Aromaticity
The Pi Molecular Orbitals of Cyclobutadiene
Last updated: May 7th, 2026 |
Cyclobutadiene: Molecular Orbital Diagram, Antiaromaticity, and Structure
Previously, we’ve seen what the molecular orbitals of benzene look like, and that the fact that they are partially duplexed (or to use the proper nomenclature, “degenerate“) helps to explain benzene’s unusual stability.
Let’s flip the coin. What about cyclobutadiene, a molecule we usually class as antiaromatic. Why is it so unusually unstable?
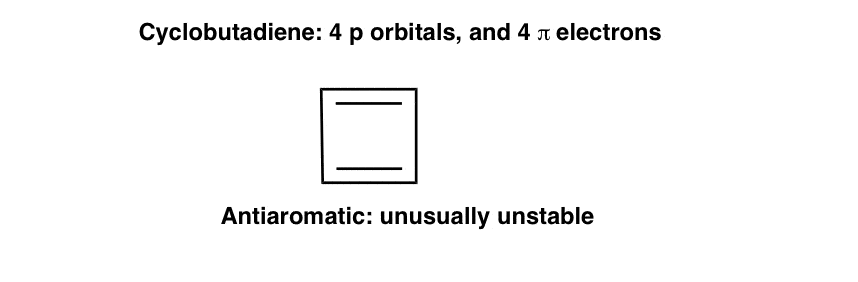
Again, examining the pi molecular orbitals will give us some useful clues.
Notably, in cyclobutadiene, the highest-occupied molecular orbitals are unpaired electrons of equal energy.
Table of Contents
- Building Up The Molecular Orbital Diagram Of Cyclobutadiene: The Lowest-Energy Molecular Orbital Has Zero Nodal Planes
- The Highest-Energy Molecular Orbital Has Two Nodal Planes
- The Two Intermediate pi Molecular Orbitals Each Have One Nodal Plane (two different ways)
- The Molecular Orbital Diagram Of Cyclobutadiene Reveals Why Cyclobutadiene Is Extremely Unstable: It Has Unpaired Electrons Of Equal Energy
- Summary: The Molecular Orbital Diagram of Cyclobutadiene
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Building Up The Molecular Orbital Diagram Of Cyclobutadiene: The Lowest-Energy Molecular Orbital Has Zero Nodal Planes
Today, let’s build up the orbitals of cyclobutadiene using the principles we’ve discussed in previous posts [e.g. see this post on butadiene] and see if we can gain some useful insights.
