Rearrangements
Rearrangement Reactions (1): Hydride Shifts
Last updated: May 29th, 2026 |
Rearrangement Reactions: Substitution Reactions With Hydride Shifts
In this post we cover several examples of reactions where carbocations form… but then a funny thing happens. An adjacent bonding pair of electrons (i.e. a C-H bond) interacts with the empty p-orbital, and before you know it, the C-H bond has moved and a new, more stable carbocation has formed! The carbocation is then attacked by the nucleophile, giving a substitution reaction (SN1) with rearrangement!
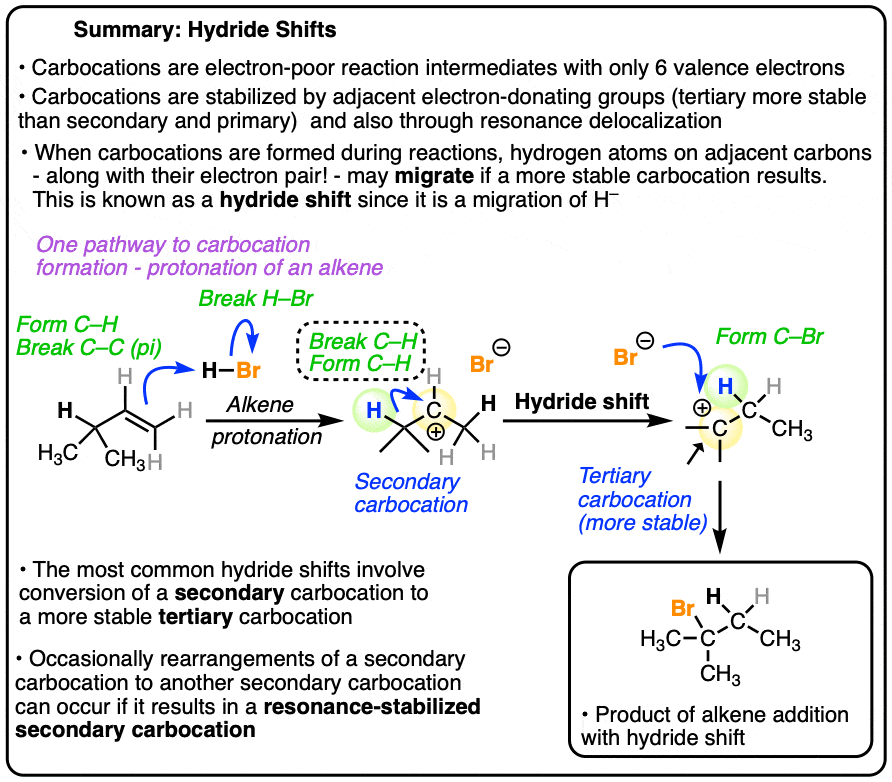
Table of Contents
- Spotting A “Substitution With Rearrangement”: An Extra Set Of C-H Bonds Forms And Breaks
- Carbocation Stability: Tertiary > Secondary >> Primary
- If A Less Stable Carbocation Can Be Transformed Into A More Stable Carbocation Through The Migration Of A C-H Bond, Then A Rearrangement Is Possible
- Examples Of “Allowed” Carbocation Rearrangement Reactions That Occur Through Hydride Shifts
- The SN1 Reaction With Hydride Shift: Arrow Pushing Mechanism
- Notes
- Quiz Yourself!
- References and Further Reading
1. Spotting A “Substitution With Rearrangement”: An Extra Set Of C-H Bonds Forms And Breaks
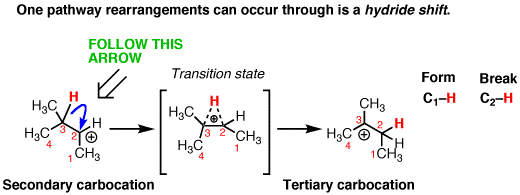
For nucleophilic substitution, the pattern of bonds that form and break is pretty straightforward. You break C-(leaving group) and you form C-(nucleophile). A straight swap. But every once in awhile you might see a “weird” substitution reaction. If you look closely at the pattern of bonds formed and bonds broken in the second reaction below, there’s an extra set!
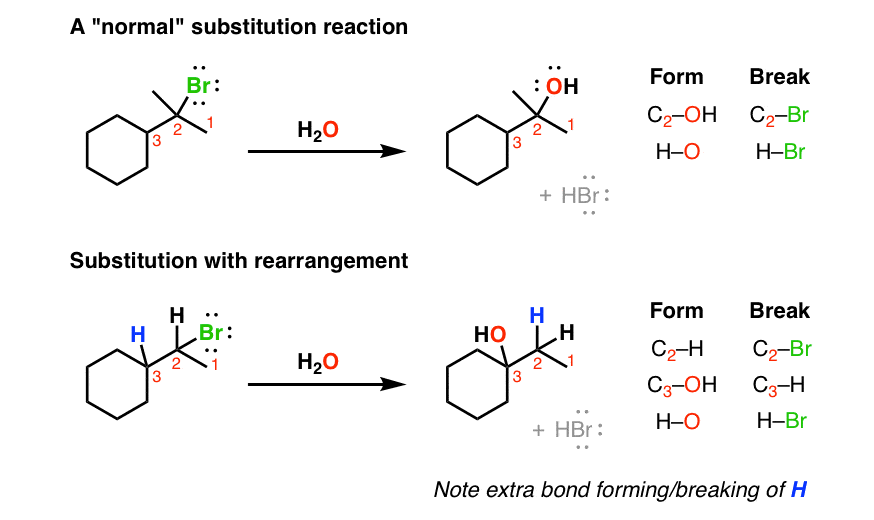
In other words it’s a substitution reaction where the hydrogen has moved. We call these movements “rearrangements”, for reasons that will become clear shortly.
The big question is, what’s going on? How did this happen?
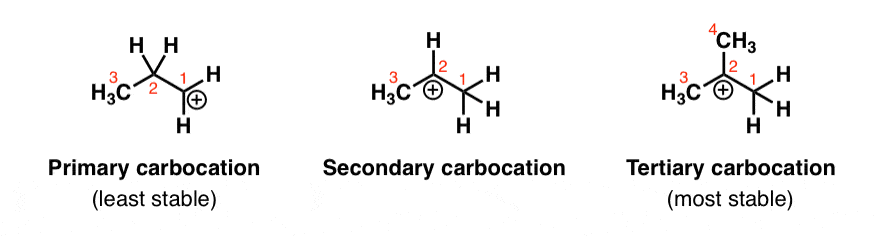
2. Carbocation Stability: Tertiary > Secondary >> Primary
As it turns out, reactions that go through carbocations can sometimes undergo rearrangements. And looking back at substitution reactions, recall that theSN1 reaction goes through a carbocation intermediate. (See post: The SN1 Mechanism)
In this post we’ll go through when you’ll expect to see a rearrangement reaction.
