Alkyne Reactions
Alkyne Halogenation: Bromination and Chlorination of Alkynes
Last updated: May 28th, 2026 |
Halogenation of Alkynes With Br2 and Cl2
- Like alkenes, alkynes can undergo halogenation with Cl2, and Br2
- When 1 equivalent of the halogen is used, the products of these reactions are trans-dihaloalkenes.
- Like halogenation of alkenes, the reaction is believed to proceed through a bridged intermediate
- Alkynes react more slowly than alkenes towards Br2 and Cl2 by 3-5 orders of magnitude
- Addition of a second equivalent of a halogen gives tetrahaloalkanes.
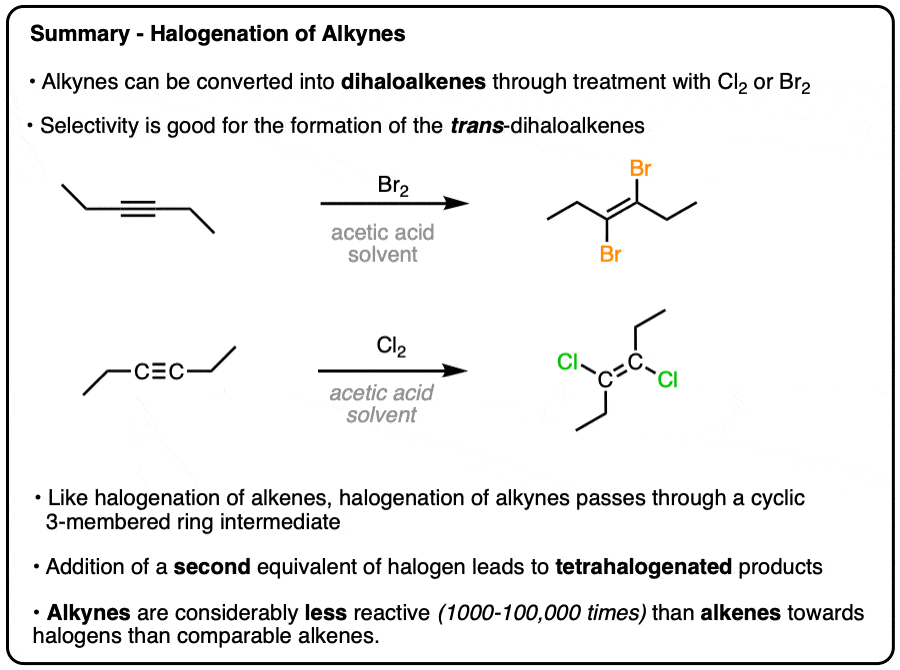
Table of Contents
1. Halogenation of Alkynes With Cl2 and Br2
If you’ll recall from the series of posts on alkenes, alkenes react with certain electrophiles (such as halogens, among others) to give positively charged bridged intermediates. (See article – Bromination of Alkenes). Common examples are the “bromonium ion” and the “mercurinium ion”. These intermediates then undergo backside attack by a nucleophile, resulting in overall anti addition across the double bond (See article – Syn and Anti) .
How do the addition of Br2 and Cl2 across alkynes compare to their reactions with alkenes with these reagents?
We might expect that alkynes, being so similar to alkenes, should also react in a similar fashion. Then again, this is organic chemistry, and sometimes changing one seemingly small variable can give an extremely different result (should you have any doubt on this, see Hydroboration of Alkynes)
Happily for us, the reaction of alkynes with electrophiles such as Cl2 and Br2 does give very similar results to what is observed with alkenes.
