Dienes and MO Theory
The Cope and Claisen Rearrangements
Last updated: May 28th, 2026 |
Cousins Of The Diels-Alder: The Cope Rearrangement and The Claisen Rearrangement
- The Cope Rearrangement and Claisen Rearrangement are pericyclic reactions in the same family as the Diels-Alder that go through a cyclic, 6-electron concerted transition states and follow the rules of orbital symmetry.
- In this article, intended for students in a course in introductory organic chemistry, we’ll introduce these two reactions with examples, mechanisms, and applications.
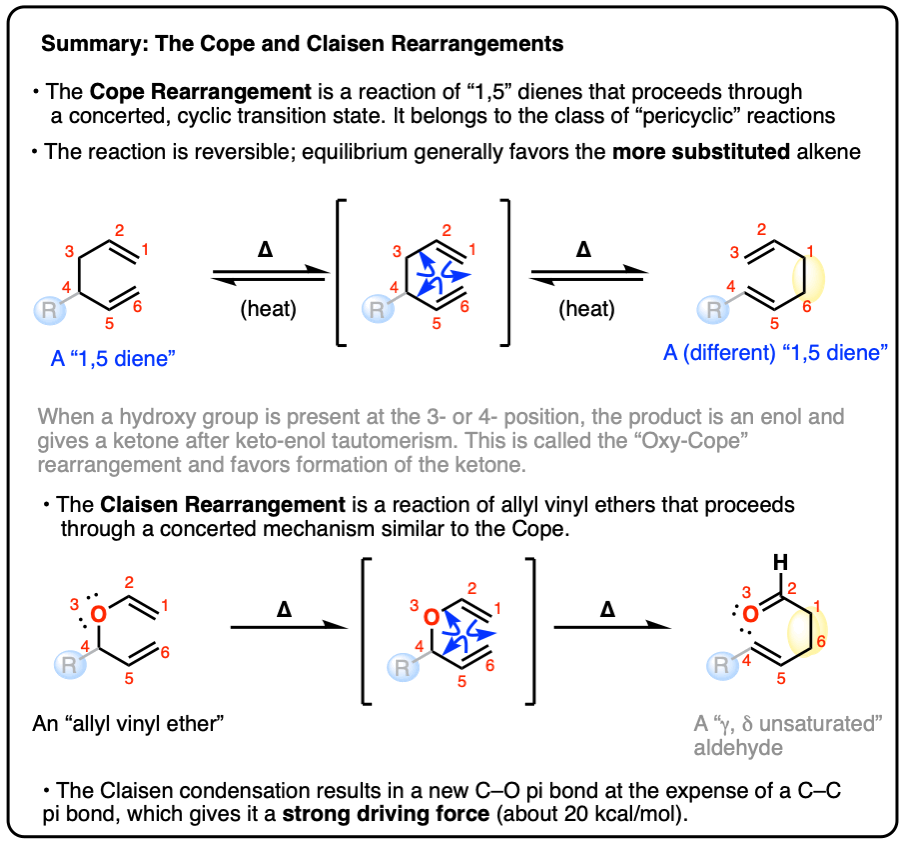
Table of Contents
- The Cope Rearrangement: Example And Mechanism
- The Cope Rearrangement Is An Equilibrium
- Cope-ing With Ring Strain
- The Oxy-Cope Rearrangement
- The Claisen Rerrangement
- Claisen Rearrangement of Allyl Phenyl Ethers
- The Claisen Rearrangement In Amino Acid Biosynthesis
- Summary: The Cope And Claisen Rearrangements
- Notes
- Quiz Yourself!
- (Advanced) Orbital Symmetry In The Cope and Claisen
- (Advanced) Synthesis of Periplanone B
- (Advanced) References and Further Reading
1. The Cope Rearrangement: Example And Mechanism
The reaction below is called the Cope Rearrangement. In this reaction, one heats a 1,5-diene to a temperature of 150° C (or higher) whereupon the starting material gives the rearranged product.
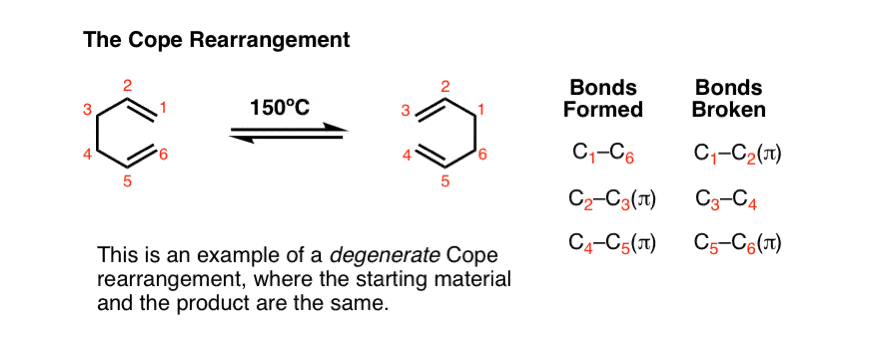
Ta-daa!!
The example above is the simplest possible example (the “degenerate” Cope rearrangement) where it is just rearranging into itself.
If you were to judge the Cope rearrangement by this one reaction (like I did, once) you would be forgiven for coming to the conclusion that it is freakin’ useless. Trust me when I say that it is not. [Note]
Let’s examine the bonds that form and break here:
- Two C-C pi bonds form (plus a C-C sigma bond)
- Two C-C pi bonds break (plus a C-C sigma bond).
So how does it work?
Like the Diels-Alder reaction, The Cope Rearrangement belongs to a large family of reactions known as “pericyclic” reactions. The Cope rearrangement passes through a concerted, cyclic transition state with no charged intermediates.
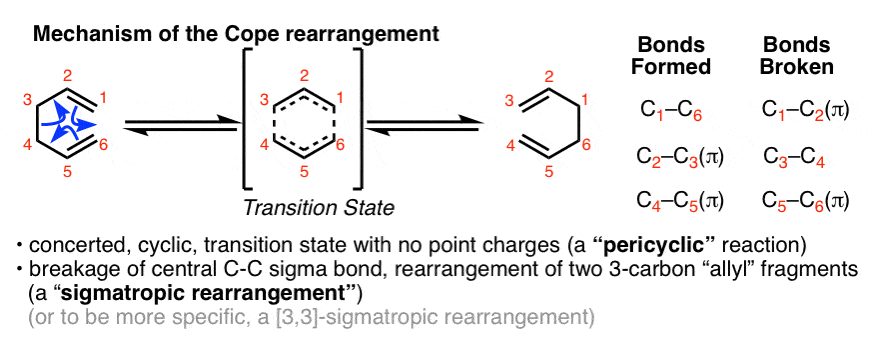
The Cope rearrangement is also an example of a “sigmatropic” rearrangement where a sigma bond (and its substituent) interchanges termini on a conjugated pi system [Note 1]
2. The Cope Rearrangement Product Is In Equilibrium With Its Starting Material
The product of a Cope rearrangement will also be a 1,5-diene, and therefore be capable of undergoing the reverse reaction.