Let’s think back to carbocations. They’re carbon atoms with six electrons bearing a positive charge. In other words, they’re electron deficient – 2 electrons short of a full octet.
So it would make sense that carbocations become more stable as you increase the number of electron donating groups attached to them. Alkyl groups are a perfect example. That’s why carbocation stability increases as you go from primary to secondary to tertiary. (See post: Carbocation Stability)
(It’s also worth pointing out that carbocations are also stabilized by resonance, which allows the positive charge to be delocalized or “spread out” over a greater area on the molecule.)
3. If A Less Stable Carbocation Can Be Transformed Into A More Stable Carbocation Through The Migration Of A C-H Bond, Then A Rearrangement Is Possible
So what does this have to do with rearrangements? As it turns out, if a situation exists where an unstable carbocation can be transformed into a more stable carbocation. then a rearrangement is possible.
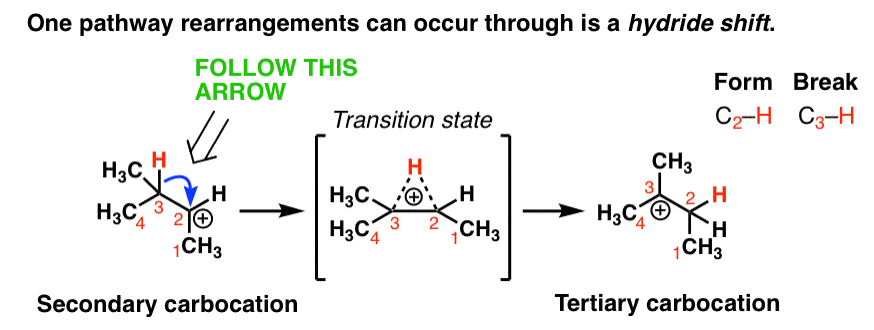
One rearrangement pathway where an unstable carbocation can be transformed into a more stable carbocation is called a hydride shift. Look at the diagram below.
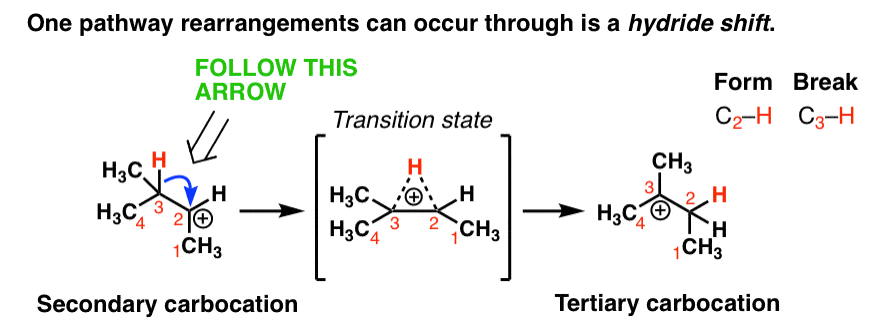
In this reaction we have a secondary carbocation on the left hand side. In this rearrangement reaction, the pair of electrons in the C-H bond is transferred to the empty p orbital on the carbocation. In the transition state of this reaction, there’s a partial C-H bond on C3 and a partial C-H bond on C2.
{kind=link}
{kind=link}
The transition state here is kind of like that split second in a relay race where one sprinter is passing the baton to another sprinter and they both have their hands on it.
Then, as the C2-H bond shortens and the C3-H bond weakens, we end up with a carbocation on C3 (a tertiary carbocation) in the product which is more stable.
Note that we only need one arrow to show this occurring!
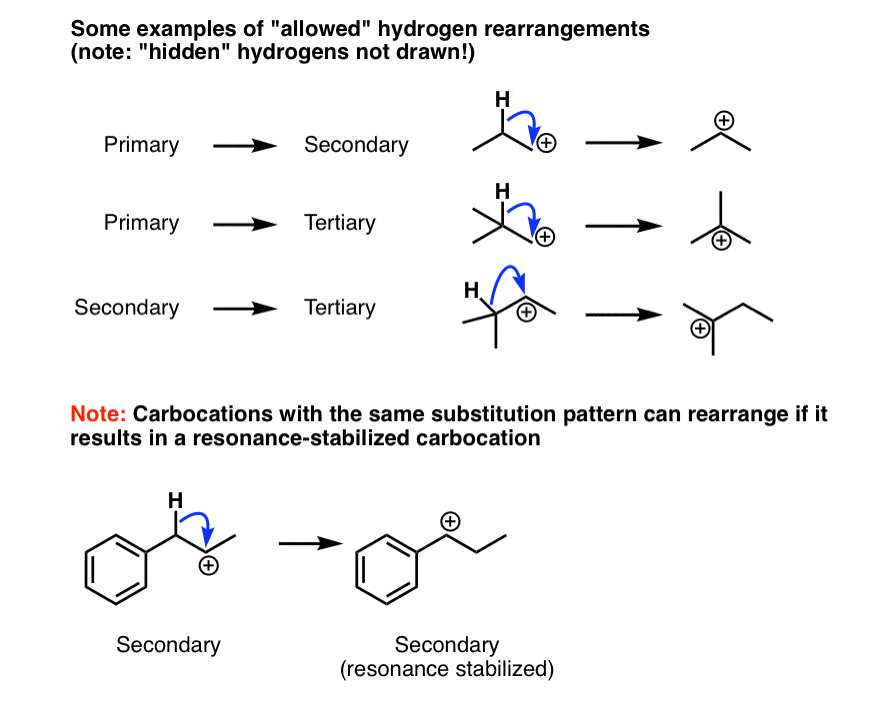
4. Examples Of “Allowed” Carbocation Rearrangement Reactions That Occur Through Hydride Shifts
Here are some examples of “allowed” rearrangement reactions. Notice how we’re always going from a less substituted carbocation to a more substituted carbocation. One exception is at the very bottom; the rearrangement is favorable because the new carbocation is resonance stabilized.