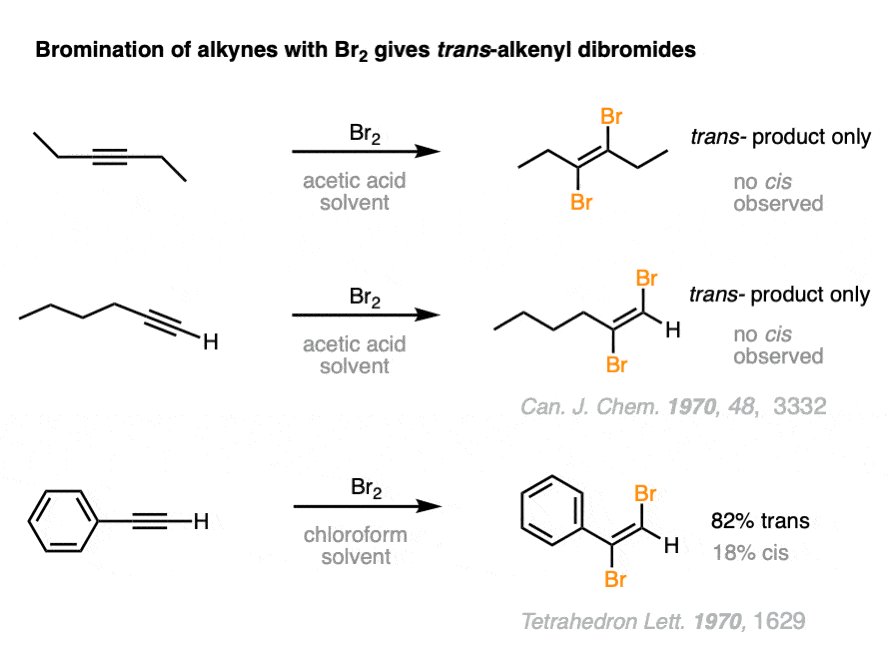
For example, treatment of an alkyne with 1 equivalent of Br2 provides a dibrominated alkene with the two bromides opposite to each other, to give us trans-dihalides. [For slightly more detail, see Note 1.]
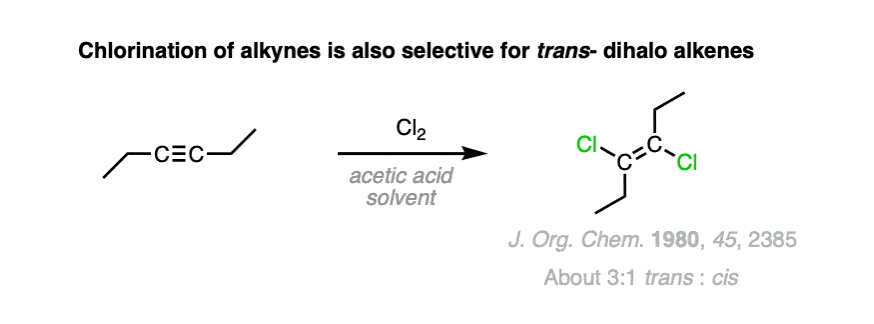
The story is similar with Cl2. Products of trans addition dominate. Although for more detail, see Note 2.
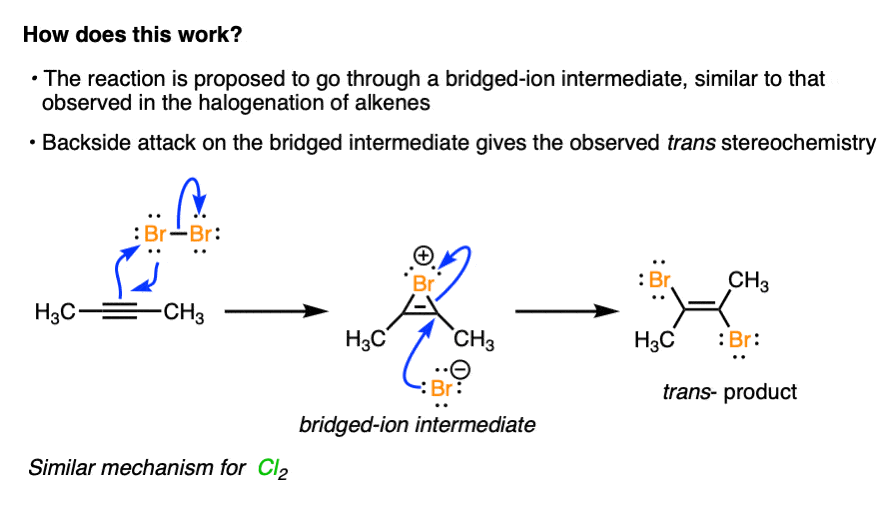
2. Halogenation of Alkynes Also Proceeds Through A Bridged-Ion Intermediate, Providing Trans Products
So how does this reaction work?
Given that the reaction predominantly gives trans dihalides, the prevailing view of the mechanism is that it passes through a bridged halonium ion intermediate similar to that observed for alkenes.
