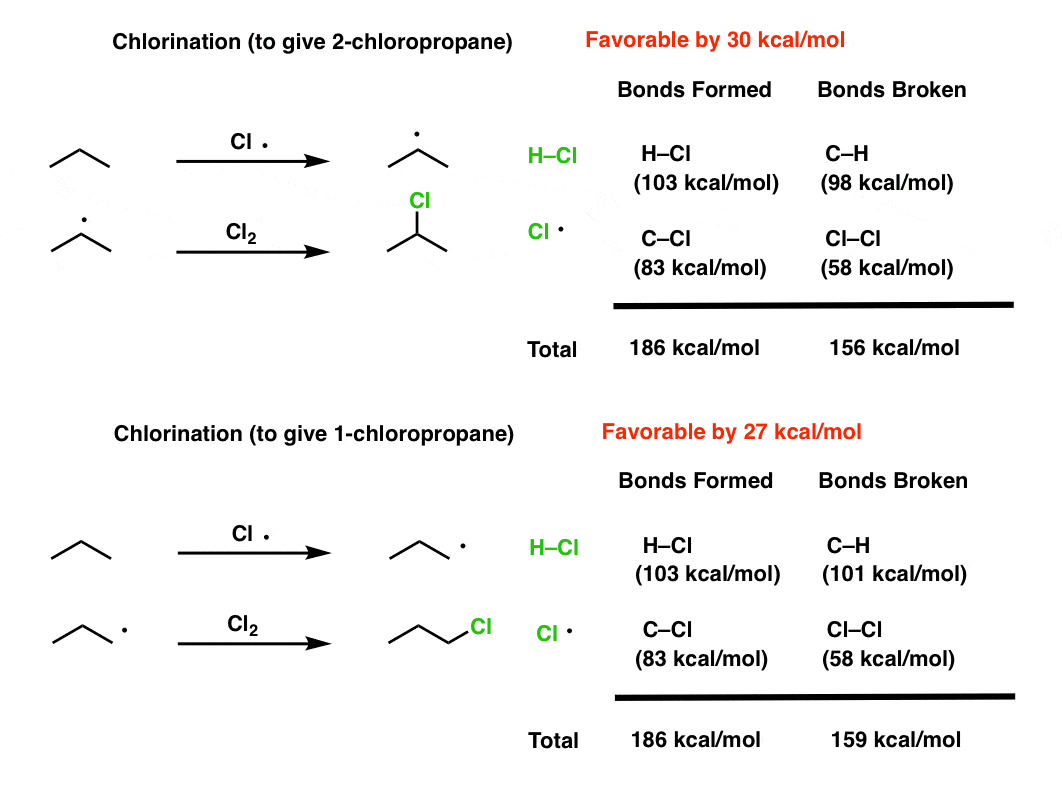
Bromination:
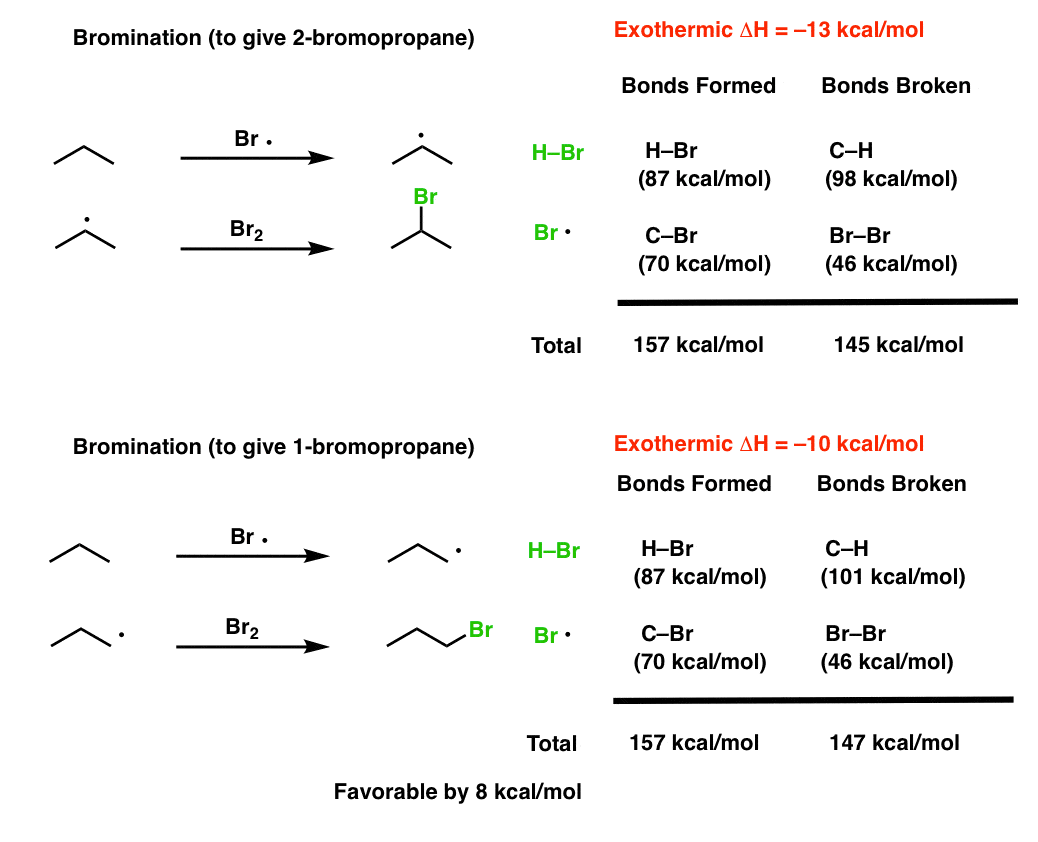
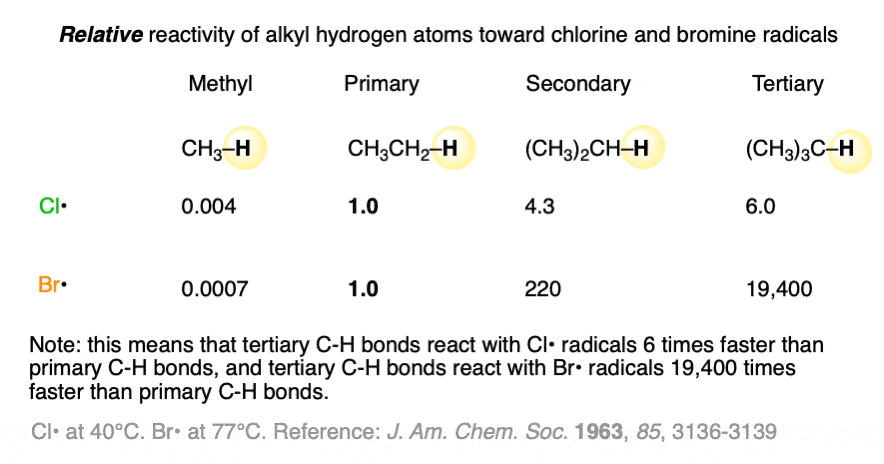
Note 2. Some data regarding the relative reactivity of primary, secondary, tertiary C-H bonds with chlorine and bromine radicals. Note how chlorine radicals are 6 times more reactive with tertiary C-H bonds than they are with primary C-H bonds, and bromine radicals are 19,400 (!!) times more reactive with tertiary C-H bonds than primary C-H bonds!
Quiz Yourself!