5. The SN1 Reaction With Hydride Shift: Arrow Pushing Mechanism
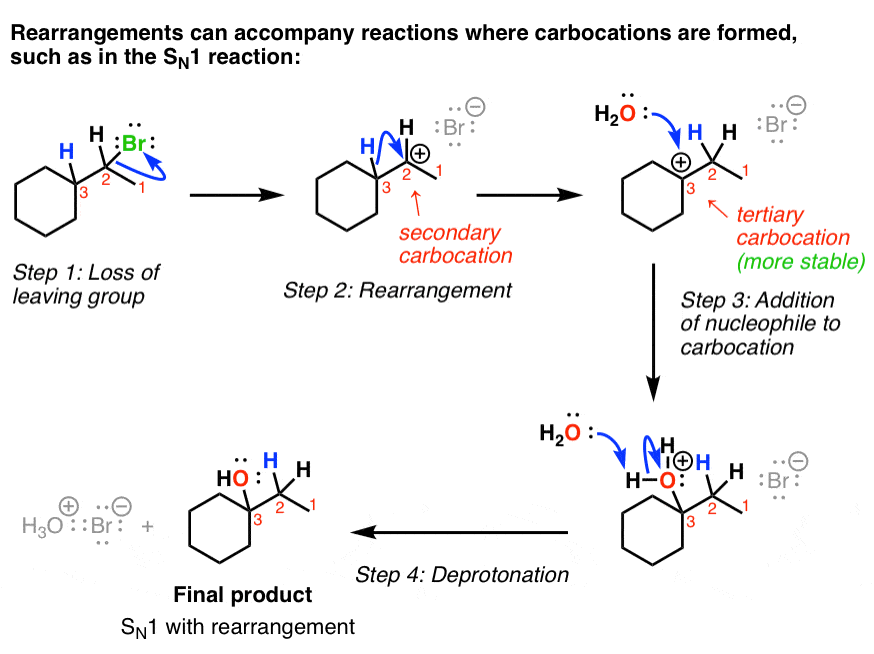
Now we’re ready to show how the rearrangement reaction occurs with the SN1. Recall that the first step in the SN1 is that the leaving group leaves to give a carbocation.
In the case below, the carbocation that is formed is secondary, and there’s a tertiary carbon next door. Therefore, a rearrangement can occur to give the more stable tertiary carbocation, which is then attacked by the nucleophile (water in this case).
Finally, the water is deprotonated to give the neutral alcohol. So this is an example of an SN1 reaction with rearrangement.