In the first step, a pi-bond from the alkyne acts as a nucleophile, attacking Br2 and giving rise to a bridged-ion intermediate.
In the next step, a halide ion attacks the carbon from the back face, leading to the trans product.
There is actually a very interesting observation to point out here, but I’ll leave that to the “Notes” section below as it is not absolutely essential for most readers’ purposes. Here’s the teaser, though: alkynes are considerably slower to react than alkenes are. [Note 3].
3. Addition of a Second Equivalent of Halogen Results in Tetrasubstituted Products
Once the dihalogenated alkene is formed, it’s possible to subject that alkene to a second halogenation, leading to the formation of a tetrahalogenated alkane.
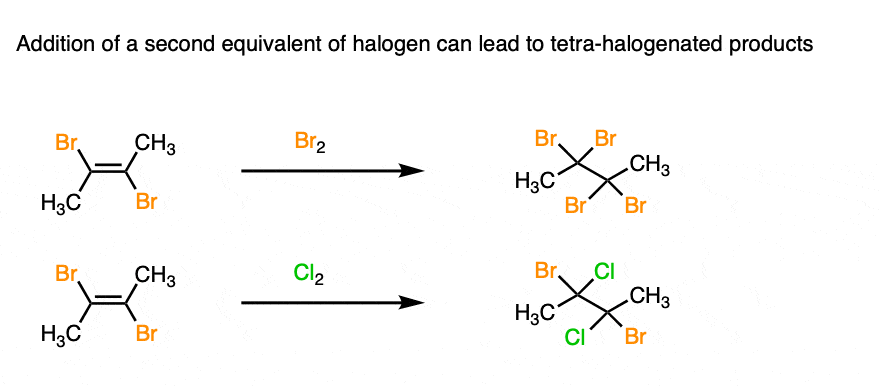
This can either be the same halogen (Br2) or a different one (Cl2) depending on your needs.
Notes
Note 1. This falls firmly into the “You Don’t Need To Know This For Org 1 / Org 2” category, so here goes. While aliphatic alkynes (i.e. alkynes attached to alkyl groups) tend to give only trans dibromides, with aryl alkynes there is considerably less selectivity for the trans product. For instance, in the example above, we saw that phenylacetylene only gave a 82:18 ratio of trans: cis whereas there were no cis products observed at all for 3-hexyne.
This loss of stereoselectivity likely reflects a slightly different mechanism may be in play. One mechanism that has been proposed is the intermediacy of a vinyl cation, which could undergo attack from either face.
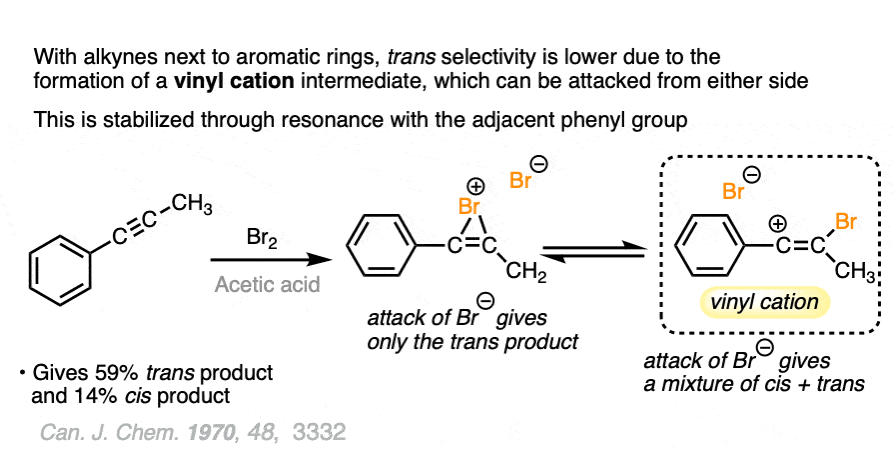
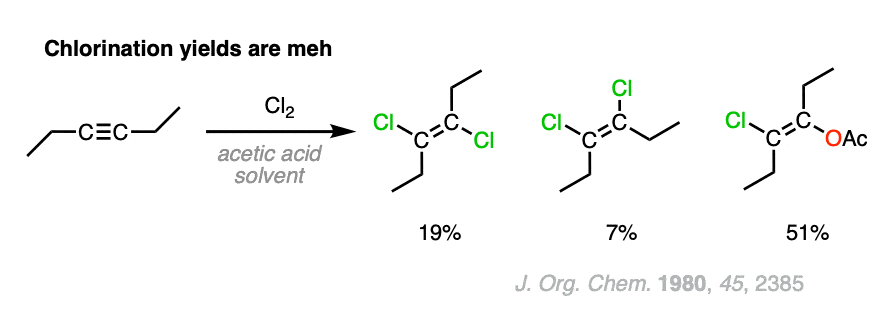
Note 2. Also falls into “You Don’t Really Need To Know This”, but chlorination is a considerably worse reaction than bromination (less selective).