The product distribution will therefore reflect an equilibrium between the starting material and the rearranged product.
In the degenerate Cope rearrangement (above) it’s impossible to see if anything is happening because the starting material is the same as the product. [Note 2 – There’s a way around this using isotopically “labelled” carbons]
So how could the equilibrium be tipped towards something actually, you know, useful?
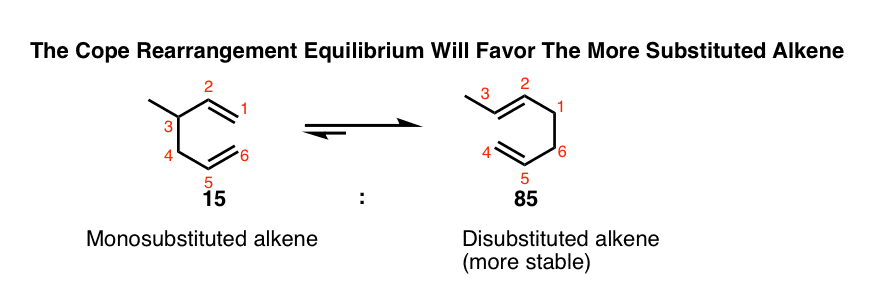
Remember Zaitsev’s rule? (i.e. “the more substituted alkene wins”). Alkene stability increases slightly (e.g. by about 1-2 kcal/mol) as successive C-H bonds on the alkene are replaced by C-C bonds.
1-2 kcal/mol might not sound like much, but it’s enough to push a reaction forward nicely!
In the simple case below, just placing a methyl group on C-3 results in a rearranged product with a disubstituted alkene instead of a monosubstituted alkene. The result is an 85:15 mixture of the product (5:1). [Note 3]
The example that started it all, by Arthur C. Cope and Elizabeth Hardy in 1940, had a trisubstituted alkene rearrange to a tetrasubstituted alkene that was also in conjugation with an ester and a nitrile. This was obtained as a single product. [Note 4]
3. Cope-ing With Ring Strain
A Cope rearrangement that really puts its elbow on the scale towards formation of a single product has a C-C sigma bond that is part of a strained ring system. This rearrangement of “cis-divinylcyclopropane” occurs below room temperature. [Note 5]
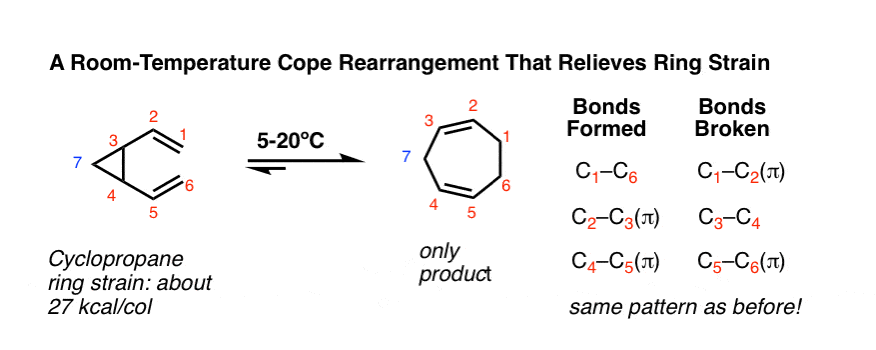
Look closely! Despite the somewhat strange appearance of the reaction, it’s the same pattern of bonds that form and break!
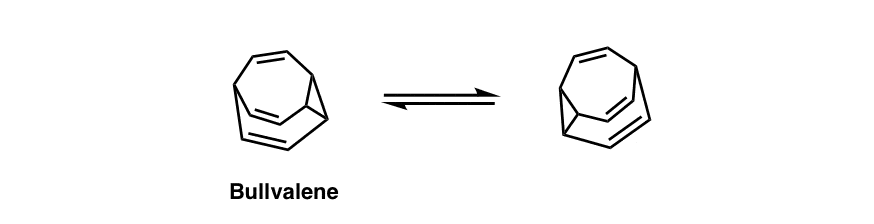
Chemists have taken these kinds of strain-driven Cope rearrangements to even crazier extremes. It’s no “bull” that there’s a molecule that has no fixed structure since it’s constantly Cope-ing with itself. [Note 6]
4. The Oxy-Cope Rearrangement
In the example above we saw that even a humble methyl group at C-3 was sufficient to give a useful 5:1 ratio of Cope Rearrangement products.
Along these lines, it was reported by the late, great Prof. Jerome Berson (and his postdoc Prof. Maitland Jones, of NYU and textbook fame) in 1964 that the Cope rearrangement of a 3-hydroxy substituted 1,5-diene led to an enol product.
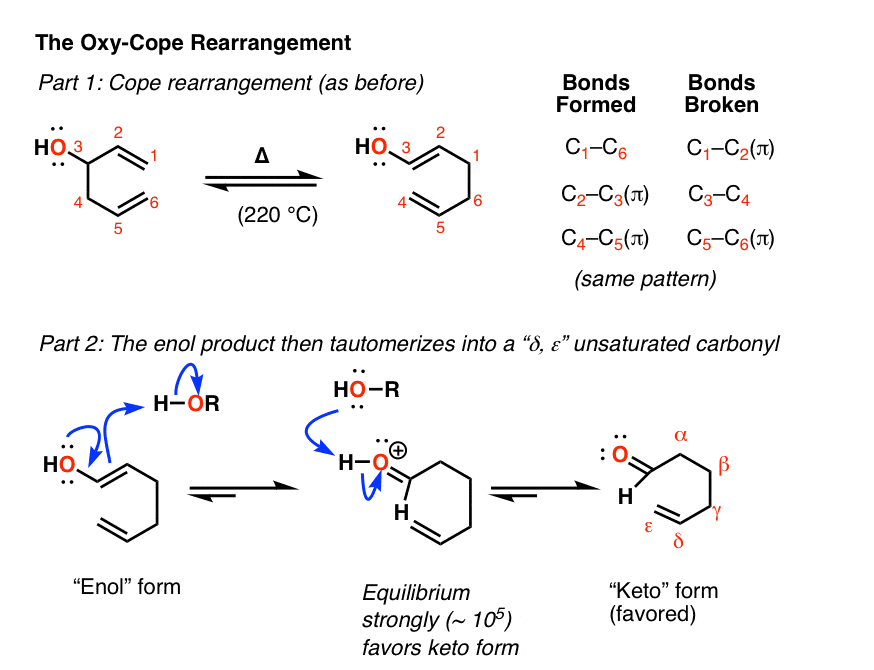
If you think back – way back – to the hydroboration and oxymercuration of alkynes, you might remember that enols tend not to stick around for very long.