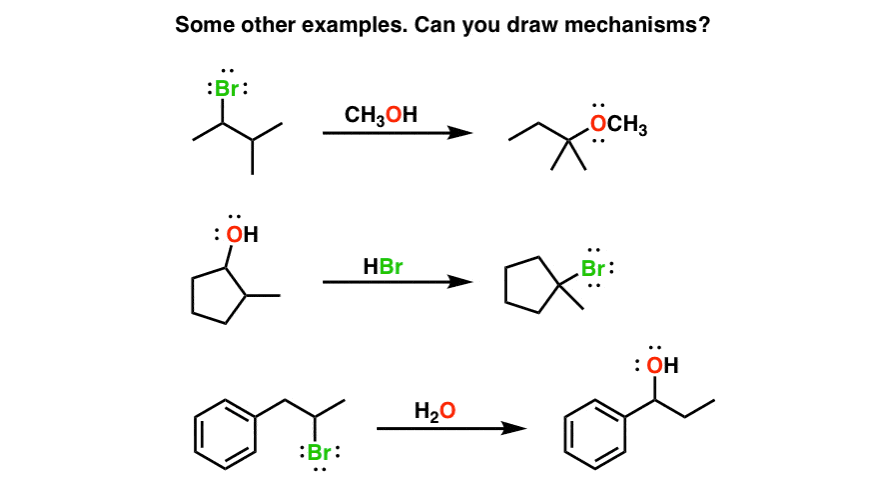
I’ve given some more examples of SN1 reactions with rearrangements below. See if you can draw the mechanisms! In the next post we’ll talk about a slightly different rearrangement pathway with substitution reactions.
Next Post: Rearrangement Reactions (2) – Alkyl Shifts
Notes
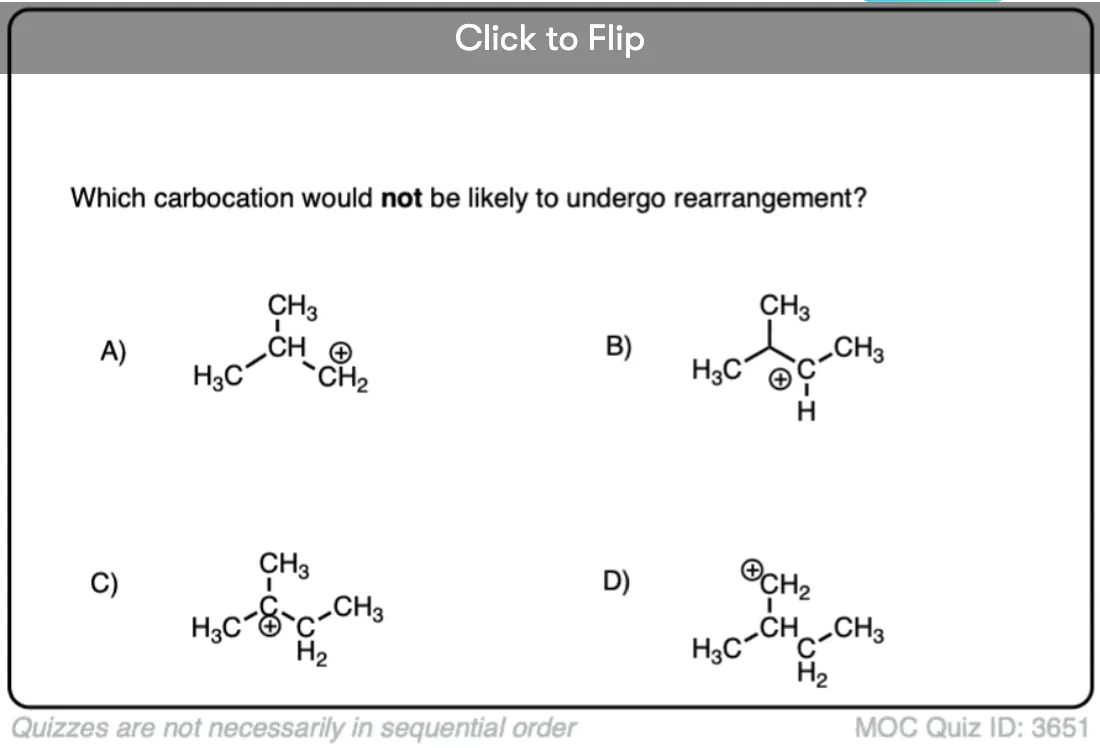
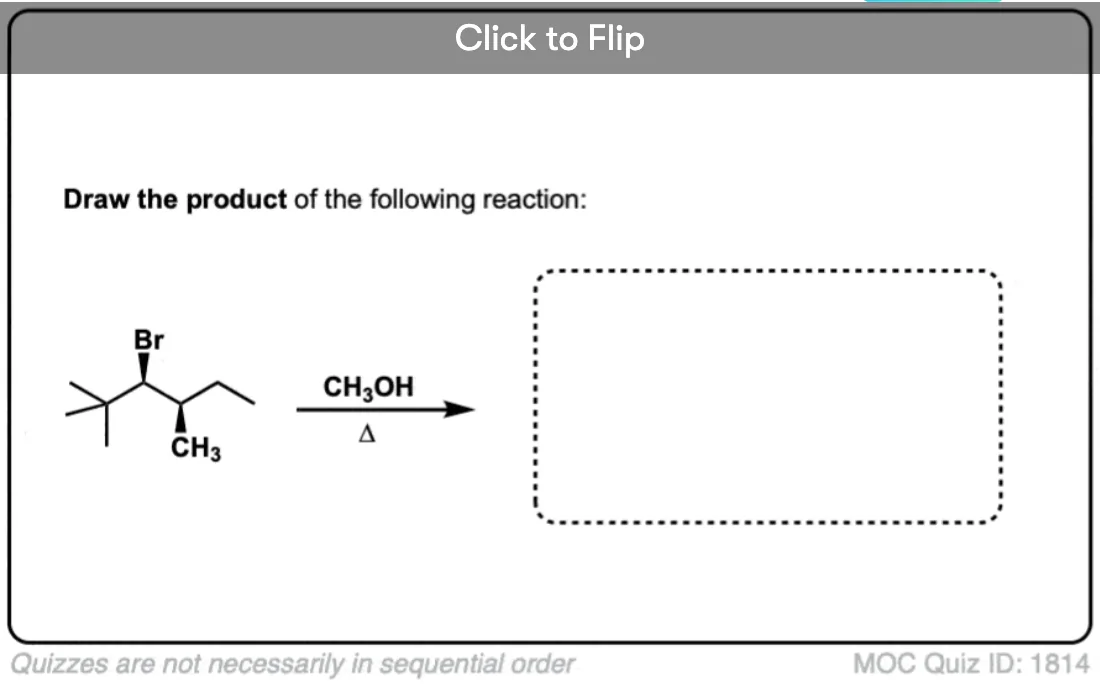
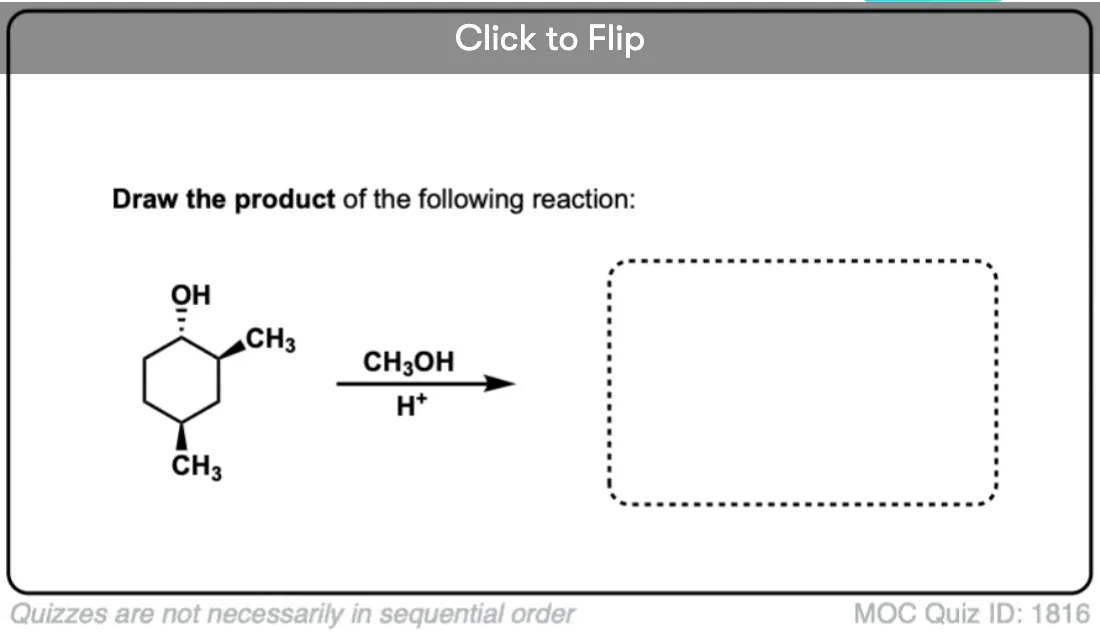
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
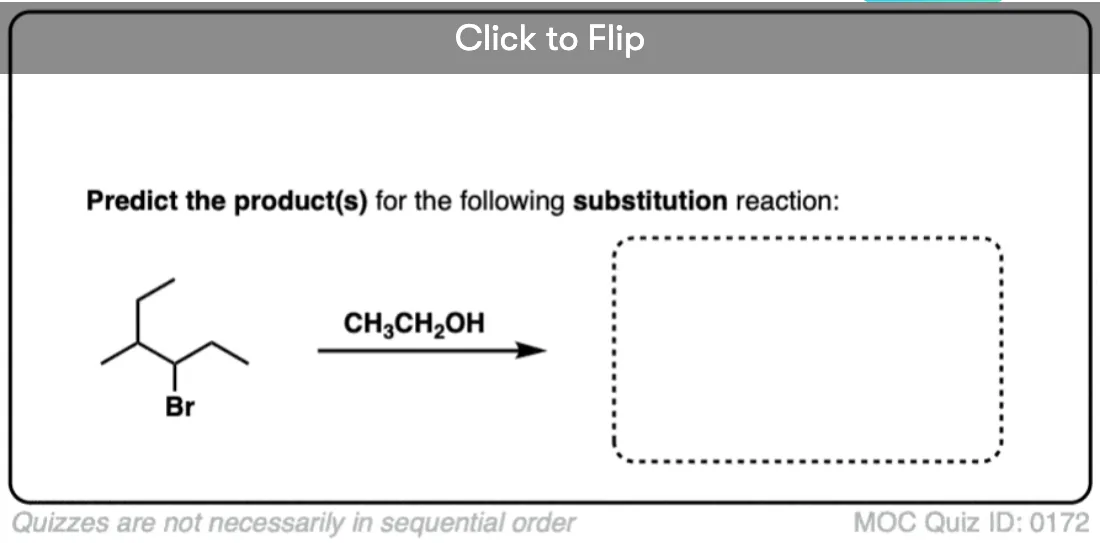
Become a MOC member to see the clickable quiz with answers on the back.