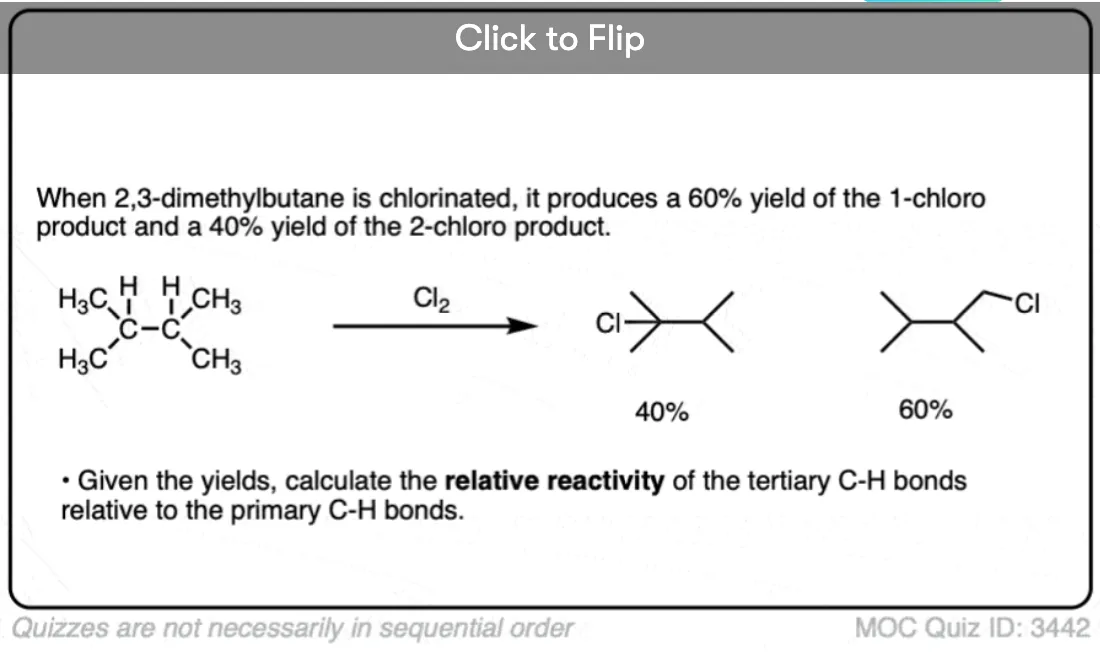
Become a MOC member to see the clickable quiz with answers on the back.
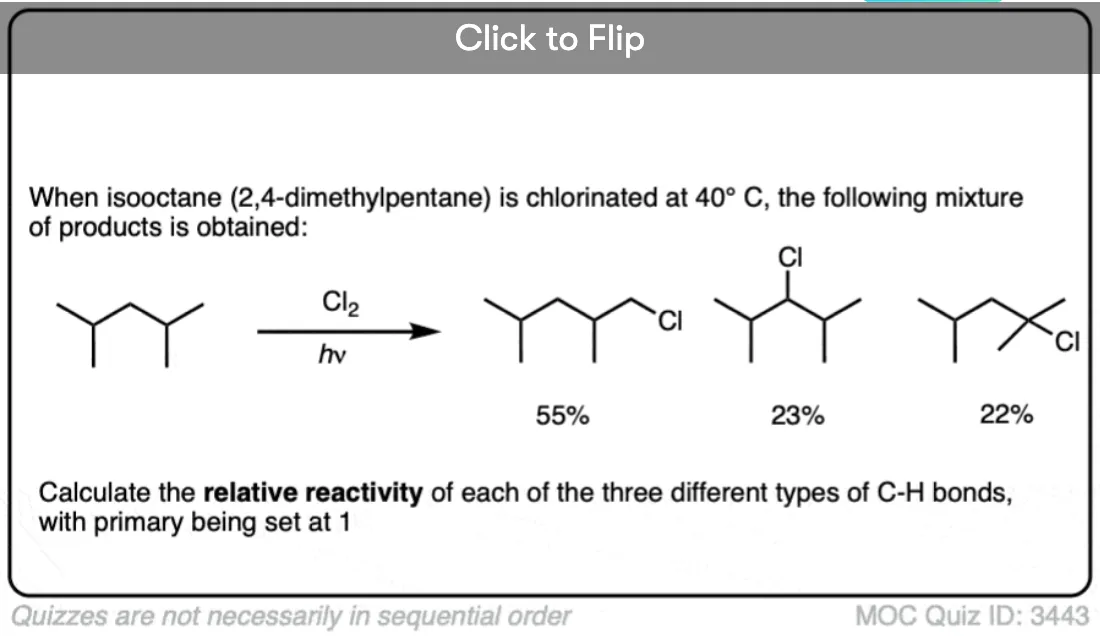
Become a MOC member to see the clickable quiz with answers on the back.