Through a process known as keto-enol tautomerism, the enol undergoes a rearangement into a ketone (or aldehyde, depending on the structure of the starting material) resulting in the irreversible formation of what we call a “gamma, delta” unsaturated aldehyde or ketone. (See post: Keto-Enol Tautomerism)
Keto-enol tautomerism is an equilibrium that generally favors the keto form due to the strength of the C-O pi bond versus the C-C pi bond. [Note 7]. In at least one case the equilibrium was measured to be about 105 favoring the keto]
Like the Cope Rearrangement, the Oxy-Cope still requires significant heating. (One of the Berson-Jones examples involved heating in a sealed tube at 320 C°.) [Note 8 on how to make this even faster]
5. The Claisen Rearrangement
A cousin of the Cope rearrangement is a slightly older process (1912) known as the Claisen rearrangement. (Yes, this is the same Ludwig Claisen responsible for the Claisen condensation). It’s also a pericyclic reaction.
In the Claisen rearrangement, a vinyl allyl ether is heated to give a gamma, delta (γ,δ) unsaturated carbonyl.
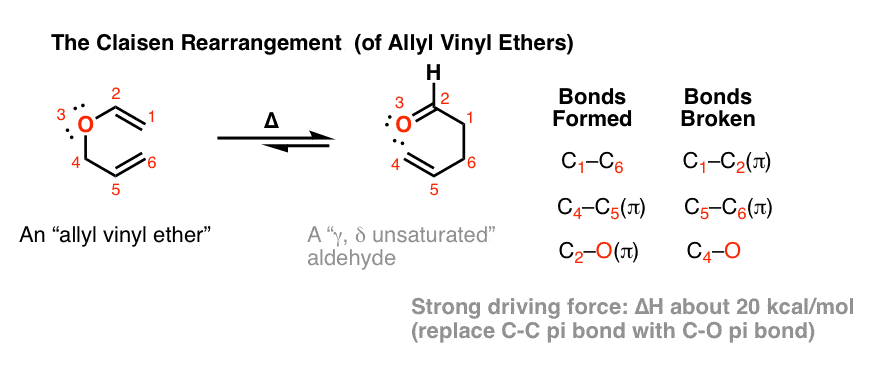
- A C-C sigma bond, a C-C pi bond, and a C-O pi bond are formed.
- Two C-C pi bonds are broken (and a C-O sigma bond).
If we compare the energies of the bonds that break with the bonds that form, we see that we are trading a relatively weak carbon-carbon pi bond (about 65 kcal/mol) for a relatively strong carbon oxygen pi bond (about 85 kcal/mol) for a gain of about 20 kcal/mol.
(The bond dissociation energies of the C-C sigma bond and the C-O sigma bond are roughly the same (about 85 kcal/mol) so they essentially cancel out )
A reaction where the product is about 20 kcal/mol (~85 kJ/mol) more stable than the reactants is for all intents and purposes irreversible. This is just one feature that makes the Claisen rearrangement so valuable.[Note 9]
So how does it work? From an arrow-pushing perspective, it looks a lot like the Cope Rearrangement!
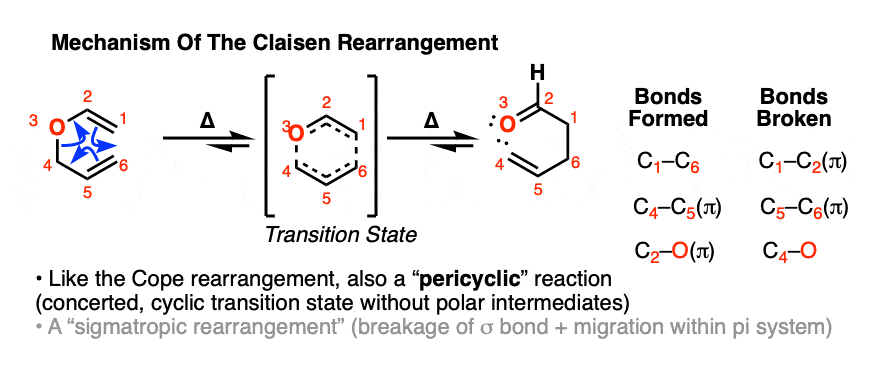
We won’t go into the stereochemistry of the Claisen rearrangement here, but suffice to say it generally follows a very reliable pattern that has made it a workhorse of stereoselective synthesis. [Note 10]
6. Claisen Rearrangement of Allyl Phenyl Ethers
Aromatic pi systems can also participate in the Claisen Rearrangement. Heating allyl phenyl ether [Note 11] leads to a Claisen rearrangement resulting in a gamma, delta unsaturated lactone.
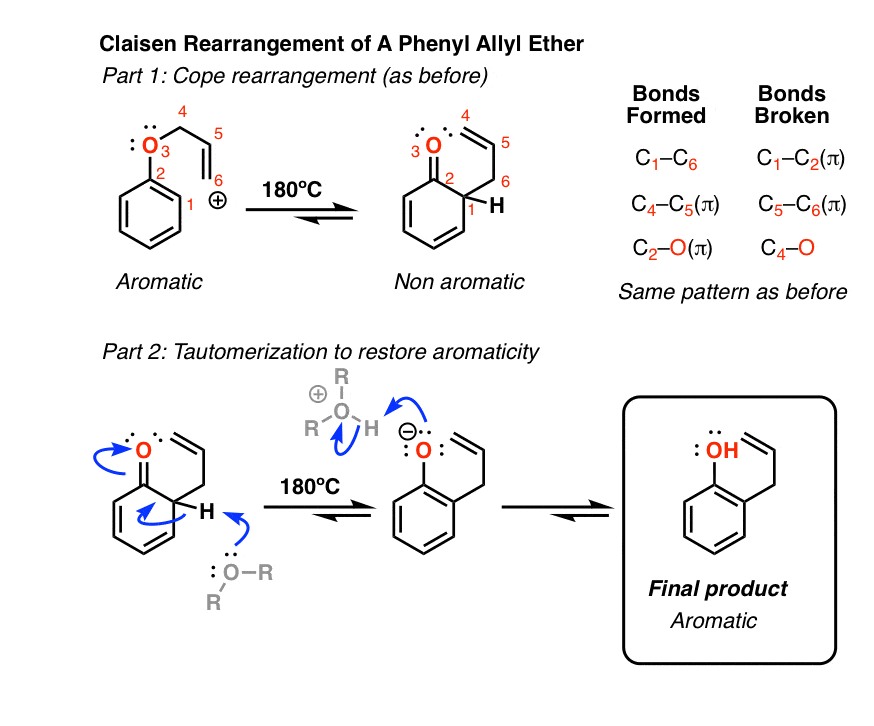
Although the Claisen rearrangement stops there, a second reaction immediately occurs (and can’t be stopped).
If you’ve covered aromaticity, then you’ll recognize that the product of the Claisen rearrangement is no longer aromatic; the sp3 carbon attached to 4 sigma bonds prevents conjugation all the way around the ring.
As it happens, this is another opportunity for keto-enol tautomerism to raise its head, although this time the driving force goes in the opposite direction (from keto to enol)! The reason is that the stability imparted by aromaticity (resonance energy >30 kcal/mol) more than compensates for the smaller difference in energy (Note 7) between the keto and enol forms.