For the reaction of Cl2 with 3-hexyne and 2-butyne, Yates and Go note, “these reactions are not clean, and many products were found”. For the chlorination of 3-hexyne shown in the scheme above, the actual yield of trans-dichlroalkene was 19% and the yield of cis-dichloroalkene was 7%. The major product (51%) incorporated acetic acid into the final product via trans addition. This shows that a cyclic bridged ion intermediate is likely still present. ( Interestingly, the reactions of terminal alkynes (1-hexyne and 1-pentyne) cleanly gives only syn addition products, which the authors speculate proceeds via a vinyl cation followed by quick trapping of the proximal chloride ion.
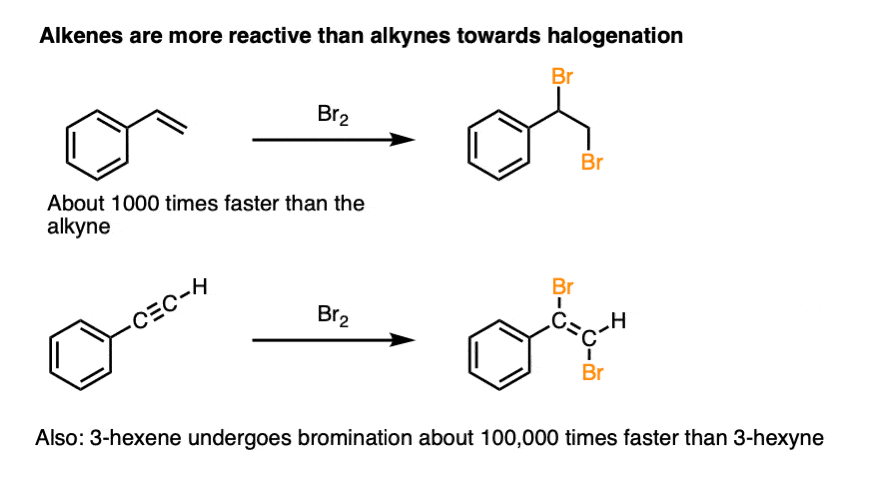
Note 3. Since the reaction goes through essentially the same pathway for alkenes as for alkynes, it presents us with an opportunity for an interesting experiment.
What reacts faster, alkenes or alkynes?
Researchers addressed this question by treating alkenes and alkynes with very similar structures (e.g. trans-3-hexene vs. 3-hexyne) and measuring the rate constants.
It was found that alkenes react with Cl2 and Br2 considerably faster than alkynes of similar structure, by factors of 1000 up to 100,000.
Another way of saying this is that the pi-bonds of alkenes are better nucleophiles than alkynes.
Remember that sp-hybridized carbons are better at stabilizing negative charge since the orbitals are closer to the nucleus? Well, that same effect means that these carbons are poorer at stabilizing positive charge (i.e. a lack of electron density) since, in effect, you are bringing an electron-deficient orbital closer to the nucleus.
You can think of alkynes as holding on to their electrons slightly more tightly than do alkenes.
Furthermore, the additional double bond leads to considerably more ring strain; sp2 hybridized carbons [ideal angle 120°] constrained into a triangle [internal angle 60°] is more unstable than an sp3 hybridized carbon [ideal angle 109°] would be.