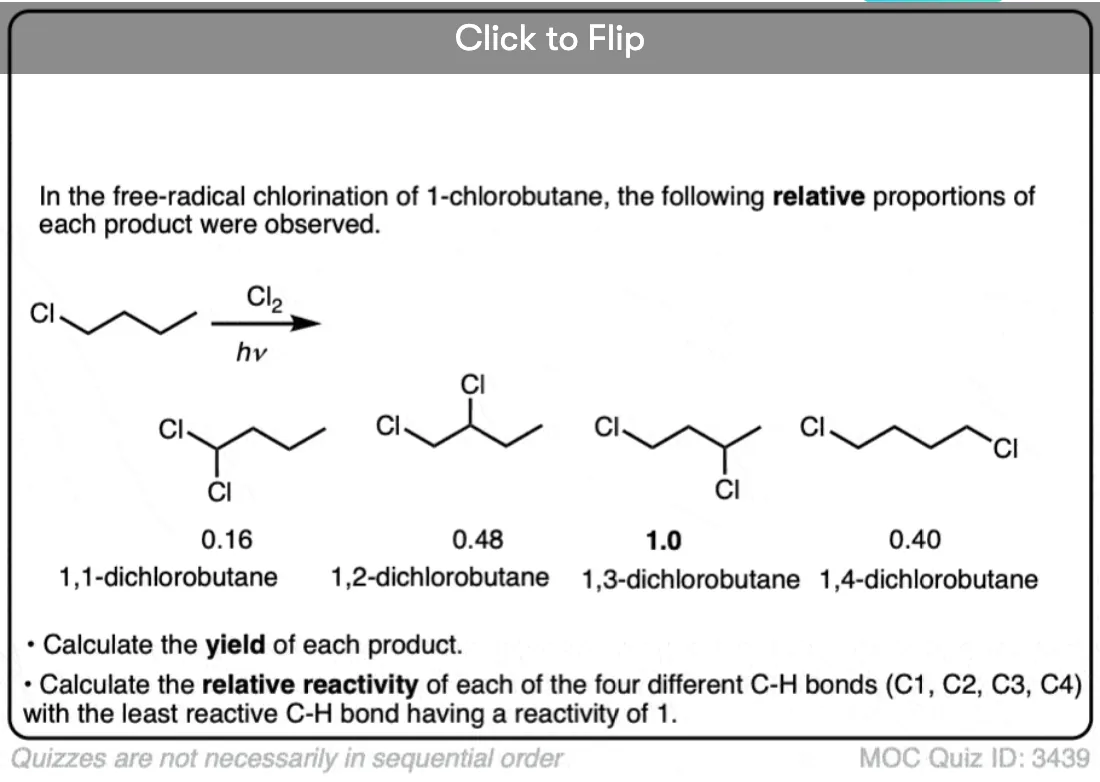
Become a MOC member to see the clickable quiz with answers on the back.
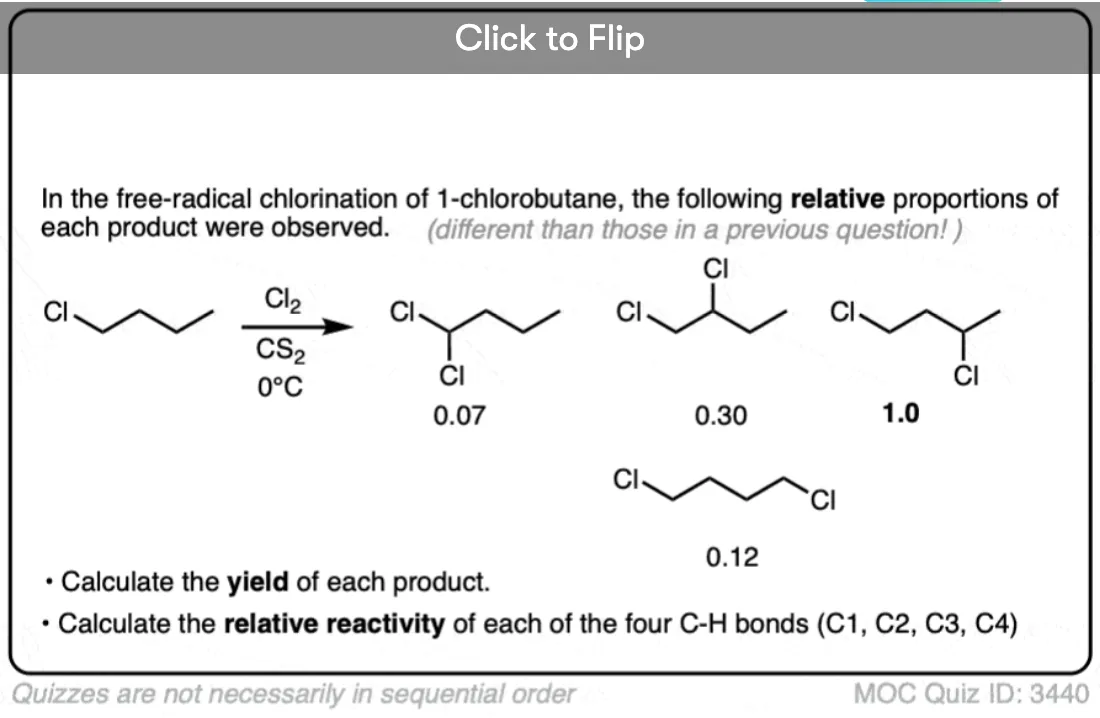
Become a MOC member to see the clickable quiz with answers on the back.
