Spectroscopy
UV-Vis Spectroscopy: Absorbance of Carbonyls
Last updated: October 31st, 2022 |
UV-Vis Spectroscopy Of Carbonyls (C=O Bonds)
UV-Visible spectroscopy is not just about C-C pi bonds. C-O pi bonds can absorb UV light as well!
Table of Contents
- A Quick Review on UV-Vis
- C=O Bonds Show An Absorbance Maximum Around 300 nm
- This is actually a n-> pi* transition, not pi to pi* (!)
- Wait. Why not pi to pi* ?
- Carbonyls also participate in conjugation
- Summary: UV-Vis Spectroscopy Of Carbonyls
- Notes
1. A Quick Review Of What We’ve Learned So Far About UV-Vis
In our last post we showed that molecules with C-C pi (π) bonds absorb light in the UV-visible region, which promotes electrons from (bonding) π orbitals to (anti bonding) π* orbitals.
We saw that
- the energy required for the transition depends mostly on the extent of conjugation (i.e. the number of consecutive pi bonds, roughly speaking).
- an alkene with little or no conjugation (e.g. ethene, CH2=CH2) possesses a large energy gap (ΔE) between the bonding and anti bonding orbitals, which requires more energetic (shorter wavelength) photons for excitation. For ethene, maximum absorbance occurs at about 170 nm, in the UV region.
- as conjugation increases, the energy gap ΔE decreases, pushing the wavelength of maximum absorbance (λmax) toward the visible (less energetic photons, longer wavelength) . For example, β-carotene (the orange pigment in carrots) with 11 conjugated pi bonds, absorbs in the visible (λmax = 470 nm).
Because the post was so damn long, we never got around to addressing a key question: does this apply to other types of pi bonds as well?
For example, do C=O pi bonds also absorb light in the UV/visible region?
- The short answer is: yes.
- The medium sized answer is: yes, but the main transition of interest is not a pi-pi* transition – it’s slightly different.
The long answer is.. well, here’s the long answer.
2. Absorbance of C=O bonds Show A Maximum Around 300 nm
Let’s start with one of the simplest compounds with a C=O bond: 2-propanone, otherwise known as acetone.
Question: Does acetone absorb UV or visible light?
Answer: You betcha. Here’s the UV-Vis absorption spectrum for 2-propanone (acetone).
[The key piece of information to glean from that spectrum is that there is an absorbance maximum at about 275 nm, in the ultraviolet.]
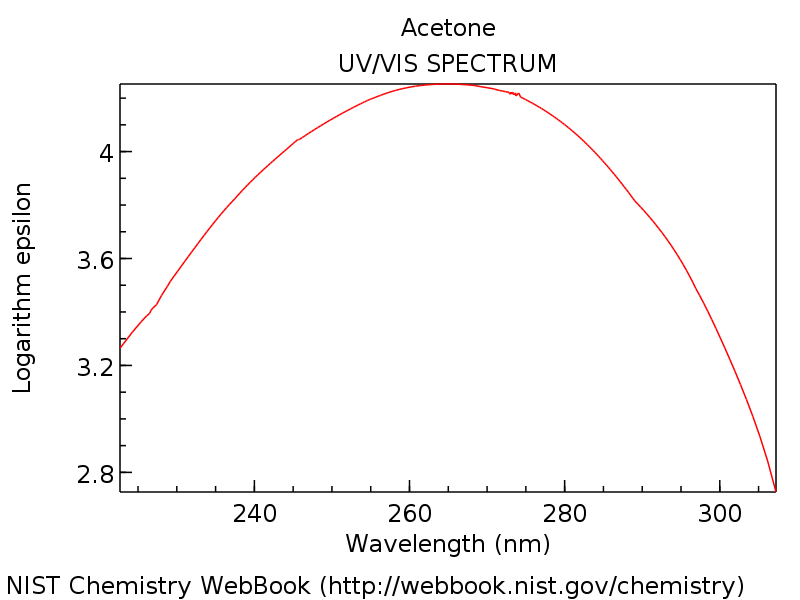
If you have an astonishingly good memory you may recall from the last post (or from my introduction above) that the absorption max for ethene (CH2=CH2) is about 170 nm.
An absorption around 275 nm means that longer wavelength and therefore less energetic photons are required for this transition.
Doesn’t that seem weird?
If anything, C=O π bonds are stronger than C=C π bonds. [You can look it up]. Wouldn’t you reasonably expect *more* energy to be required to promote an electron from pi(π) to pi* (π*)?
What gives?