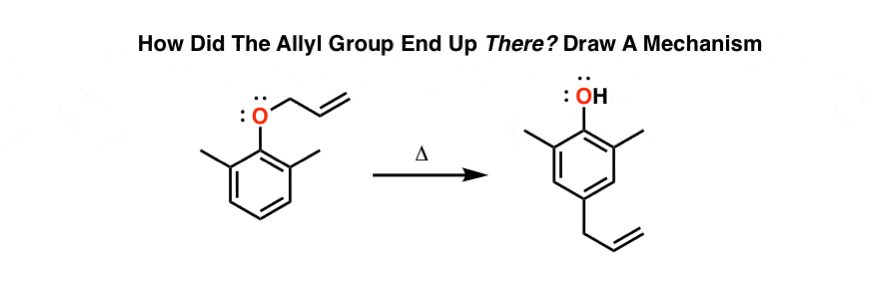
What happens if the ortho position is blocked? See if you can figure out this fun (heh) mechanism.
7. The Claisen Rearrangement In Nature
As it turns out, the Claisen Rearrangement occurs in living organisms. As you might expect, rather than endure heating to 150°C, Nature has discovered a way to catalyze the reaction with an enzyme.
Below is drawn the rearrangement of chorismate to prephenate. This reaction is catalyzed by the enzyme chorismate mutase, which leads to a rate acceleration of about 106. Note the pattern of bonds formed and broken – despite the somewhat funky appearance of these molecules, the exact same pattern of bond-breaking and bond-forming as is found in the reaction above.
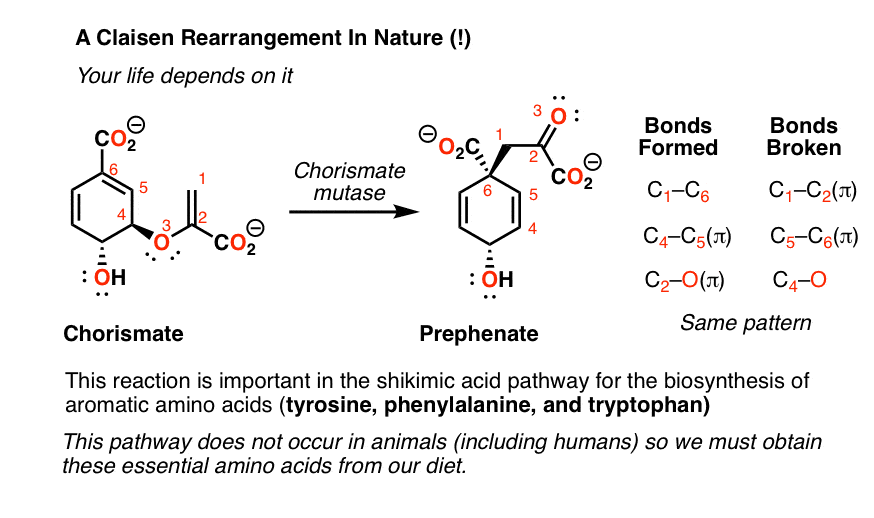
Why is this biosynthetic pathway important? It’s a key step in the shikimic acid pathway responsible for the biosynthesis of the essential amino acids tryptophan, phenylalanine and tyrosine. This pathway does not operate in animals (humans included) – that’s why we must obtain these amino acids from dietary sources.
8. Summary: The Cope And Claisen Rearrangements
We have really just barely dented the surface of the Cope and Claisen rearrangements here, but for most students in introductory courses this will be enough.
- Like all pericyclic reactions, the Cope and Claisen rearrangements are subject to the Woodward-Hoffman rules concerning orbital symmetry. The orbital symmetry of the Cope and Claisen rearrangements is probably beyond most introductory course material but is dealt with in the appendix below
- Likewise the details of stereochemistry, including the chair (and sometimes boat!) geometry of the transition states has been ignored here. For a good time, check out the classic Doering-Roth experiments. Maitland Jones also discusses them in his textbook.
- The applications of the Cope and Claisen in synthesis are too numerous to mention. Over 15 variants of the Claisen rearrangement exist, and some have only recently been developed. Organic chemistry is very much a living and active discipline where new discoveries are constantly being published!
Notes
The activation energy of the Cope is about 33 kcal/mol. The activation energy of the Claisen rearrangement is about 30 kcal/mol. [Refs]
Note 1. IUPAC defines a “sigmatropic rearrangement” as a pericyclic reaction involving both breaking and formation of a new σ bond in which the total number of π and σ bonds do not change.
Note 2. One could observe the “degenerate” Cope rearrangement by 13C NMR if one were to install a 13C label on one of the terminal alkene atoms of the 1,5-diene (say, C-1 in our figure). The position of the equilibrium of the Cope could then be monitored by observing the appearance of a 13C labelled methylene (CH2) group.
Note 3. Using the Gibbs free energy relationship (Δ G = –RT ln K), a ratio of 85:15 at 150°C corresponds to an energy difference of about 1.5 kcal/mol. It doesn’t take much of an energy difference to push the equilibrium towards a useful ratio!
Note 4. The original report on the Cope rearrangement by Arthur C. Cope and Elizabeth Hardy:
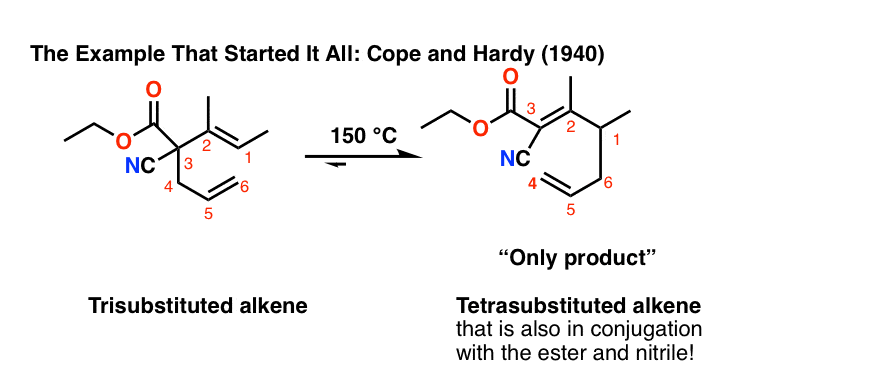
Note 5. Only cis-divinylcyclopropane rearranges easily. The trans-divinyl cyclopropane has to be heated up to about 190°C in order to rearrange since the cyclopropane needs to fragment first. [JACS 1972 94 5910]
Note 6. Bullvalene (below) is a molecule with no fixed structure. Its life is a series of never-ending Cope rearrangements; all the carbons are chemically shift equivalent.
Note 7. Marvell measured the equilibrium constant to be about 105 so by the Gibbs equation this would put the energy difference at about 7 kcal/mol.
Note 8. This would seem to put a damper on applications of the reaction, but about 10 years later Evans and Golob discovered that treating the starting material with potassium hydride puts the Oxy-Cope rearrangement into hyperdrive, lowering the activation barrier so much (by about 10-15 kcal/mol) that the reaction can happen at room temperature or below. This is now known as the “anionic Oxy-Cope rearrangement”.For a fun example of the Oxy-Cope leading to the synthesis of cockroach sex pheromone periplanone B, see the appendix.