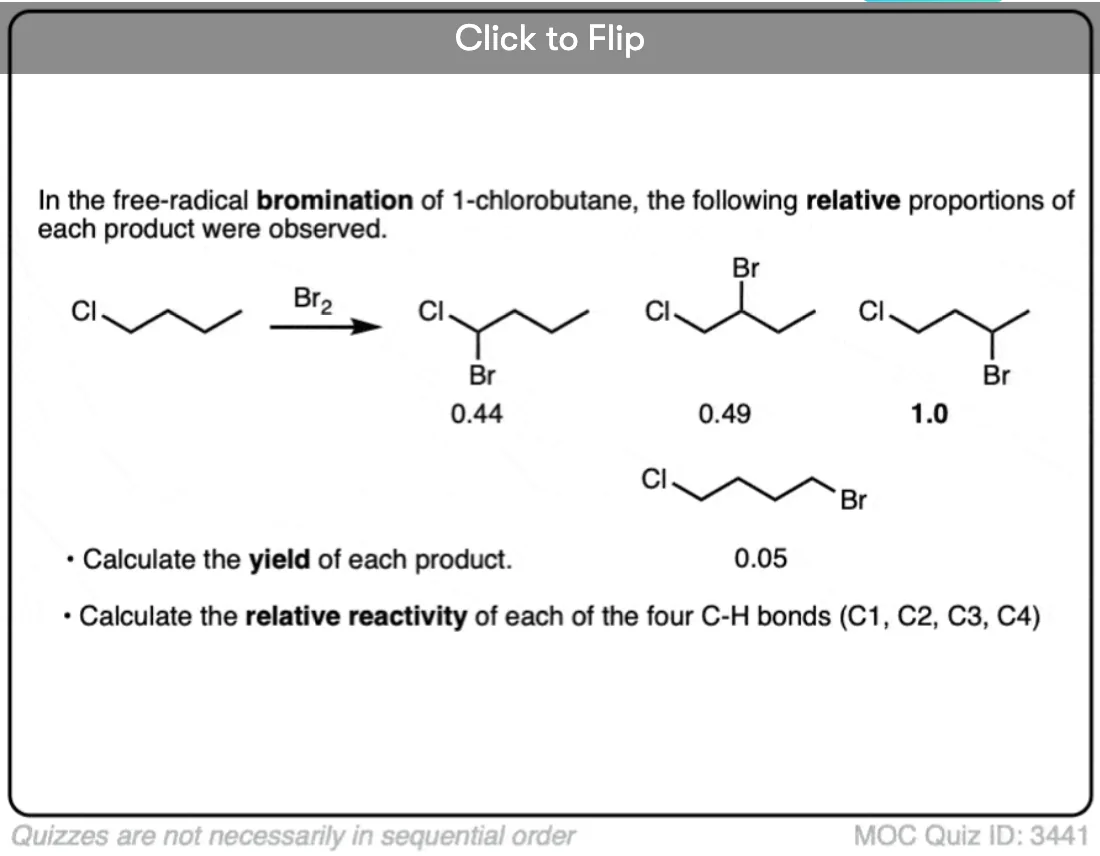
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
The following book chapter is extremely helpful on the subject of selective halogenations:
“Chapter 7. Reactivity, Selectivity, and Polar Effects In Hydrogen Atom Transfer Reactions” by Glen A. Russell (Iowa State University) in Free Radicals, Vol. 1, (Jay Kochi, ed.) Wiley Interscience, 1973. pp 275-331. Internet Archive Library link.