Now: as we’ll see in a minute, there is a pi to pi* ( π→π*) transition for acetone in the UV, but that peak at 275 nm is NOT a pi to pi* transition. It’s a transition from a non-bonding orbital (n) to the pi* orbital (n→π*).
3. Carbonyl (C=O) Groups Tend To Show Weak Absorbances At (Roughly) 300 nm That Correspond To Transitions Between Non-Bonding Orbitals and Pi* Orbitals
Huh? Let’s look at a simple molecular orbital drawing of acetone.
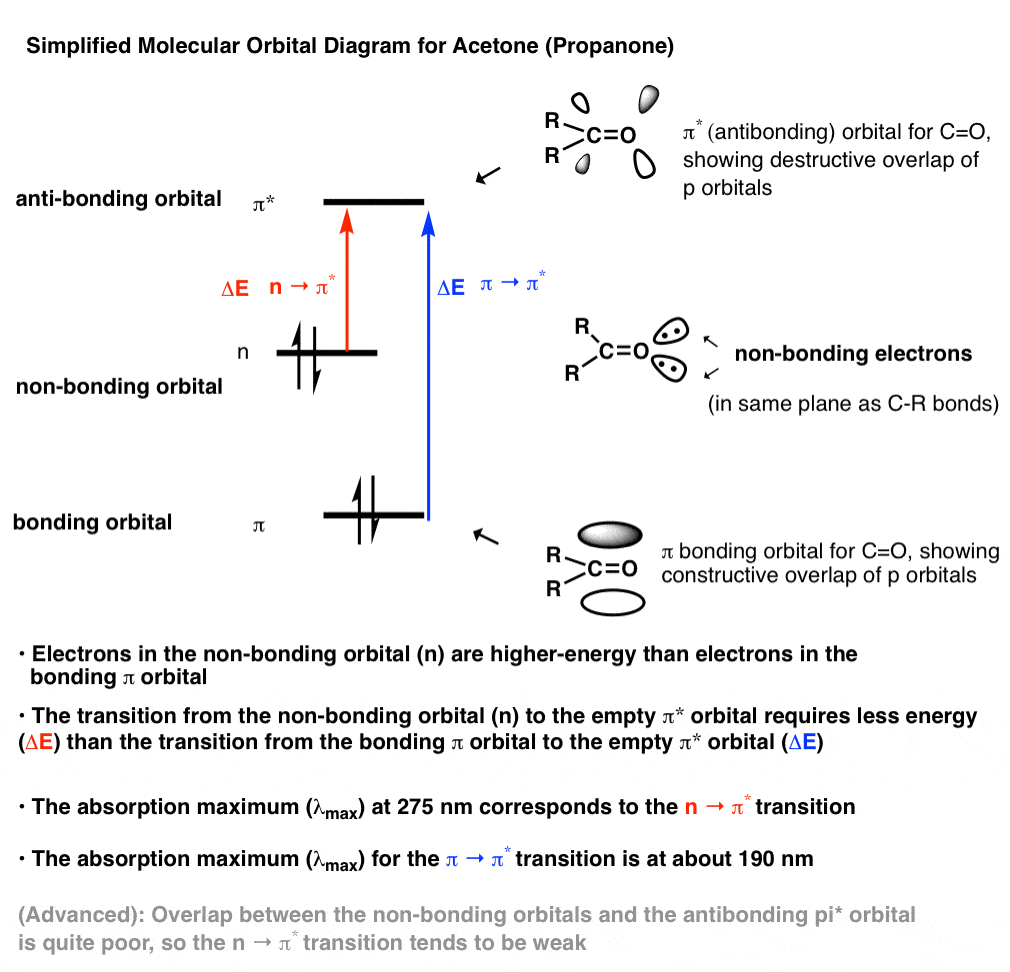
[This is a somewhat simplified picture. For a more detailed MO diagram for that also includes a more thorough discussion about the nature of the non-bonding orbitals, I highly recommend Reusch’s online textbook entry here. ]
A few important things to note:
- Carbonyl groups contain non-bonding electrons that are in an orbital intermediate in energy between the bonding pi orbital and the anti bonding pi* orbital. These orbitals are absent in typical alkenes such as ethylene [CH2=CH2] [note 1]
- Being higher in energy, transitions between electrons in the non-bonding orbital to the pi* orbital have a smaller ΔE and therefore absorb at longer wavelength.
- It is this (n→π*) transition which is responsible for the peak at around 275 nm.
4. What About Pi to Pi* Transitions for C=O?
So what about the pi to pi* transition? Doesn’t that happen too?