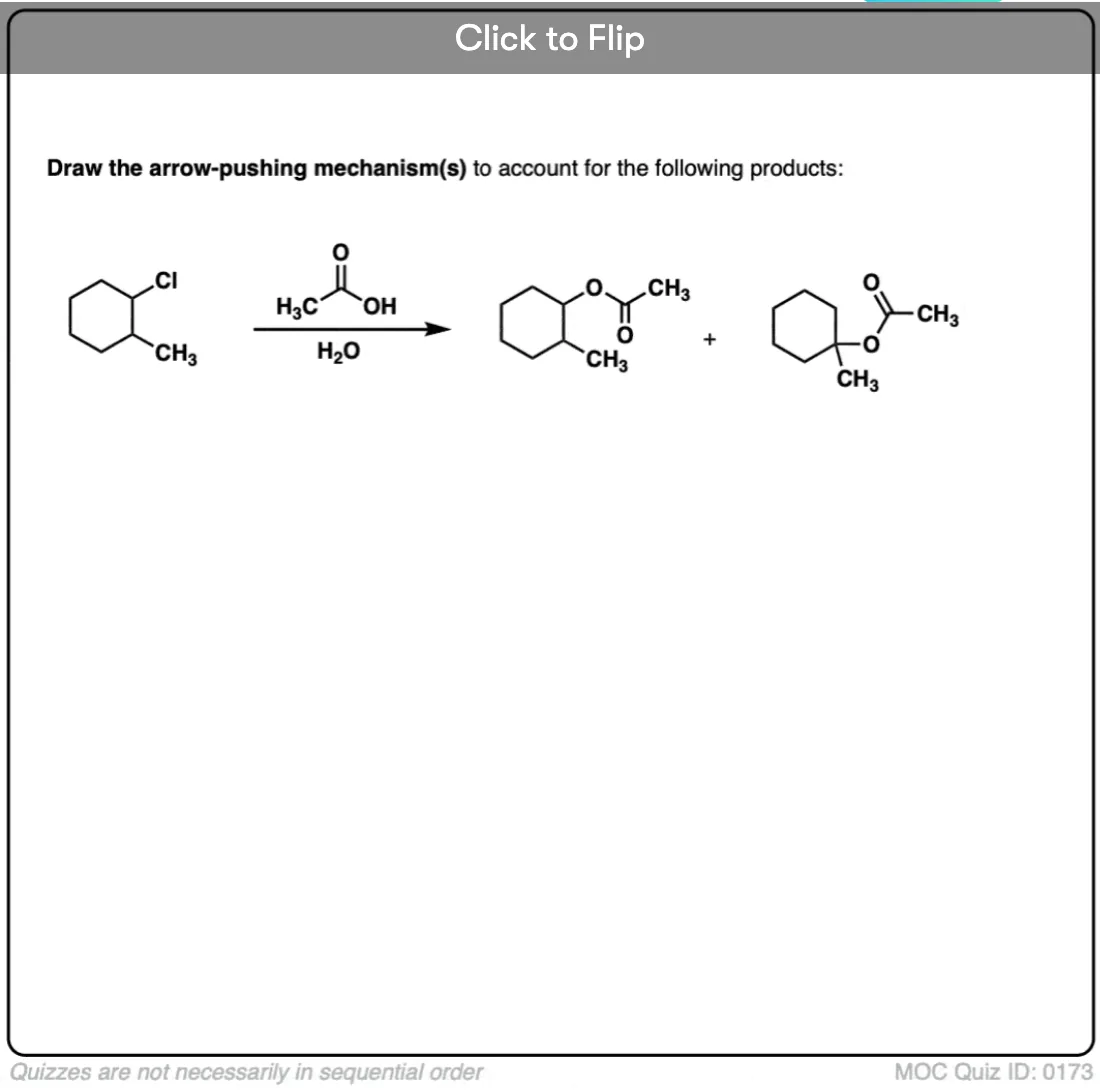
Become a MOC member to see the clickable quiz with answers on the back.
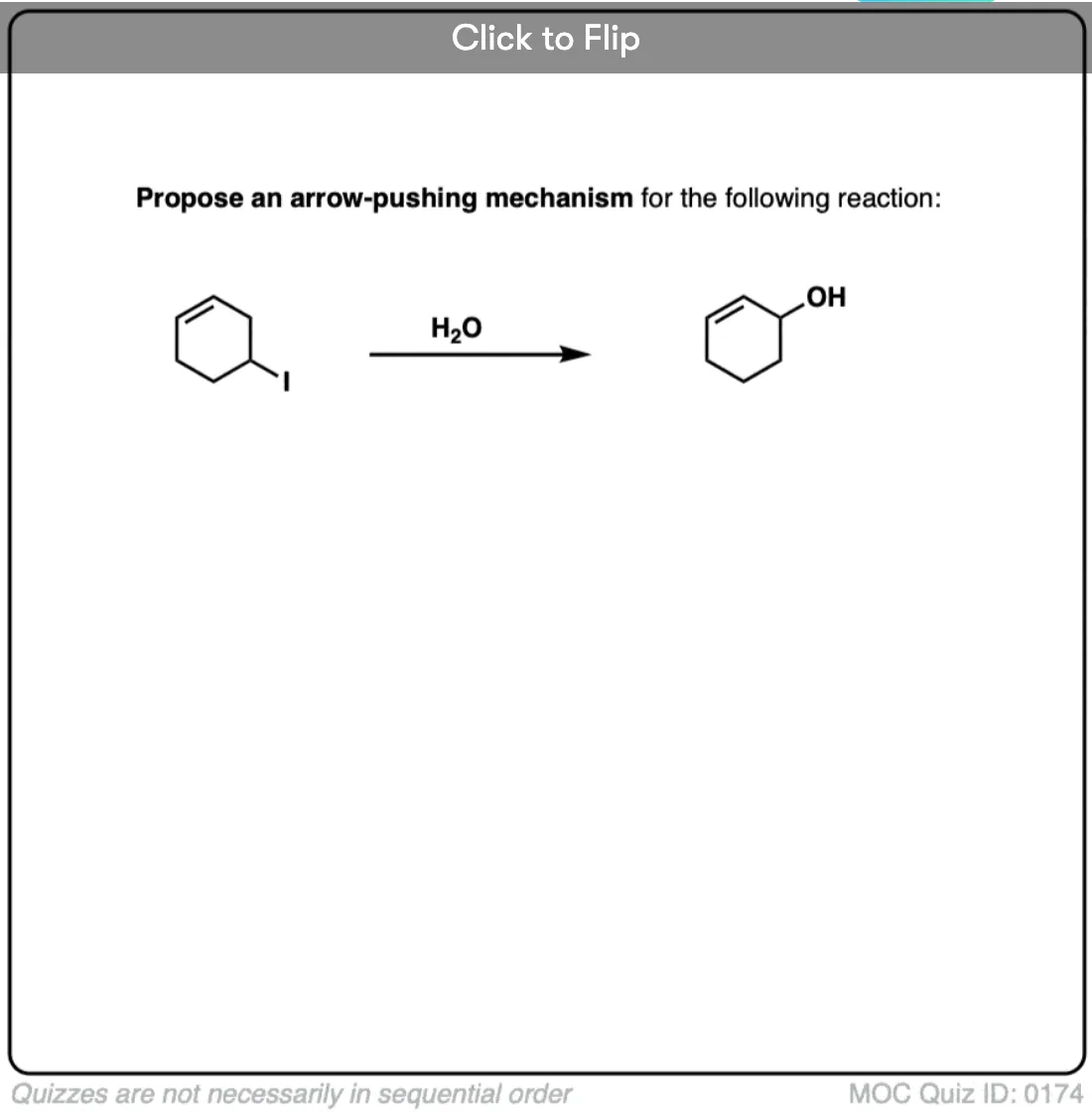
Become a MOC member to see the clickable quiz with answers on the back.