- The interaction of free radicals with saturated aliphatic compounds
J. M. Tedder
Q. Rev. Chem. Soc., 1960, 14, 336-356
DOI: 10.1039/QR9601400336
This review, though dated, contains a wealth of useful information, including the derivation of the activation energy of free-radical aliphatic bromination (13.3 kcal/mol), and the proposal that NBS can brominate alkanes by providing a steady low concentration of Br2. - Competitive chlorination reactions in the gas phase: hydrogen and C1—C5 saturated hydrocarbons
John H. Knox and Robert L. Nelson
Faraday Soc., 1959, 55, 937-946
DOI: 10.1039/TF9595500937
This paper is concerned with the free-radical chlorination of C1-C5 alkanes, and is a detailed kinetic analysis of the chlorination reaction using the Arrhenius equation. The experimentally determined activation energies are all rather low, on the order of 0.02-4.1 kcal/mol. - Substitutions at Saturated Carbon-Hydrogen Bonds Utilizing Molecular Bromine or Bromotrichloromethane
Glen A. Russell and Charles DeBoer
Journal of the American Chemical Society 1963, 85 (20), 3136-3139
DOI: 1021/ja00903a019
This paper studies benzylic bromination via a free-radical mechanism. As expected, reaction rates increase with increasing substitution at the benzylic position, but this paper provides experimental evidence for that. - Directive Effects in Aliphatic Substitutions. XIX. Photobromination with N-Bromosuccinimide
Glen A. Russell and Kathleen M. Desmond
Journal of the American Chemical Society 1963, 85 (20), 3139-3141
DOI: 1021/ja00903a020
This paper immediately follows Ref. 3 in the same issue of JACS, and provides evidence that NBS can also brominate via a free-radical mechanism (i.e. the Wohl-Ziegler reaction). - Solvent Effects in the Reactions of Free Radicals and Atoms. VIII. The Photochlorination of Aralkyl Hydrocarbons
Glen A. Russell, Akihiko. Ito, and Dale G. Hendry
Journal of the American Chemical Society 1963, 85 (19), 2976-2983
DOI: 1021/ja00902a022
Significant solvent effects are observed in the free-radical photochlorination of benzylic compounds. Complexation by solvent can significantly attenuate the reactivity of the chlorine radical, making it react more selectively. - Reactions of radicals. 42. Hydrogen abstraction by the p-nitrophenyl radical
A. Pryor, K. Smith, J. T. Echols, and D. L. Fuller
The Journal of Organic Chemistry 1972, 37 (11), 1753-1758
DOI: 10.1021/jo00976a019
Besides bromine and chlorine, other radicals can be generated and used in free-radical reactions. Organic radicals can also be used, and in this case, the phenyl and p-nitrophenyl radicals can be generated from decomposition of the respective azo precursors. The p-nitrophenyl radical is observed to be more selective than the phenyl radical in free-radical substitution reactions. - Inertia and driving force of chemical reactions
M. G. Evans and M. Polanyi
Trans. Faraday Soc., 1938, 34, 11-24
DOI: 10.1039/TF9383400011
This is a very important paper, introducing what is now known as the Bell-Evans-Polanyi principle. This observes that the difference in activation energy between two reactions of the same family is proportional to the difference of their enthalpy of reaction, thus allowing comparisons of similar reactions. Interestingly, while Michael Polanyi (this paper), was elected to the Royal Society, he did not receive a Nobel Prize, but his son, J. C. Polanyi, received the Nobel Prize in Chemistry in 1986 for his work in chemical physics. - A Correlation of Reaction Rates
George S. Hammond
Journal of the American Chemical Society 1955, 77 (2), 334-338
DOI: 1021/ja01607a027
Hammond’s Postulate is commonly taught to chemistry undergraduates worldwide and is a common mental tool which is also employed by experienced scientists. Exergonic reactions will have ‘early’ transition states resembling the starting materials, while endergonic reactions will have ‘late’ transition states resembling the products.