Glad you asked. If you take a quick look back at the UV-Vis absorption spectrum of acetone, above, you’ll note that the X-axis gets cut off around 240 nm or so. There’s a reason for that (mwah-ah-ah).
If you zoom out, you’ll see that there’s a much stronger transition around 190 nm. [I went looking for a decent full-size UV spectrum of acetone, and the diagram below is the best I could find. I didn’t make this image and it is not my intellectual property. I found it here. ]
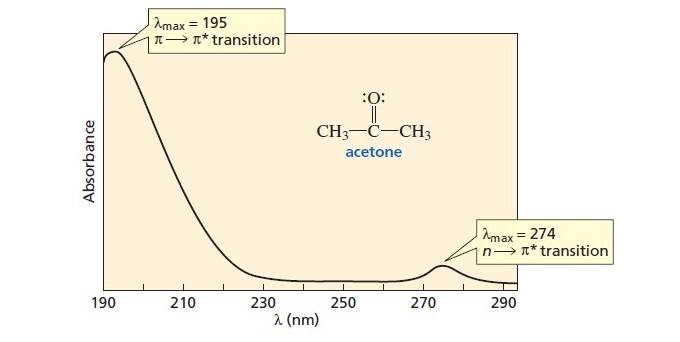
So as it turns out, that “peak” at 275 nm (n→π*) we were looking at turns out to be a molehill, next to the (π→π*) mountain at about 195 nm in the deeper UV.
In other words, the (n→π*) transition at 275 nm that we’ve spent so much time talking about is very weak relative to the (π→π*) transition. [sometimes a term called “epsilon, ε” is used to denote this difference in magnitude of absorption].
Why might that be? It has to do with differences in orbital overlap. In order for an electron to transition from one orbital to another, two conditions must be met.
- First, as previously discussed, the orbital has to interact with a photon of appropriate energy ΔE.
- Second, there has to be significant overlap between the orbitals in space. We generally don’t discuss this for (π→π*) and (σ→σ*) transitions because each pair of bonding and anti bonding orbitals occupies the same region of space. If you look back to the diagram for the location of the n orbitals in acetone compared to the location of the pi* orbitals, you might notice that they are essentially at right angles to each other. Poor orbital overlap means that even if the electron has sufficient energy ΔE to make the transition, the transition is considerably less likely to occur since the excited electron will be less likely to be occupying an area of space corresponding to the higher-energy orbital. [Extra detail: you might recall that orbitals are 3-dimensional volumes where the probability of encountering an electron is 95%. Therefore, there is some electron density outside of the volumes we typically consider “orbitals”]
5. Carbonyls Also Participate in Conjugation
Carbonyls can also participate in conjugation with C-C pi bonds. This leads to an increase in the overall λmax of the molecule. For instance, the absorbance of the alkene 2-methyl pent-2-ene is below 200 nm, as is the π→π* absorbance of 4-methyl pentane-2-one (below).
In mesityl oxide, where the alkene and C=O group are in conjugation with each other, the absorption maximum moves to longer wavelength at 228 nm. This is similar to the difference between the absorbance of ethene (174 nm) and butadiene (217 nm).
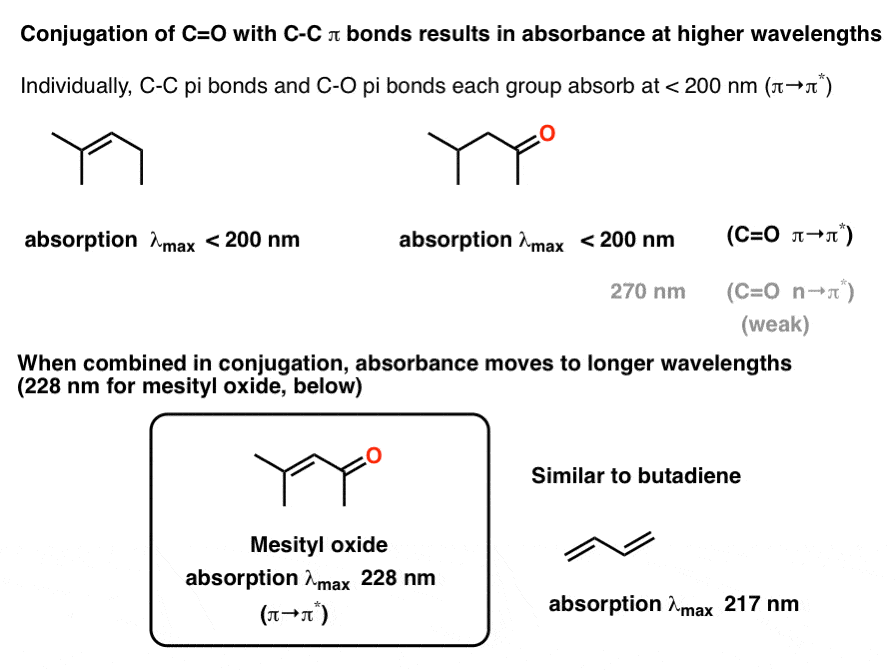
NoteThe absorbance maximum can be sensitive to the identity of the solvent as well as the identity of the substituents on the alkene. [Note 2].