Note 9. From the standpoint of synthesis, a subtle feature that makes the Claisen rearrangement useful is that it trades a bond that is relatively easy to make (C-O) for a bond that is relatively hard to make (C-C) in a very predictable fashion. (For instance we’ve learned several ways to make C-O bonds through the Williamson ether synthesis, addition of alcohols to alkenes and alkynes, and various other processes. )
Note 10. A fascinating detail about the Claisen rearrangement is the transfer of stereochemical information. If the C-O bond and the double bond(s) are constructed in a stereoselective manner (E or Z) then this geometry can be relayed into the formation of new chiral centers with very predictable stereochemistry. This has been particularly well developed for the Ireland ester enolate Claisen rearrangement.
Note 11. Easily made from a Williamson ether synthesis, right?
Quiz Yourself!
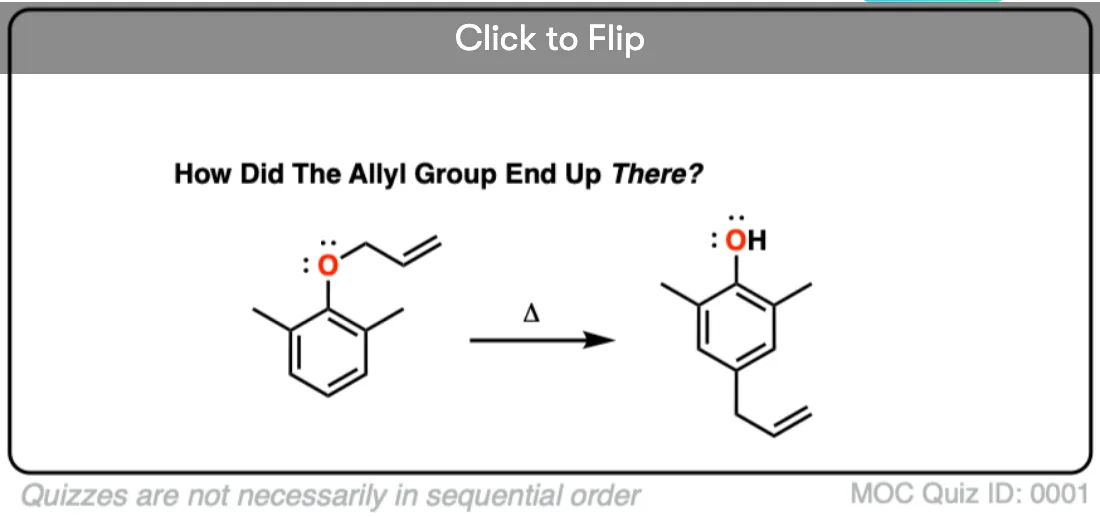
Become a MOC member to see the clickable quiz with answers on the back.
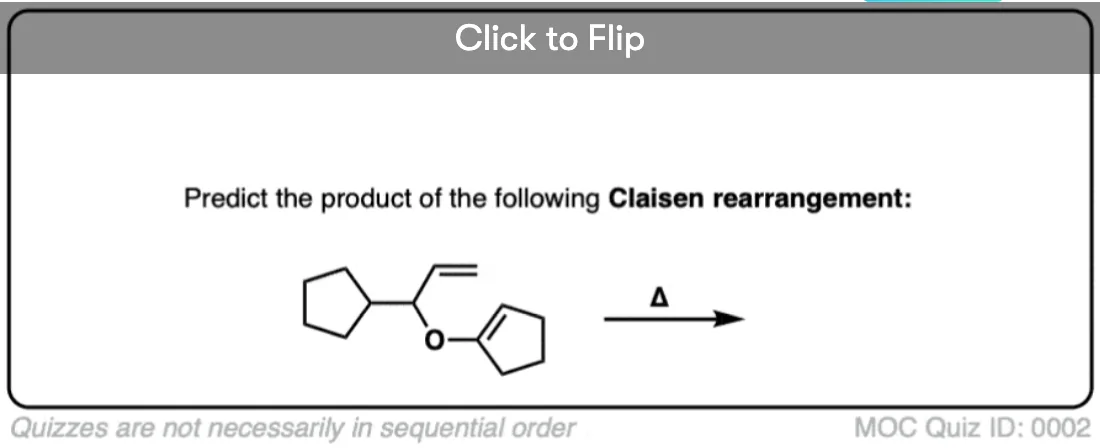
Become a MOC member to see the clickable quiz with answers on the back.
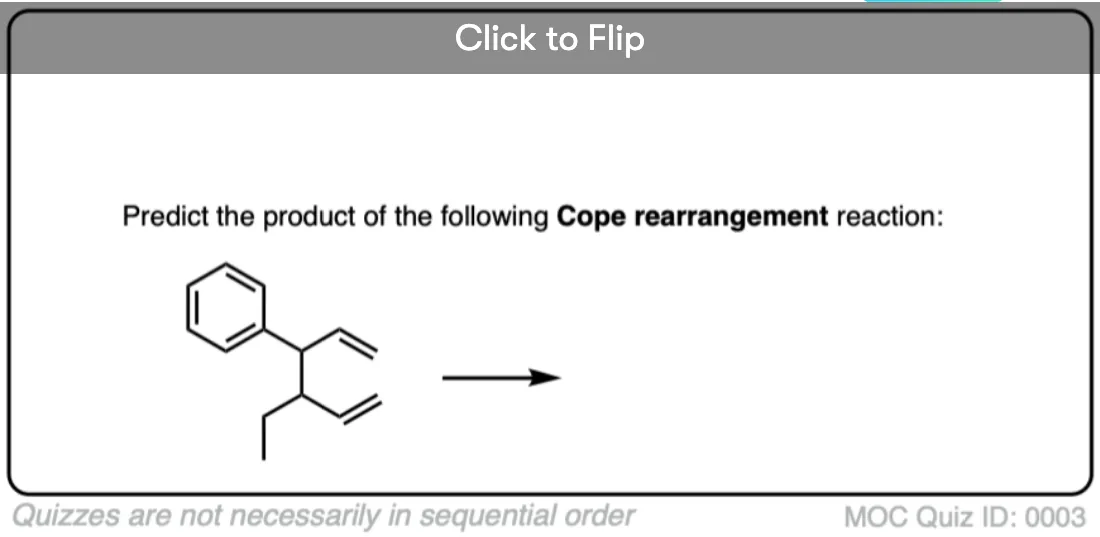
Become a MOC member to see the clickable quiz with answers on the back.