There’s a second point which doesn’t become apparent for most students until second-semester organic chemistry. The 3-membered ring intermediate formed has antiaromatic character. (See post: Antiaromaticity)
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
Good reviews are to be found in Carey and Sundberg A. Chapter 6 (Polar Addition and Elimination Reactions) p. 374 in the 4th ed. Also, Patai’s Chemistry of Triple Bonded Functional Groups part 1, p. 539 has a good discussion of the mechanism (vinyl cations with phenyl substituents, bridged intermediates with alkyl acetylenes).
- Untersuchungen über Alloisomerie. II
Arthur Michael
J. Prakt. Chem. 1892, 46 (1), 209-210
DOI: 10.1002/prac.18920460115
An early paper on the bromination of alkynes. This paper mentions that bromination of dicarboxyacetylene gave 70% of the trans isomer! - Kinetics and mechanism of electrophilic bromination of acetylenes
James A. Pincock, Keith Yates
Canadian Journal of Chemistry, 1970, 48 (21): 3332-3348
DOI: 1139/v70-561
Stereoselective anti addition was found in the bromination of 3-hexyne, but both cis and trans products were obtained in the bromination of phenylacetylene. Notably, the reaction was found to be about 105 faster for 3-hexene than for 3-hexyne in acetic acid. - Vinyl cation intermediates in electrophilic additions to triple bonds. 2. Chlorination of alkylacetylenes
Keith Yates and T. Andrew Go
The Journal of Organic Chemistry 1980 45 (12), 2385-2391
DOI: 10.1021/jo01300a024
Electrophilic chlorination of alkyl acetylenes is less stereospecific than for bromination. Yields are lower, and there is more incorporation of solvent. - The Stereochemistry of Electrophilic Additions to Olefins and Acetylenes
Robert C. Fahey
Topics in Stereochemistry 1968, 3, 237-342
DOI: 1002/9780470147122.ch4
This review is more weighted towards alkene reactions, but does contain sections on the addition of Cl2 and Br2 to acetylenes. On pg. 291, the author states, “[…] bromine additions to acetylenes […] in acetic acid follow kinetics similar to those found for olefins, but that acetylenes are 100- to 50,000-fold less reactive than the corresponding olefins”. - Electron transmission study of the splitting of the p* molecular orbitals of angle-strained cyclic acetylenes: implications for the electrophilicity of alkynes
Lily Ng, Kenneth D. Jordan, Adolf Krebs, and Wolfgang Rueger
Journal of the American Chemical Society 1982, 104 (26), 7414-7416
DOI: 1021/ja00390a005
Another possible explanation for the lower reactivity of alkynes relative to alkenes has to do with the availability of the unfilled orbital in the alkyne. It has been shown that a p* orbital of bent alkynes (e.g. cyclooctyne) has a lower energy than the p* orbital of alkenes, and it has been suggested that linear alkynes can achieve a bent structure in their transition states when reacting with an electrophile.
Re: Note 3. Wouldn’t the bigger issue with halogenation of alkynes, and the bigger contributor to slow speeds, be the anti-aromatic character of the bridged ion? (Compare to oxirene.)
Is it possible the addition follows a more concerted but also more convoluted pathway to avoid an anti-aromatic intermediate?
Yes, that’s also an issue, but due to the poorer overlap of 1st-row carbon 2p with the 3p (for chlorine) or 4p (for bromine) orbitals and the corresponding longer bond lengths it’s considerably less of an obstacle than with oxirene.
Alkynes are just generally less nucleophilic than alkenes.
Synthesis of the first five alkynes from bromo ethane
Waiting for your reply
Thank you so much
Thanks sir 👍
One of three lone pairs of cl has contribution to it
Hi ,
Why in Note 2 you mentioned that the antiaromatic character of cyclic brominium ion of alkyne halogenation?
Is the 1 pie bond makes it aromatic ??
Kindly explain me sir
*anti* atomatic. One pi bond from the alkene, and another from a lone pair of the bromine.
For an example of a three-membered ring that has antiaromatic character, see oxirene:
https://en.wikipedia.org/wiki/Oxirene
Thank you
But how is C2H6 an alkyne?
Very unselective
I have seen somewhere that you even get a cis product,through what other mechanism can you get a it
Wrong halohydrin rxn with alkyne
two halo atoms get attached
“The 3-membered ring intermediate formed has antiaromatic character. That is, there are 4 π electrons constrained in a conjugated ring,” does this mean that one of the two lone pairs of Cl is part of the antiaromatic system?
Yes, exactly!