6. Summary: UV-Vis Spectroscopy Of Carbonyls
Absorbance in the rough neighbourhood of 270-300 nm is common for molecules containing a C=O group (such as ketones and aldehydes) and this corresponds to a (n→π*) transition.
These absorbances tend to be weak, relative to (π→π*) transitions. Still, observing this absorbance can be an important clue in the structure determination of unknown compounds.
In the next post we’ll go into practical details of using UV-Vis in structure determination.
[Again, for a more in depth look on the subject of C=O absorbance, go to Reusch. We’re really skimming the surface here, but it is enough for our purposes.]
Notes
Note 1. It should be noted that non-bonding orbitals are present in species such as the allyl cation, allyl anion, and other ions of odd-numbered pi systems.
Note 2. For carbonyls, generally more polar solvents lead to higher λmax values, as does the presence of substituents (such as methyl groups) on the alkene.
Bonus Topic: Azo Dyes
Since we’re on the subject, let’s briefly explore another system where both n→π* and π→π* transitions are observed: azo dyes.
Azo dyes are the kind of thing that you’ve likely seen a million times without specifically knowing what they are.
For example, the yellow color of highway markings? That’s Pigment Yellow 10.

Azo dyes are commonly used in colouring textiles, plastics, and many other substances not intended for human consumption (they’re generally banned as food additives).
The key structural feature of an azo compound is a N=N linkage. One of the simplest azo compounds is azobenzene, where each nitrogen is connected to an aromatic ring. Slight modifications to the benzene ring can dramatically modify the color of the molecule. Aniline Yellow, discovered in 1861, was the first azo compound to find commercial use as a dye, and countless derivatives of azobenzene have been synthesized since then. [The synthesis is via diazo coupling – we won’t get into that here].
Here’s the UV-Vis spectrum of Aniline Yellow, as calculated by ChemTube 3D.
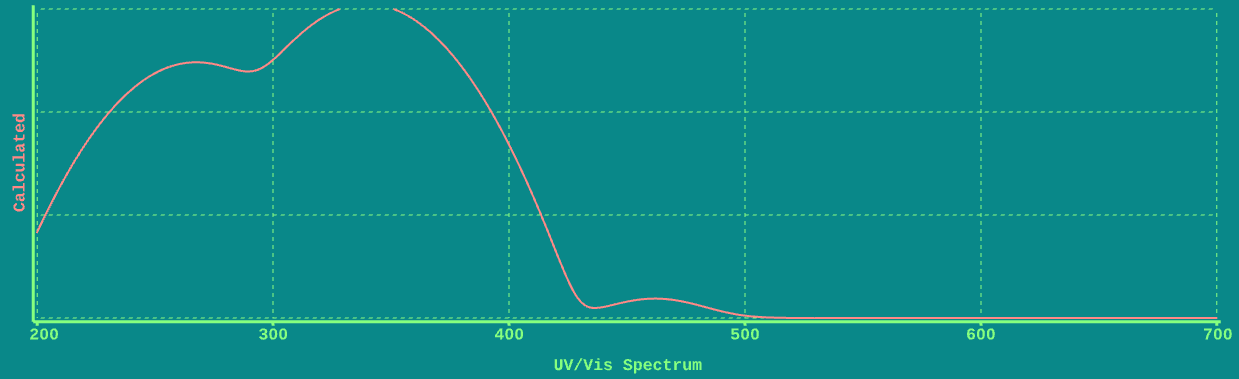
Note how it is qualitatively similar to that of acetone: a strong absorbance on the left (towards the UV) and a weak absorbance on the right (towards the visible).
In contrast to acetone, however, where the weak absorbance is at 260 nm, the weak absorbance in Aniline Yellow is in the visible region of the spectrum at about 460 nm. It is this absorbance at 460 nm that is responsible for the color of Aniline Yellow.