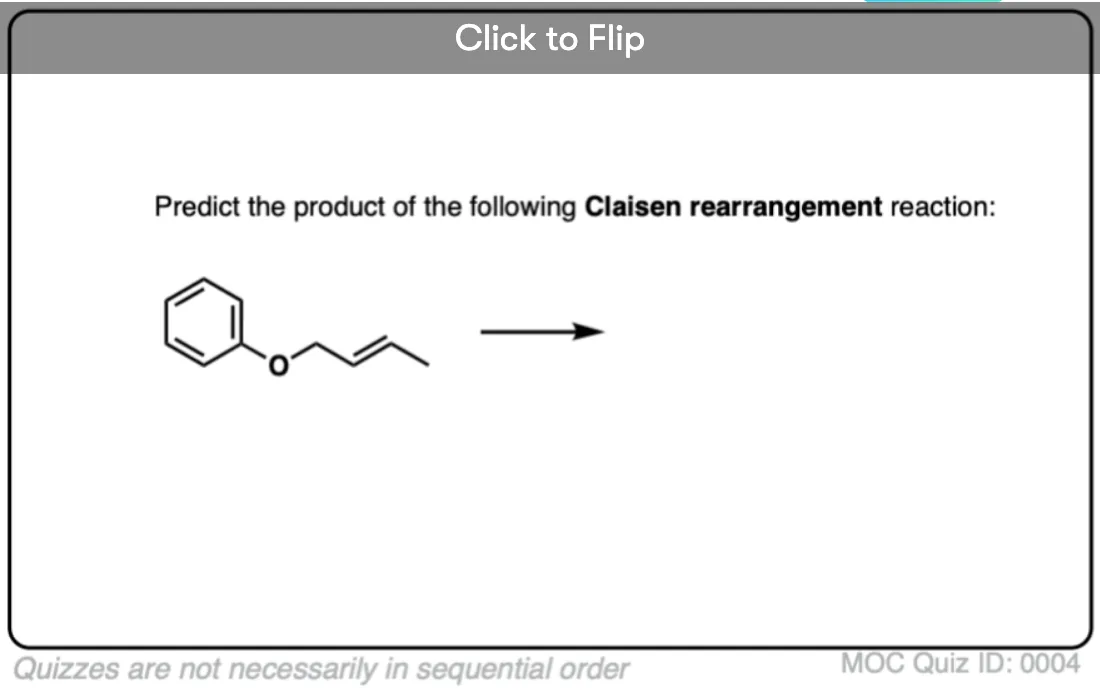
Become a MOC member to see the clickable quiz with answers on the back.
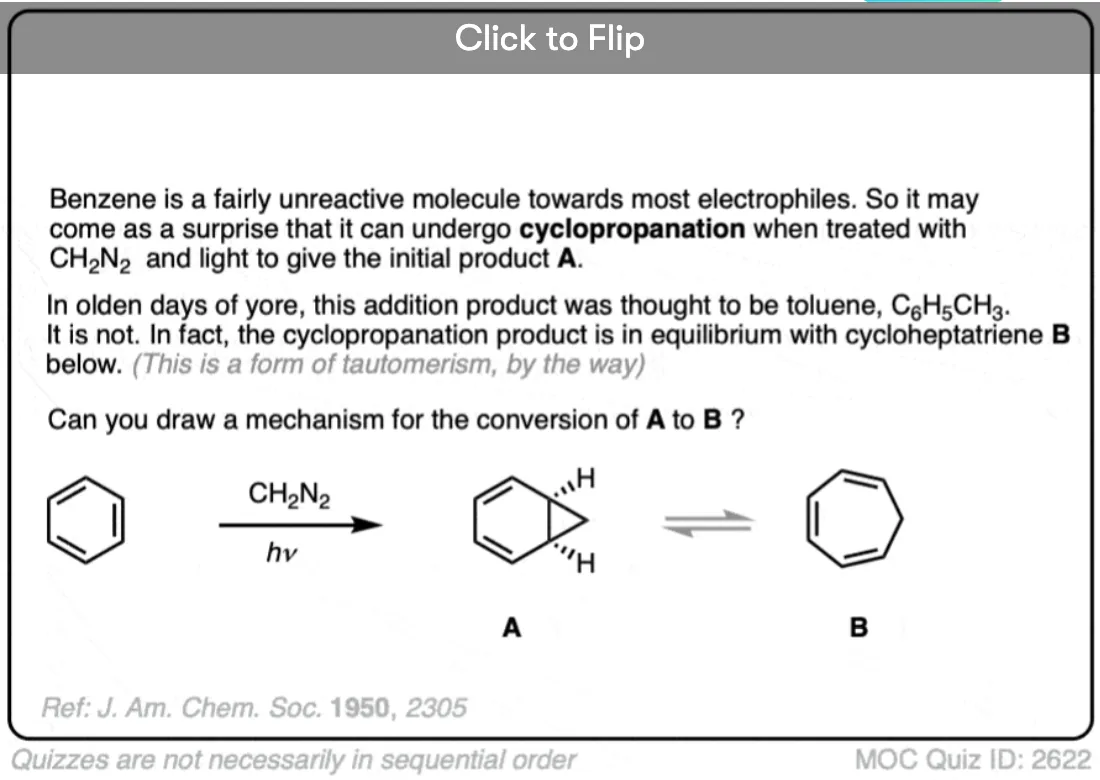
Become a MOC member to see the clickable quiz with answers on the back.
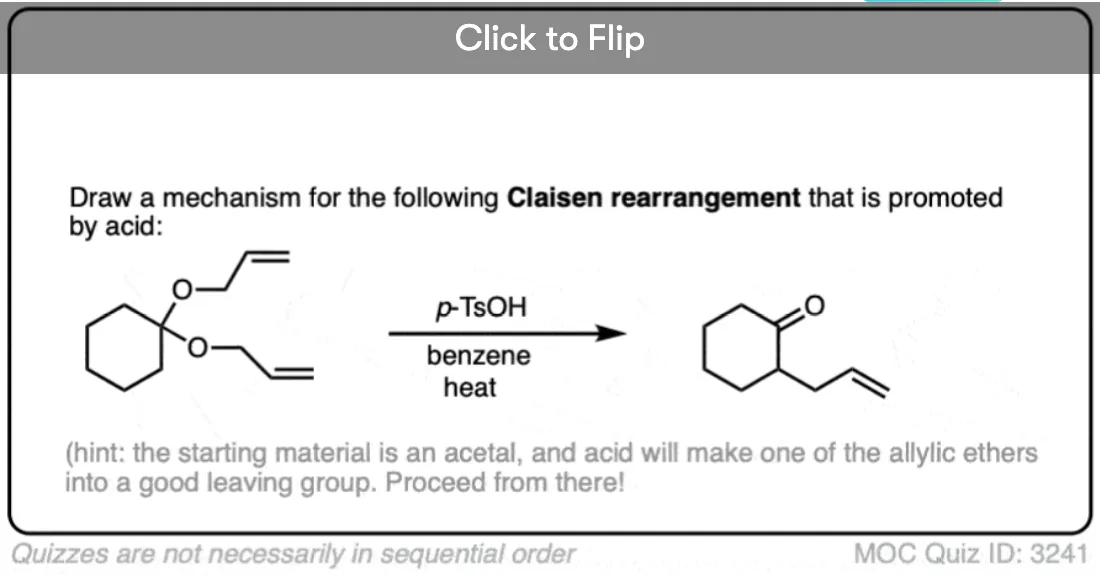
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) Orbital Symmetry In The Cope and Claisen Rearrangements
Being pericyclic reactions, and therefore relatives of the Diels-Alder reaction, you’d figure that orbital symmetry had to appear somewhere to allow us to rationalize why the Cope and Claisen rearrangements are “thermally allowed”.
