Reactions of Aromatic Molecules
The Wolff-Kishner, Clemmensen, And Other Carbonyl Reductions
Last updated: May 28th, 2026 |
The Wolff-Kishner, Clemmensen, and Other Fun Reductions
In this post we go through 4 ways of reducing C=O to CH2 including:
- The Wolff-Kishner reaction
- The Clemmensen reduction
- Catalytic hydrogenation
- Thioacetal formation and reduction
But first: why might you want to do this? Well, it’s a key component in a common little synthesis problem I like to call “The Great Friedel-Crafts Workaround”.
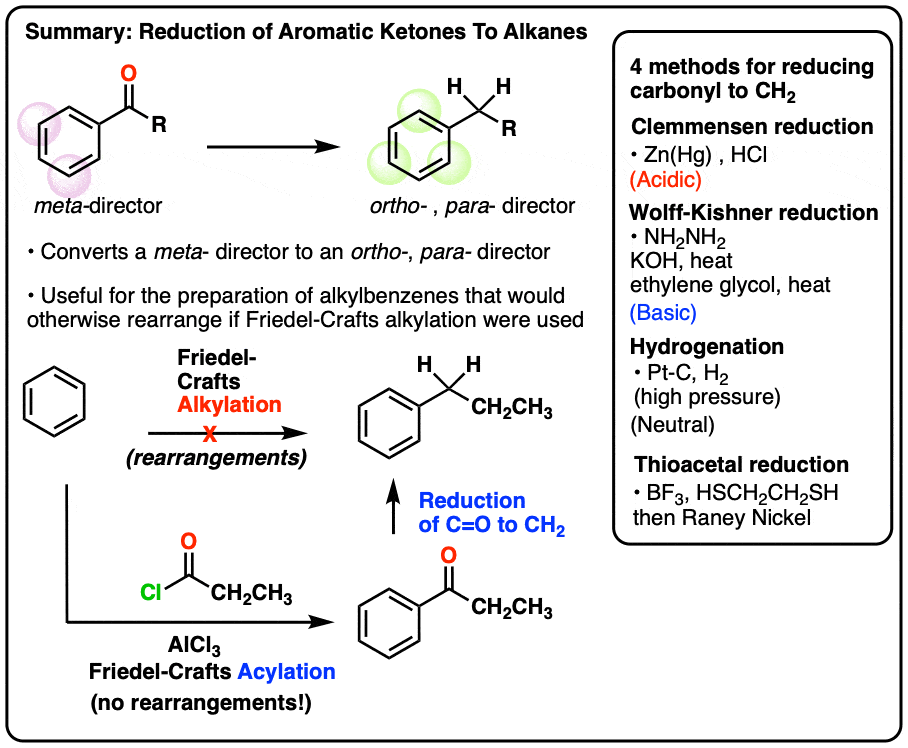
Table of Contents
- The Great Friedel-Crafts Workaround
- The Wolff-Kishner Reduction Of Ketones
- Mechanism Of The Wolff-Kishner Reaction
- The Clemmensen Reduction of Ketones
- Why Would You Prefer The Wolff-Kishner Over the Clemmensen, Or Vice Versa?
- A Third Method For Carbonyl Reduction: Catalytic Hydrogenation
- Reduction of Thioacetals
- A Workaround Example
- A Final Note: Reversing Polarity
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. The Great Friedel-Crafts Workaround
A recent post covered oxidations on the “benzylic” carbon (i.e. on the carbon adjacent to the aromatic ring). We showed that benzylic C-H bonds are unusually weak, and can be converted relatively easily (and selectively!) to C–Br or C–O bonds.
Today we’re going to go in the reverse direction and address reduction of the benzylic carbon, notably reduction of ketones (C=O) to alkyl (CH2).
This is particularly important because of the Great Friedel-Crafts Workaround.
What’s that, you ask?
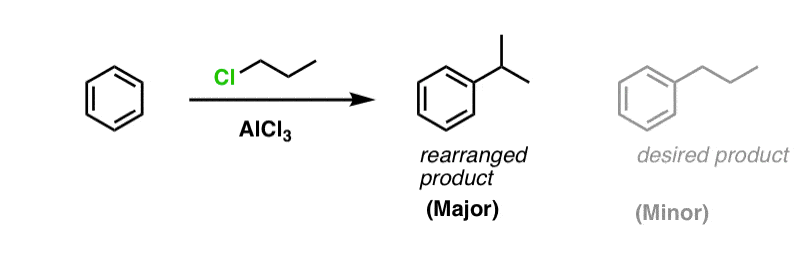
You may recall that Friedel-Crafts alkylation of aromatic rings with primary alkyl halides can result in carbocation rearrangements. For example, attempting a Friedel-Crafts alkylation of benzene with propyl chloride results in isopropylbenzene, not propyl benzene.
The Great Friedel-Crafts Workaround solves this issue. We begin with a Friedel-Crafts acylation, which proceeds without rearrangement, and follow by reducing the ketone down to CH2.