By analogy to acetone, the weak transition is an (n→π*) transition and the strong transition around 360 nm is a π→π* transition.
Photoisomerization
What’s even more interesting about azobenzenes and their derivatives (e.g. Aniline Yellow) is the phenomenon of photoisomerization, where absorption of specific frequencies of light can lead to isomerization of trans isomers to cis isomers and vice-versa.
Absorbance of UV light by trans-azobenzene (a π→π* transition) leads to isomerization to cis-azobenzene. Contrariwise, absorbance of visible light (blue light) by cis-azobenzene (the n→π* transition) results in conversion back to the trans-isomer [so does leaving cis-azobenzene in the dark, a process known as thermal relaxation]. The mechanism for this process is still not completely settled.
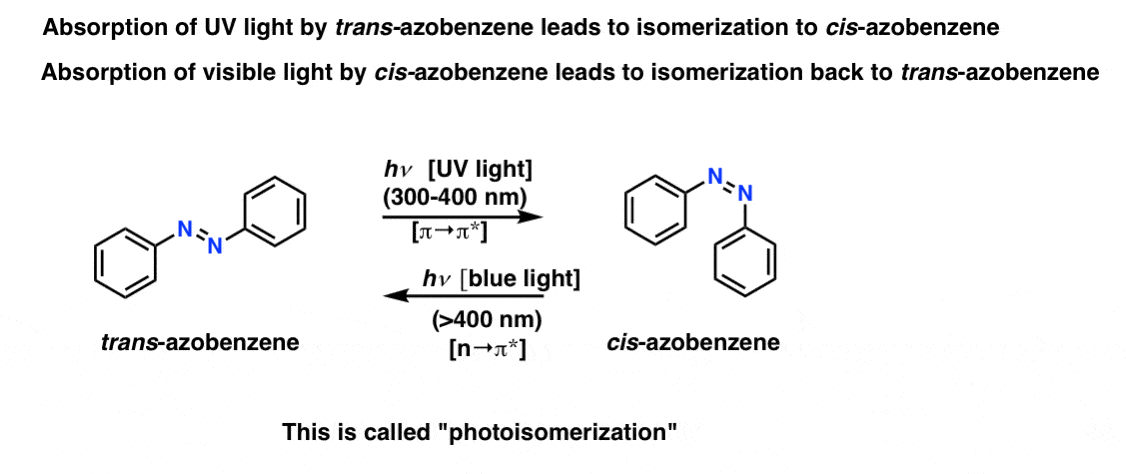
Pretty neat that you can target a specific isomer merely by changing the frequency of light.
Very good. Made the learning fun.
The link for the acetone spectrum showing lambda max at 195nm does not seem exist. It goes to a general page. Do you have an updated link or any link that shows absorbance at 195nm?
Like I said I could not find the original, and I linked to a page which was not my intellectual property.
Another source could be here, on the NIST webpage:
https://webbook.nist.gov/cgi/cbook.cgi?ID=C67641&Units=SI&Mask=400#UV-Vis-Spec
Hello
i have a Question plx…in the uv vis soectrum of acetobe we only consider two types of transition which are pi to pi* or n to pi*. My question is that sigma bonds are also present in carbonyls, similarly there may be transition between n to sigma*. Then why we consider only two possible transitions? why we don’t consider other transitions due to sigma bonds?
Nice explanation.
Related question;
How do conjugation helps in increasing the lambda max of conjugated compounds?
See previous post in the series: https://www.masterorganicchemistry.com/2016/09/16/introduction-to-uv-vis-spectroscopy/
Great discussion of the topic!!
Question on Azo yellow dye. When I look up the absorption spectrum of cis azo yellow on the internet, I see that the absorption peak at ~420 is much weaker than the peak further into the UV, which would be expected for an n->pi star transition for the reasons you explained, but the lambda max for the stronger shorter wavelength absorption of the cis is actually at shorter wavelength than the trans. I would expect the opposite given the steric strain with the two phenyl groups in the cis conformation and poorer overlap of the p orbitals
Related question: I presume that the n > pi star transition results in weakening of the N=N double bond just like a pi>pi star transition, thus allowing the cis trans photo isomerization?
Thanks!
Very helpful in my revision on organic spectroscopy…
Glad to hear it Tyson. Thanks!
Thank you very much. It is the most clear course I read. Beautiful work!
This is a very helpful lesson on how the UV absorbance is related to orbital transitions. I am a high school chemistry teacher, and I learned a lot!
Awesome Steve! Thanks for letting me know!