Cyclobutadiene has a pi system comprised of 4 individual atomic p orbitals and thus should have a total of 4 pi molecular orbitals.
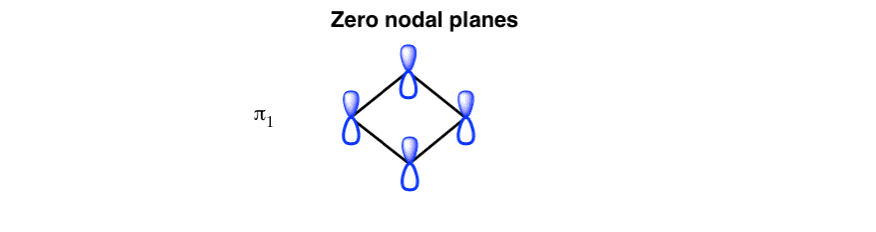
The lowest-energy molecular orbital: zero nodal planes
Following our “apartment building” analogy from last time, the lowest-energy molecular orbital (the “ground floor” of cyclobutadiene, if you will) should have all phases of the p-orbitals aligned and zero nodal planes, like this:
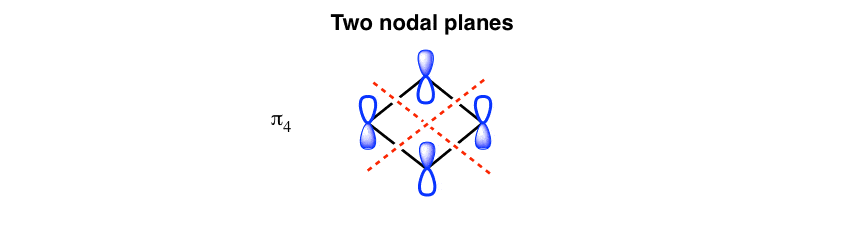
2. The Highest-Energy Molecular Orbital Has Two Nodal Planes
Conversely, the highest-energy pi orbitals (the “penthouse”) will have all phases alternating, and thus have two nodal planes. (As we said last time, the “penthouse” is not exactly desirable real estate for electrons)
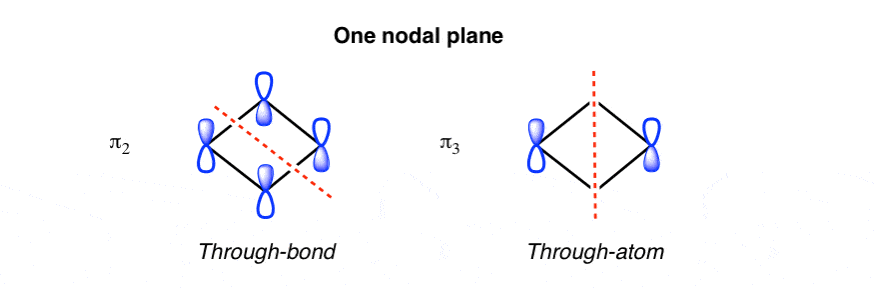
3. The Two Intermediate pi Molecular Orbitals Each Have One Nodal Plane (two different ways)
That leaves us with the intermediate pi orbitals, which each have a single nodal plane. As with benzene, there are two ways to place a single nodal plane on cyclobutadiene, either through the bonds, or through the atoms:
That gives us our four molecular orbitals. Now lets populate them with the “tenants”: the pi electrons.
4. The Molecular Orbital Diagram Of Cyclobutadiene Reveals Why Cyclobutadiene Is Extremely Unstable: It Has Unpaired Electrons Of Equal Energy
Cyclobutadiene has a total of 4 pi electrons. So ranking all the pi molecular orbitals by energy, and populating the orbitals according to Hunds rule, we get the following picture:
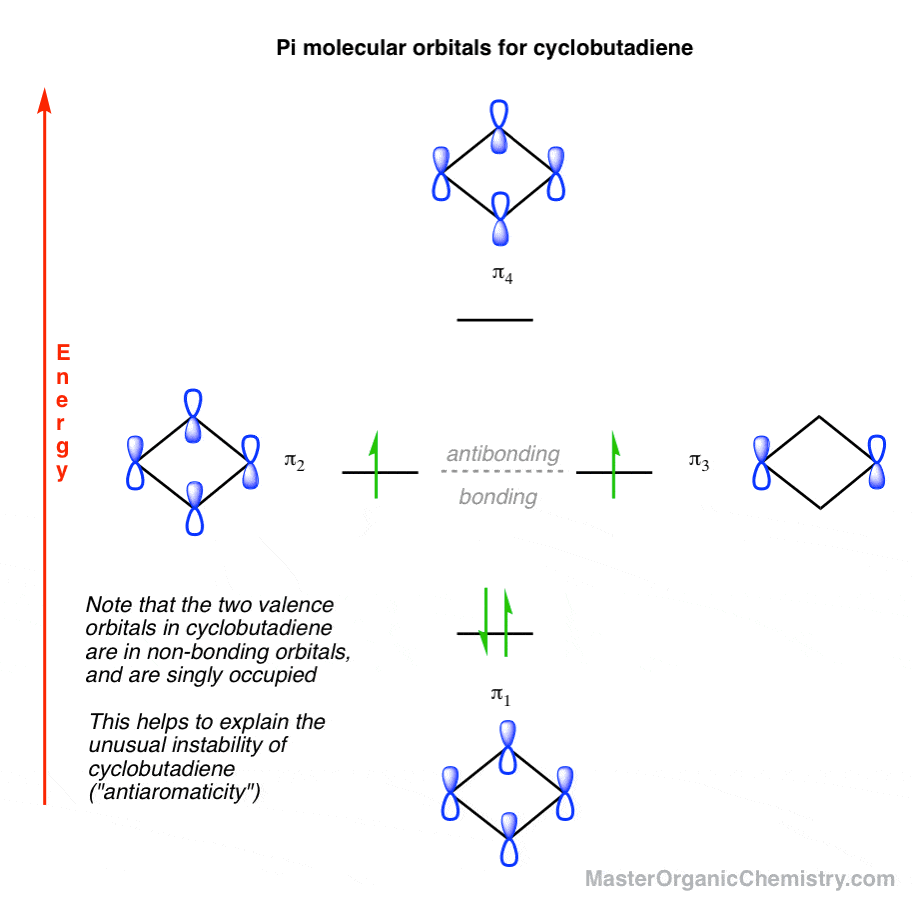
Can you see why cyclobutadiene might be unstable?