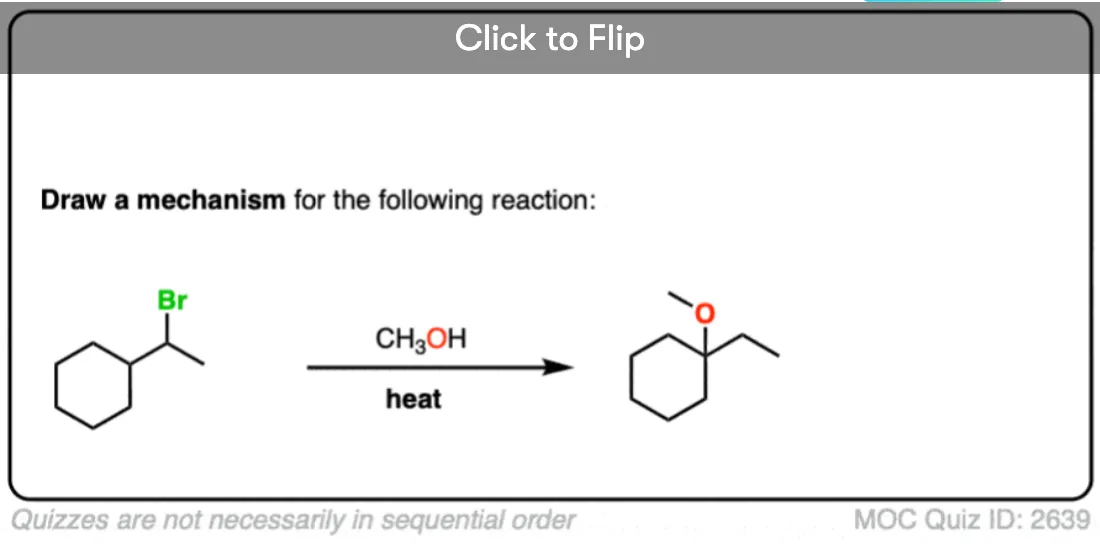
Become a MOC member to see the clickable quiz with answers on the back.
References and Further Reading
- THE COMMON BASIS OF INTRAMOLECULAR REARRANGEMENTS
Frank C. Whitmore
Journal of the American Chemical Society 1932 54 (8), 3274-3283
DOI: 10.1021/ja01347a037
‘,’Rearrangement Reactions (1) – Hydride Shifts
how will the rearrangement happen in 3rd reaction
In the third reaction, a secondary carbocation is formed. Hydride shift from the adjacent carbon results in a *resonance-stabilized*, benzylic carbocation, which is considerably more stable. This provides the driving force for the rearrangement.
Sir / maam ,
If there is a carbocation at 5th carbon of 2,2- dimethyl hexane ,then is it possible that the carbocation rearranges to and does methyl shift ???
Generally only shifts from adjacent carbons occur (1,2-shifts). Rearrangement from a carbocation on the 5th carbon of 2,2-dimethylhexane would still only result in a secondary carbocation, so there would not be a significant driving force.
Hello, why does HBr turn into Br- before the nucleophilic attack but CH3OH and H2O don’t turn into CH3O- and OH- respectively before the nucleophilic attack on the carbocation ?
Acidity. HBr is extremely acidic (pKa about -8) whereas water and alcohols are about 22 orders of magnitude less acidic (pKa 14-16).
Hi James. Here is my little question, If I have a structure that looks like (CH3)3-CH2-CH2+, does it allow me to move the cation from right to the left to get tertiary cation or not? it takes two times to rearrangement. Thank you.
There are examples where multiple hydride and/or alkyl shifts can occur. The most famous example is in the biosynthesis of lanosterol. https://en.wikipedia.org/wiki/Lanosterol
Do Methyl and Hydride shifts stabilize SN1 reactions? Like, for example, if an SN1 reaction originally wouldn’t occur due to the leaving group being attached to a primary carbon, would it have a higher chance of occurring if that carbocation could rearrange to be a secondary or tertiary carbocation?
Some of the classic experiments on these shifts were done with neopentyl alcohol and neopentyl halides by Whitmore. https://pubs.acs.org/doi/abs/10.1021/ja01347a037
How we can judge that whether alkyl shift or hydride shift would occur?
Is there a more stable carbocation that could be formed via an alkyl or hydride shift? Then a rearrangement could be possible. Look for a secondary carbocation adjacent to a tertiary or quaternary carbon.
Is rearrangement of carbocation in sn1 is part of Rate Determing Process or not
Rate determining step is formation of the initial carbocation.
Could you share a reference for the carbocation rearrangement you’ve shown in section 5, where it goes from outside the cyclohexane ring to inside?
AFAIK this would not be possible and instead ring expansion would occur, since the rearrangement you’ve drawn will pass through an unstable spiro transition state, although I may be wrong. I’ve never seen such a rearrangement before; came here after seeing this on another page of yours.