Reply to Stefan,
I was wondering the same thing… my guess is this is what James was referring to in the notes of a prior post, “Bond Strength and Radical Stability” where he writes:
“the general trends in this post are valid because we discuss bonds to H, but use caution when comparing any other type of bond other than hydrogen…. Using the bond strengths (BDE’s) of unstrained bonds to hydrogen is a reasonable method for discerning trends in radical stabilities, as discussed in this post. However, BDE’s in and of themselves are not reliable for discerning absolute radical stabilities in cases where the bond may be weakened by strain, repulsion between lone pairs, or other factors. For example the BDE for hydrogen peroxide is 51 kcal/mol, which does NOT imply that the HO• radical is stable, but rather that the O–O bond is destabilized by repulsion between the lone pairs.”
Now when I look at the BDE table in Wade’s Organic Chemistry the only cases I find where tertiary BDE are significantly lower than primary BDE was in breaking of C-H or C-C bonds. There was little BDE difference between tertiary and primary halides (C-F, C-Cl, C-Br, C-I) or C-O bonds.
Since breaking of the C-X halide bond still produces a C* radical (and the halide is now gone), that C* radical ITSELF must be more stable if it is tertiary rather than primary. To me, that suggests the only way BDE can be equal (for tertiary and primary C-X halides) is if the tertiary C-X halide bond is more stable than a primary C-X halide bond. But why would that be?
My guess (only a guess): the CH3- groups are electron donating and the halide is electron withdrawing, so perhaps that stabilizes the C-X halide bond in tertiary carbons (halides like electrons!) and make the halide more loathe to leave as a radical (where it only takes a single electron from the bond) … but I’m not a chem-major… so it would be good if James weighed in…
So what says you James?!?
Thanks!
Is iodine more reactive than bromine in monohalogenation?
Iodine is not reactive at all in free-radical halogenation of alkanes. The H-I bond strength is too weak.
Leroy wade said in a write-up for hammonds Postulate –
“The transition state is always the point of highest energy on the energy diagram.
Its structure resembles either the reactants or the products, whichever ones are
higher in energy. In an endothermic reaction, the products are higher in energy, and the
transition state is product-like. In an exothermic reaction, the reactants are higher in energy, and the transition state is reactant-like. Thus, the Hammond postulate helps us understand why exothermic processes tend to be less selective than similar endothermic processes.”
How is the last line obvious in light of understanding Selectivities ofChlorination vs Bromination in light of Hammonds Postulate?
Great quote, and very relevant to this blog post. Was this a question?
Hey guys! awesome material you have presented here. Would you mind providing literature sources for the data?
March, Advanced Organic Chemistry, 5th ed. is my standard reference.
Hello,
Just pointing out R=1.987 cal/ K mol (not kilo) written at the “Calculating the selectivities based on differences in activation energy” image and the paragraph above it.
Also, could you please explain “[…] in chlorination, the key propagation step is exothermic and in bromination, the key propagation step is endothermic.”
What makes the first propagation step the “key” step? Why not look at the net energy difference for both as you did in the image at the bottom?
The first propagation step is the key step because it results in formation of a radical at the alkane – this is the slow step (breakage of C-H). The second step is fast because a considerably weaker bond is being broken (Cl-Cl) .
Thanks for the correction on cal vs kcal. Fixed!
Very well explained topic! There’s only one thing I don’t get (although it’s not important for the regioselective step):
Why is the formation of the bond between the halogen and carbon energetically identical for the primary and secondary carbons? (83 kcal/mol in case of Cl, 70 kcal/mol in case of Br?) I would assume that there’s also a slight difference? Unfortunately I couldn’t find any thermodynamical data.
I would appreciate your short answer.
Ohhh my my my !!!! Hats off to you…..I am speechless…..its too detailed and lucid to understand.
Wow, really detailed explanations. I really appreciate your website.
Great job!
Seriously I loved reading the concepts explained so nicely!
Nice way of explaining the concept of physical organic chemistry
This is awesome from a physical organic chemistry perspective. How on earth are you not a professor at some school. You actually know your chemistry. I wish you were a professor at my school. This is a great write-up!