- First, the highest-occupied molecular orbitals of cyclobutadiene are non-bonding orbitals, intermediate in energy between the lowest (π1, bonding) and highest (π4, antibonding) energy orbitals. “Non-bonding” implies that filling these orbitals with electrons does not result in any stabilization of the molecule.
- Second, note that each of the non-bonding orbitals are singly occupied. Therefore this orbital picture predicts that cyclobutadiene should have a diradical nature. We’re used to thinking of free-radicals as highly reactive intermediates… so you can imagine that a species containing two free radicals is even more reactive! [Note 1 ]
5. Summary: The Molecular Orbital Diagram of Cyclobutadiene
The bottom line here is that the pi molecular orbital picture of cyclobutadiene is in agreement with our observations that cyclobutadiene is unusually unstable. (As previously noted, cyclobutadiene has only ever been isolated as a “matrix-isolated species” – that is, a species frozen in an inert gas at extremely low temperatures. Warming to a balmy –80° results in self-destruction. Note 2 )
Hopefully these two posts have helped to show that molecular orbital diagrams can provide extremely useful clues about molecular stability!
In the next post we’ll cover a very convenient short-cut that will help us quickly draw molecular orbital diagrams in seconds (yes, really!) called Frost Circles. Or, more blandly, the Polygon method.
Notes
Note 1. More advanced calculations, far beyond what we will discuss, predict that cyclobutadiene distorts to a rectangular shape which results in the two singly-occupied orbitals resolving into two orbitals of slightly different energy, one doubly-occupied and the other empty. The bond lengths of cyclobutadiene have been measured, confirming the rectangular shape.

Note that the pi electrons are not “delocalized” like they are in benzene.
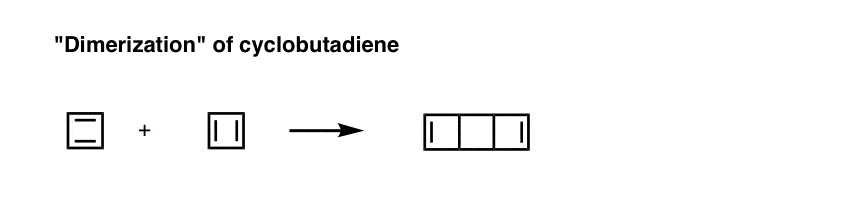
Note 2. Upon warming (–80° is considered “warm” for these purposes), cyclobutadiene reacts with itself through a Diels-Alder process to give “dimeric” species.
Note 3. If benzene is about 36 kcal/mol more stable than (theoretical) cyclohexatriene, exactly how unstable is cyclobutadiene? The negative resonance energy of cyclobutadiene is calculated to be –54.7 kcal/mol, relative to 1,3-butadiene. In addition, 30.7 kcal/mol of strain is found, giving a total destabilization of 85.4 kcal/mol. [Ref]
Quiz Yourself!

Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- Cyclobutadiene
Watts, J. D. Fitzpatrick, and R. Pettit
Journal of the American Chemical Society 1965, 87 (14), 3253-3254
DOI: 10.1021/ja01092a049
Interestingly, this paper precedes an article on the reactivity of cyclobutadiene-iron tricarbonyl. Cyclobutadiene is prepared by the oxidation of that organometallic complex using Ce4+, which is then trapped in situ with an alkyne. - Cyclobutadiene
Thomas Bally, Satoru Masamune
Tetrahedron 1980, 36 (3), 343-370
DOI: 1016/0040-4020(80)87003-7
This paper from 1980 reviews work done on cyclobutadiene up to that time. This is divided into 2 parts – experimental synthetic efforts and theoretical calculations.Elaborate MO treatments and theoretical calculations indicate that the most stable geometry for cyclobutadiene is rectangular. - Potential energy surfaces of cyclobutadiene: ab initio SCF and CI calculations for the low-lying singlet and triplet states
A. Jafri and M. D. Newton
Journal of the American Chemical Society 1978, 100 (16), 5012-5017
DOI: 10.1021/ja00484a016 - The potential surfaces for the lowest singlet and triplet states of cyclobutadiene
Weston Thatcher Borden, Ernest R. Davidson, and Paul Hart
Journal of the American Chemical Society 1978, 100 (2), 388-392
DOI: 10.1021/ja00470a006 - A theoretical study of the structure of cyclobutadiene
Kollmar and V. Staemmler
Journal of the American Chemical Society 1977, 99 (11), 3583-3587