Like many rearrangement questions, these are completely made up.
Honestly I wish I could find more literature examples that correspond to actual exam questions. Then I would show them. They are hard to find.
Rearrangements are much more likely to happen in undergraduate midterm problems than they are in any reaction vessel.
why unsaturated carbocation system do not undergo rearrangement even if they gain stability
When type of substrate gives rearrange product in substitution nucleophilic reactions?
Look for a secondary (or possibly primary) carbocation adjacent to a tertiary or quaternary carbon.
Can a hydride or methyl shift happen to both SN1 and SN2 reaction mechanism?
Hydride and methyl shifts only occur in the SN1 reaction mechanism.
Can a primary carbocation even form? I am asking this because I’ve seen some problems where the primary carbocation forms and then a methyl or hydride shift occurs. I was under the impression that a reaction would need lots of energy put into it for a primary carbocation to form.
Good question. They are known, but only under very exotic conditions. See for example https://pubs.acs.org/doi/pdf/10.1021/ja00326a006
It is much more likely (as you imply) that rearrangement is accompanied by loss of the leaving group in more of a concerted process.
Can a hydride shift occur between a tertiary carbocation to another nearby tertiary carbocation?
The simple answer is “no”, because there’s no significant energetic driving force between two tertiary carbocations (as opposed to, say, a secondary carbocation being transformed to a tertiary carbocation).
The more complicated (real-life) answer is, “sometimes”, as there can be an equilibrium between two carbocations of roughly equal stability. For the reaction to be useful, however, there has to be a strong driving force that eventually leads to a thermodynamically more stable product.
Thanks for replying. The exact question was that which of the following will show greater reactivity towards SN1 reaction:
i. Benzyl chloride
ii. (2-Chloro ethyl) benzene
So now initial carbocation of ii. is not resonance stabilised whereas that of i. is in resonance with benzene.
But after rearrangement of carbocation in ii. it becomes a benzylic carbocation(highly resonance stabilised) and also has an electron donating group (methyl) stabilising it.
So I thought ii. Should be more reactive as it’s carbocation intermediate is more stable than i.
But my book says that benzyl chloride will be more reactive towards SN1.
What is your take on this??
Which of the following alkyl halide will have greater reactivity towards SN1 reaction?
The one in which carbocation is benzylic
Or
The one in which a benzylic carbocation forms AFTER carbocation rearrangement (from primary carbocation to a benzylic carbocation)???
Please reply
I’d have to see the structure to be sure, but if rearrangement is happening, it’s likely a non-resonance-stabilized secondary carbocation, which is less likely to ionize than a benzylic carbocation.
Why is a tertiary carbocation more stable than the primary and secondary? What does having more C atoms attached have to do with the stability?
Carbocations are electron-poor species that lack a full octet. They are somewhat stabilized by donation of electron density from neighboring atoms. Hydrogens are poor donors since the only electrons they possess are tied up in the C-H sigma bond. On the other hand alkyl groups (i.e. carbon substituents) have a full octet of electrons, and these electrons can be partially donated to the electron-poor carbocation which result in a more stable carbocation species.
The more adjacent carbon-containing neighbors a carbocation has (primary < secondary < tertiary) the more electron-density can be donated to the electron-poor carbocation. You can think of it a bit like a poor person who is given a loan by a rich neighbor.
Can hydride shifts occur in a reaction of epoxide under acidic conditions?
Yes, check out the Meinwald rearrangement of epoxides to aldehydes catalyzed by lewis acids.
Thank you so much James. You make every concept very easy
OK, thank you Adithya.
Sir..can rearrangement occur within tertiary carbocation
Yes, but typically there has to be a driving force otherwise you’ll get a mixture of products.
Can rearrangement occur within a tertiary carbocation…one tertiary carbocation to another tertiary carbocation..
It can happen… like for example in the cascade of shifts that occur after cyclization of squalene oxide to lanosterol. https://en.wikipedia.org/wiki/Lanosterol
If we’re adding H-Br to an alkene and on both sides of the alkene carbons we have carbons that can potentially form more stable carbocations but one involves a methyl shift and the other involves a hydride shift, which pathway would we follow when doing the mechanism and adding the proton in the first step?
Thank you
All else being equal, hydrogen will migrate faster than anything else.
greetings james, is it general for 1,3 Hydride shifts to occur?
It most certainly is not!
James,
Thanks, thanks, thanks a million!!!!!
Can hydride shift feom 1st to 4th carbon
Ph-CH2-CH+=CH2
WHAT WILL N REARRENGEMENT
There’s something wrong with your structure.
Out of methyl and hydride shift which will be preferred?
Hydride shifts are faster.
Why double bond of cyclohexene won’t break on addition of Br₂, UV light and heat? The product I expected was bromocyclohexane but it was 3-bromocyclohex-1-ene!
Sounds like conditions for allylic bromination. https://www.masterorganicchemistry.com/2013/11/25/allylic-bromination/
Hi,
Please help me with following questions
A carbon and hydrogen bond energy is around 413kJ/mol. Is it feasible for such hydride shifts to occur? Because I feel more energy is spent on breaking C-H bond than the energy released by making carbocation stable.
Secondly, how we could justify shifting of hydride from one molecule to other as in Meervein-pondroff-verley reaction?
Thanks
You have to balance out the energetic “cost” of breaking the bond with the energetic “gain” of forming a new bond. The bond strength of the C-H that’s being broken is usually within 10 kcal/mol of the bond strength of the C-H that’s being formed.
What we call “instability” of various carbocations (e.g. secondary, aryl, etc.) is really just another word for high electron affinity, which means that the more “unstable” the neighboring carbocation, the more energy will be released when it completes its octet. If the carbocation resulting from the shift has a relatively low electron affinity (e.g. tertiary or allylic) then the net energy gain can be quite considerable. For instance going from a typical secondary carbocation to a typical tertiary carbocation can be worth about 18 kcal/mol which is a LOT. (Based on gas phase R-H bond dissociation energies. See March’s Advanced Organic Chemistry 5th ed p. 224 table 5.2. )
If I have 1-methyl cyclohexane carbocation ( positive charge on 3rd C of cyclohexane) , will there be a 1-4 or 1-3 hydride shift to furninsh a tertiary carbocation?
It’s possible for sequential C-H shifts to occur, so a 1,2- shift could be followed by another 1,2- shift. In very rare examples it’s possible that transannular shifts can occur which would be examples of 1,4 or 1,5 shifts but this is unlikely in cyclohexane.