The easiest way to come up with the orbitals for these reactions is to imagine them as two allyl fragments combining with each other. If we think back to the HOMO of the allyl radical (we talked about it here) the HOMO is the molecular orbital where the terminal lobes are of opposite phase and the center is a weird-looking dot (that’s because a node passes through the central carbon).
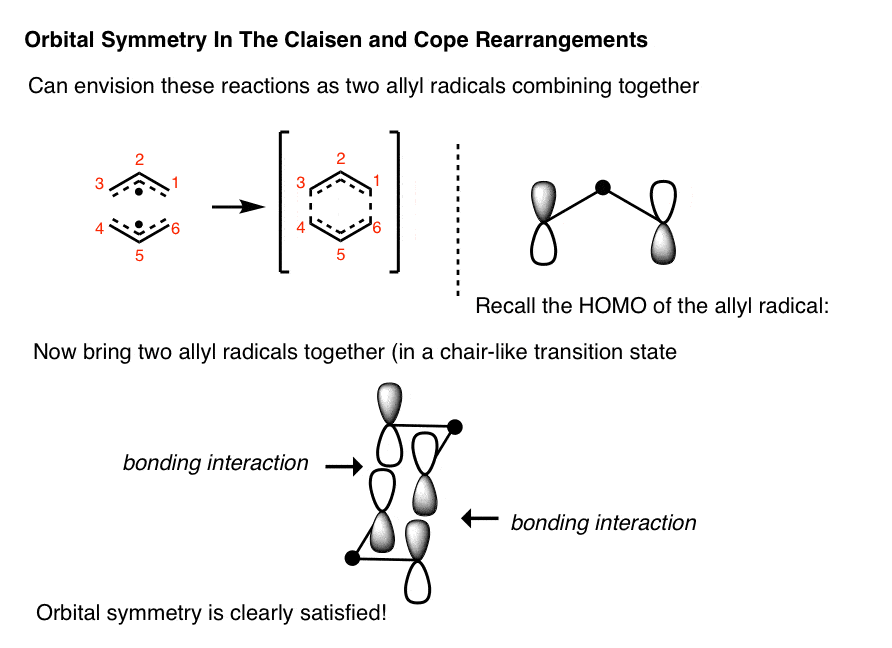
If you combine two allyl radicals this way the orbitals line up nicely. We didn’t go into details about the geometry of the Cope transition state, but it tends to be a chair-like species, which is why it’s been drawn this way.
We have kept things really simple here. The mechanism of the Cope is still being probed, and there are pathways where the transition state really does resemble the combination of two allyl radicals!
(Advanced) Application Of The Oxy Cope In Synthesis: Cockroach Sex Pheromone
As mentioned above, David Evans at Harvard University and his collaborator A. Golob reported in 1975 that oxy-Cope rearrangements were significantly accelerated if the hydroxyl group was deprotonated under conditions where the alkoxide ion was weakly coordinated with the resulting counterion. This has led to many exciting natural product synthesis adventures.
One fairly straightforward way to set this up is to add a vinyl Grignard reagent to a beta, gamma unsaturated ketone, followed by mild heating. In the case below, 60°C was sufficient!
This synthesis of the cockroach pheromone periplanone B by Prof. Stuart L. Schreiber (then of Yale, now Harvard) and graduate student Conrad Santini is a master class in the Woodward-Hoffman rules.
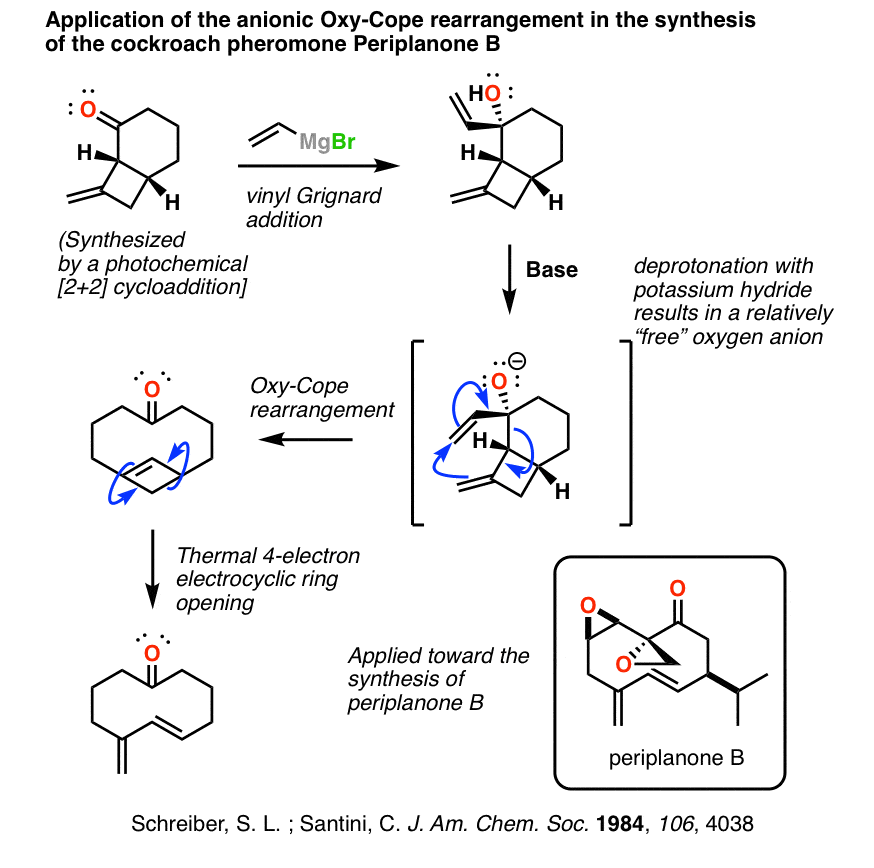
It starts with a photochemical [2+2] cycloaddition to make the bicyclic ring system. Adding a vinyl Grignard sets up the anionic Oxy-Cope [3,3] sigmatropic rearrangement resulting in a highly strained cyclobutene. This then undergoes thermal electrocyclic ring opening to give a diene which is subsequently epoxidized into the cockroach pheromone.
Now for the fun part.
Having synthesized the pheromone, Schreiber and Santini then set about searching for cockroaches in the basement of the Yale chemistry building:

What a feeling that must have been.
(Advanced) References and Further Reading
Lots of references, just because both of these reactions are truly interesting and useful.