DOI: 10.1021/ja00453a009 - Ground states of molecules. 36. The cyclobutadiene problem and MINDO/3 calculations of molecular vibration frequencies
Michael J. S. Dewar and Andrew Komornicki
Journal of the American Chemical Society 1977, 99 (19), 6174-6179
DOI: 10.1021/ja00461a002
Analysis of the IR spectra of the product and deuterated analogs generated from labeled precursors has confirmed the theoretical conclusion that cyclobutadiene is a rectangular molecule: - Cyclobutadiene is not square
Satoru Masamune, Fernando A. Souto-Bachiller, Takahisa Machiguchi, and John E. Bertie
Journal of the American Chemical Society 1978, 100 (15), 4889-4891
DOI: 1021/ja00483a043
Ah, for the days when papers had simple, punchy titles (Refs. 1 and 2 above). - Ab initio second-order Moller-Plesset calculation of the vibrational spectra of cyclobutadiene and its isotopic derivatives
Andes Hess Jr., P. Carsky, and L. J. Schaad
Journal of the American Chemical Society 1983, 105 (4), 695-701
DOI: 10.1021/ja00342a003 - The Dimerization of Cyclobutadiene. An ab Initio CASSCF Theoretical Study
Yi Li and and K. N. Houk
Journal of the American Chemical Society 1996, 118 (4), 880-885
DOI: 10.1021/ja921663m - Ab Initio Calculation of Resonance Energies. Benzene and Cyclobutadiene
A. Hess, Jr. and L. J. Schaad
Journal of the American Chemical Society 1983, 105 (26), 7500-7505
DOI: 10.1021/ja00364a600
A paper from the 80’s using computational methods to quantify the antiaromatic destabilization of cyclobutadiene. These authors obtain a value of -54.7 kcal/mol for the negative resonance energy of cyclobutadiene. - Experimental Determination of the Antiaromaticity of Cyclobutadiene
Ashok A. Deniz, Kevin S. Peters, Gary J. Snyder
Science 1999, 286 (5442), 1119-1122
DOI: 1126/science.286.5442.1119
This is a very rigorous paper that uses novel spectroscopic techniques to determine antiaromatic destabilization of cyclobutadiene. Relative to a hypothetical strain-less, conjugated diene reference, cyclobutadiene is destabilized by a total of 87 kcal/mol, 32 kcal/mol of which can be attributed to ring strain and 55 kcal/mol to antiaromaticity (compared with 21 kcal/mol for the aromatic stabilization of benzene). - Quantentheoretische Beiträge zum Benzolproblem
Die Elektronenkonfiguration des Benzols und verwandter Verbindungen
Erich Hückel
Zeitschrift für Physik 1931, 70, 204–286
DOI: 10.1007/BF01339530
Erich Hückel achieved recognition by elaborating, together with Peter Debye, the theory of strong electrolytes in 1923 and later by applying a simplified version of quantum theory to p-electrons in conjugated molecules, which became known as Hückel molecular orbital (HMO) theory. Although he never explicitly formulated a “4n + 2 rule”, this was obvious from his work. Hückel showed that monocyclic systems with continuous conjugation having 6, 10, 14, etc. p-electrons benefited from extra stabilization and were aromatic. But it is more accurate to refer to the “Hückel 4n + 2 p-electron rule,” rather than to “Hückel’s rule.” - A Mnemonic Device for Molecular Orbital Energies
Arthur A. Frost and Boris Musulin
J. Chem. Phys. 1953, 21, 572
DOI: 10.1063/1.1698970
The origin of the “Frost Circle” mnemonic device for determining the MO’s of electrocyclic systems.
sir please draw the digram of carbon 7 pi molecular orbital picture
Cyclic or acyclic?
Hello!
I’m confused as to where to locate the Pi bonds when looking at the MO orbital. Since the two valence orbitals in cyclobutadiene are in non bonding orbitals and are singly occupied, why then do we see two pi bonds? I understand how Pi 1 represents a double bond but I don’t see where the second bond is. Thank you!
Hi Danielle – You can think of the situation where both carbons have free radicals as a “resonance form” of a pi bond.
(my answer here could, and should, go a lot deeper, but I’m just going to keep it simple in this case).