Order of migration tendency b/w hydride, methyl $ phenyl
Hi I’m a little confused about carbocation rearrangements. How do you know when it will be a hydride or methyl shift?
So long as you’re going from a less stable carbocation to a more stable carbocation, a good rule of thumb is that hydride shifts are faster than alkyl shifts.
In the case of an sn1 reaction why would the leaving group not already be attached to what would be the most stable carbocation before the addition of the substituting group given the if an sn1 reaction were going to take place then the leaving group would already have been able to leave the substrate on it own?
Say if we are having an intermediate Br-CH2-CH (+)-CH3. Will the positive charge shift towards CH2 as it will be stabilised by mesomeric effect of bromine? (There are many questions in my book in which this is not done!!)
There is no way that exists as a free carbocation. The Br will form a 3-membered bromonium ion and will not be prone to rearrangement.
In some cases – such as if you replaced the CH3 in your example with Ph – then the resulting carbocation would be quite stable, and it is during such situations that breakdown of the typically observed ‘anti’ selectivity for bromination is observed.
As aPHD organic chemist
I personally in the lab was able to remove hydride from activated carbon at 1100C.
The issue here with this PHD Chemist is the carbon originally was in a two electron covalent bond, Therefore removing a hydride leaves an electron on the carbon.
1 That means the carbon has 7 electrons.
2 Chemist are weird! They call this a carbocation.
Wow
Wayne Harlan PHD
So shifts only occur when a secondary carbocation is formed? I ask this because if a primary carbocations aren’t generated in the E2 mechanisms for dehydration or dehalogenation, then how can if undergo a shift if it isn’t generated? Therefore I thought that shifts only occur in E1 dehydration and dehalogenation reactions.
*I’m only talking about elimination reactions right now.
More likely that the shift accompanies loss of the leaving group. You can think of the migrating group as being a bit like a nucleophile that moves over to the adjacent carbon, displacing the leaving group. This results in a new carbocation which can then undergo elimination.
That’s one pathway
What if the base/electrophile to needs to neutralize the carbocation is quite large and sterically hindered? Would the rearrangement still be favorable? Such as H2PO4- on a tertiary carbocation post-rearrangment…
Removal of the proton is the easiest step, and these protons are very acidic (pKa less than 0). Shouldn’t be a problem. Remember you’re not deprotonating the tertiary position, you’re deprotonating the carbon adjacent to it.
Is the hydride//methyl shift a characteristic of only SN1 reactions or could this also happen in E1?
It can happen anytime a carbocation is generated. That can happen via loss of a leaving group as the beginning step of E1/SN1 or it can also happen during addition of acids to alkenes (if you’ve covered that).
why does the reaction between 2- methylbromopropane and NaOH form two products??
Do shifts only occur between adjacent carbons? For example, if a shift between carbons 1 and 3 created a more stable carbocation, but a shift between 1 and 2 created a less stable carbocation, would the shift from 1 to 3 still occur? Or the rearrangement only occurs if a shift to an adjacent carbon produces a more stable carbocation? Thanks
The shift between 1 and 3 or 1 and 4 is prohibited becos for shift to take place the orbital overlap has to take place between the c-h and the empty orbital on C+.
Which would be difficult with 3rd and 4th..
But as the chain gets longer then it can bend and C+ comes near C-H. So, 1,5 shifts take place…
Exactly, thank you Harmeet.
I had this same question. The answer is no, here’s a brief explanation: http://web.pdx.edu/~wamserc/C334F10/rearrangements.pdf
That’s a really nice page, thanks!
which is more stable between PhCH2+ and (CH3)C+
Benzyl carbo cation is always more stable than alkyl due to resonance ..
So (Ph)-CH2+. Is more stable ….but at the same time remember that (Ph)+ is most unstable also..
Hii!! Is it possible that if a primary carbocation is generated and to its side we have a 2 degree carbon and to that 2 degree carbon’s side we have a 4 degree carbon somewhat like (ch3)3- c -ch2-ch2+ .. Then is it possible that directly a 3 degree carbocation is generated ( somewhat like simultaneous shift ) ??? Thankyou
It’s possible to have multiple hydride shifts, if that’s what you’re asking. In fact there are multiple hydride shifts in the biosynthesis of the important molecule lanosterol: http://www.chem.qmul.ac.uk/iubmb/enzyme/reaction/terp/lanost.html
What i think is that for a shift you need to have a hydride atom on the neighbour carbon as well…..so we cannot have a tertiary shift in this though we can have a secondary shift
Although hydride shifts seem like I good method to offer shortcuts in synthesis, I’ve often wondered why you don’t see them that often and also why I learned about them relatively late in my chemical education. I think it’s because they often occur from carbocations which only form when they are stabilized. And hydride shifts occur to increase the stability of the carbocations, so there’s not an awful lot of scope for them. For aliphatic compounds this only really permits the carbocation to change from secondary to tertiary.
They’re also pretty unselective, so if there’s multiple neighbours where a hydride can be donated from, it’s difficult to control for which one will happen. Plus there are side reactions (like elimination)
Might be a dumb question, but are multiple hydride and or methyl shifts allowed? If so what is the justification/if not why as well?
Been thinking as I was solving some tricky sn1 with ring openings and stuff (how do I even see that?!)
Multiple shifts are certainly possible, and they could happen, but generally will only happen if each shift generates a successively more stable carbocation.
For example, you probably wouldn’t see a shift if it involved turning a tertiary carbocation into a secondary carbocation.
Sir,
What happens if a chiral centre is generated?
For eg,
(C2H5)(CH3)CH—–C((Cl)(CH3)(C3H8)———–> (C2H5)(CH3)CH—–C+(CH3)(C3H8)
——————>(1,2-hydride shift) (C2H5)(CH3)C+—–CH(CH3)(C3H8)
What is R/S configuration of rearranged carbocation(inversion or retention or both)?
Since a carbocation is planer the resulting stereocenter following reaction with a nucleophile would be racemic or an even mixture of (R) and (S) at the position of the carbocation. This can become more complex with the influence of additional stereochemical considerations from other portions of the molecule that don’t involve the cation or molecules with limited degrees of freedom.
can we do hydride and alkyl shift multiple times in order to get a stable carbocation or is it allowed only once?
It’s possible for multiple hydride/alkyl shifts to occur. One amazing example is in the biosynthesis of lanosterol: http://en.wikipedia.org/wiki/Lanosterol
can we do multple shifts in order to get a stable carbocation or shift is allowed only once?
Multiple shifts can certainly occur. For a particularly amazing example of multiple shifts, check Wikipedia for how lanosterol is made from squalene.