- The Introduction of Substituted Vinyl Groups. V. A Rearrangement Involving the Migration of an Allyl Group in a Three-Carbon System
Arthur C. Cope and Elizabeth M. Hardy
Journal of the American Chemical Society 1940, 62 (2), 441-444
DOI: 10.1021/ja01859a055
The original report on the reaction that came to be known as the Cope Rearrangement. The Cope Rearrangements and its variants (e.g. the oxy-Cope and anionic oxy-Cope rearrangements) are [3,3]-sigmatropic rearrangements. It is a concerted reaction and is an equilibrium process. This is named after Prof. Arthur Clay Cope, who was a professor at MIT. The American Chemical Society has named prestigious awards, the Arthur Cope and Cope Scholar awards, after him. - The overlap of two allyl radicals or a four-centered transition state in the Cope Rearrangement
W. von E. Doering, W. R Roth
Tetrahedron, 1962, 18, 67-74
DOI: 10.1016/0040-4020(62)80025-8
In this classic study, von Doering and Roth proposed a six-membered ring chair-like transition state of the Cope rearrangement based on the rearrangements of diastereomeric precursors.The following papers are on the Oxy-Cope rearrangement. This variant provides a thermodynamic sink, since the rearrangement results in an enol, which tautomerizes to the ketone/aldehyde, providing a thermodynamic driving force. - A Synthesis of Ketones by the Thermal Isomerization of 3-Hydroxy-1,5-hexadienes. The Oxy-Cope Rearrangement
Jerome A. Berson and Maitland Jones
Journal of the American Chemical Society 1964, 86 (22), 5019-5020
DOI: 10.1021/ja01076a067
This is the first paper describing the rearrangement, and is the genesis of the term “Oxy-Cope”, as proposed by the authors here. - Stepwise Mechanisms in the Oxy-Cope Rearrangement
Jerome A. Berson and Maitland. Jones
Journal of the American Chemical Society 1964, 86 (22), 5017-5018
DOI: 10.1021/ja01076a066
A variant of the Oxy-Cope is the Anionic Oxy-Cope rearrangement, which uses a base to generate an alkoxide, which generates an enolate ion after rearrangement, allowing significantly lower temperatures compared to traditional Cope or Oxy-Cope reactions. - [3,3] Sigmatropic rearrangements of 1,5-diene alkoxides. Powerful accelerating effects of the alkoxide substituent
D. A. Evans and A. M. Golob
Journal of the American Chemical Society 1975, 97 (16), 4765-4766
DOI: 10.1021/ja00849a054
The first publication on the anionic Oxy-Cope rearrangement. Seminal. - Macroexpansion methodology : Medium ring synthesis based on an eight unit ring expansion process
Paul A. Wender, Scott McN. Sieburth, Joseph J. Petraitis, Sunil K. Singh
Tetrahedron 1981, 37 (23), 3967-3975
DOI: 10.1016/S0040-4020(01)93271-5
An application of the anionic Oxy-Cope rearrangement in the synthesis of macrocycles, from the lab of Prof. Paul A. Wender (now at Stanford). - Über Umlagerung von Phenol‐allyläthern in C‐Allyl‐phenole
L. Claisen
Chem. Ber. 1912, 45 (3), 3157-3166
DOI: 10.1002/cber.19120450348
The first report on the [3,3] thermal rearrangement of allyl phenyl ethers, which subsequently became named the Claisen Rearrangement after its discover, Ludwig Claisen. - The Rearrangement of Vinyl Allyl Ethers
Charles D. Hurd and Maxwell A. PollackJournal of the American Chemical Society 1938 60 (8), 1905-1911
DOI: 10.1021/ja01275a051
Using thermochemical measurements, the rearrangement of allyl vinyl ether to give allylacetaldehyde was found to be favorable by about 24 kcal/mol (100.6 kJ/mol) in this study. - The Mechanism of the Claisen Rearrangement: Déjà Vu All Over Again
Bruce Ganem
Angew. Chem. Int. Ed. 1996, 35 (9), 936-945
DOI: 10.1021/anie.199609361
A review on the mechanism of the Claisen Rearrangement, covering solvent effects, catalysis, stereoelectronic effects, and ending with enzymatic catalysis. - Claisen Rearrangement over the Past Nine Decades
Ana M. Martín Castro
Chemical Reviews 2004, 104 (6), 2939-3002
DOI: 10.1021/cr020703u
A comprehensive review on the Claisen rearrangement, with plenty of examples demonstrating the substrate scope and applications of this reaction in chemistry. - 5β-CHOLEST-3-ENE-5-ACETALDEHYDE
R. E. Ireland and D. J. Dawson
Org. Synth. 1974, 54, 71
DOI: 10.15227/orgsyn.054.0071
An example of a procedure involving tandem Claisen rearrangements. This is from Organic Syntheses, a reliable source of independently tested organic chemistry procedures.Several variants of the classic Claisen rearrangement have been developed – two of the most popular are below: - Simple stereoselective version of the Claisen rearrangement leading to trans-trisubstituted olefinic bonds. Synthesis of squalene
William Summer Johnson, Lucius Werthemann, William R. Bartlett, Timothy J. Brocksom, Tsung-Tee Li, D. John Faulkner, and Michael R. Petersen
Journal of the American Chemical Society 1970, 92 (3), 741-743
DOI: 10.1021/ja00706a07411
Heating an allylic alcohol with an excess of trialkyl orthoacetate in the presence of trace amounts of a weak acid gives a mixed orthoester. Mechanistically, the orthoester loses alcohol to generate the ketene acetal, which undergoes a [3,3]-sigmatropic rearrangement to give a g,d-unsaturated ester. This is known as the Johnson-Claisen orthoester rearrangement, after its developer, Prof. W. S. Johnson, who built up Stanford’s chemistry department. - Claisen rearrangement of allyl esters
Robert E. Ireland and Richard H. Mueller
Journal of the American Chemical Society 1972, 94 (16), 5897-5898
DOI: 10.1021/ja00771a062
The rearrangement of allyl trimethylsilyl ketene acetals, prepared by the reaction of allylic ester enolates with TMSCl. This is known as the Ireland-Claisen (silyl ketene acetal) rearrangement, after its developer, Prof. Robert Ireland. The advantage with this reaction is that much milder conditions are possible. - Enantioselective Claisen Rearrangements: Development of a First Generation Asymmetric Acyl-Claisen Reaction
Tehshik P. Yoon and and David W. C. MacMillan
Journal of the American Chemical Society 2001, 123 (12), 2911-2912
DOI: 10.1021/ja015612d
This is an example of modern developments in the Claisen rearrangement. This is one of several papers that Prof. MacMillan (Princeton) has published in the area of asymmetric Claisen rearrangements.
Hello James, regarding the first quiz (where the ortho position is blocked), in the first arrow-pushing diagram, the arrow shows the O–C bond breaking and forming a new C=C pi bond. Since oxygen is more electronegative, my textbook, By Brown et al., also shows such arrow pushing. But shouldn’t the electron pair always move from C-C toward oxygen to form a C–O bond instead?
That is a great point. Thanks – on my fix list, will get fixed within 48h.
Thank you James for your concise and easy to understand notes on important topics that seems difficult for most undergrads. You’re doing a great job man.
God bless you with more wisdom,energy and every good thing to continue creating amazing content for us.Stay blessed.
Thanks so much for this publishing this lesson. I have learnt alot.