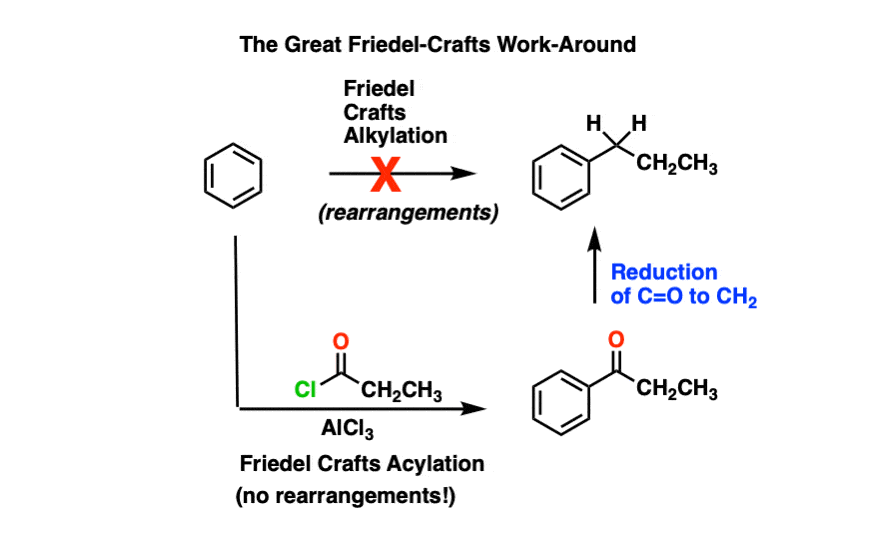
So how can we reduce the ketone down to an alkane? Four ways.
2. The Wolff-Kishner Reduction Of Ketones
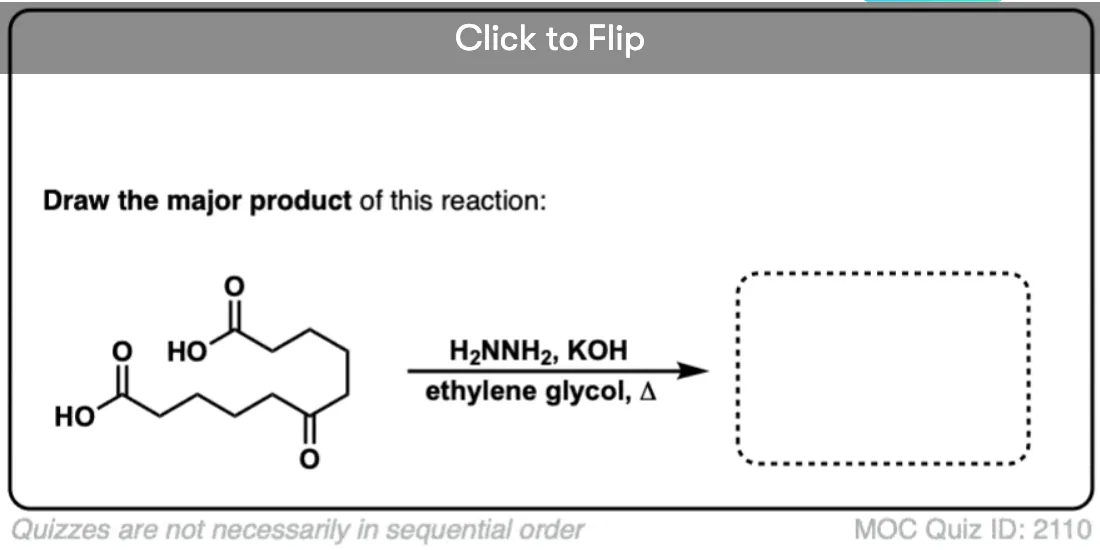
The Wolff Kishner reduction of ketones utilizes hydrazine (NH2NH2) as the reducing agent in the presence of strong base (KOH) in a high-boiling protic solvent (ethylene glycol, HO-CH2CH2-OH, boiling point 197 °C).
The driving force for the reaction is the conversion of hydrazine to nitrogen gas.
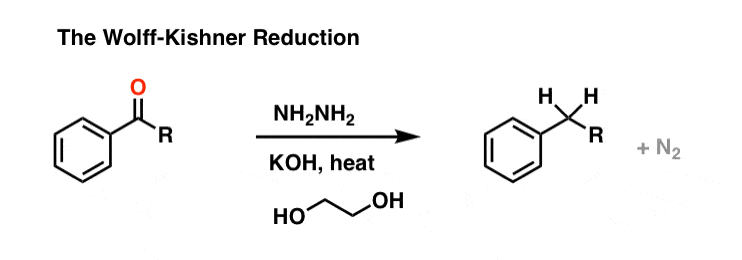
This is not exactly a gentle process; heating to almost 200 °C is required to make the reaction occur at a reasonable rate. [Note 1]
The first step is formation of a hydrazone from the ketone (hydrazones are a cousin of imines, which we cover later in the course). Hydrazine (NH2NH2) adds to the carbonyl, and following a series of proton transfer steps, water is expelled. Click here to see an image of the mechanism for hydrazone formation. (link to image)
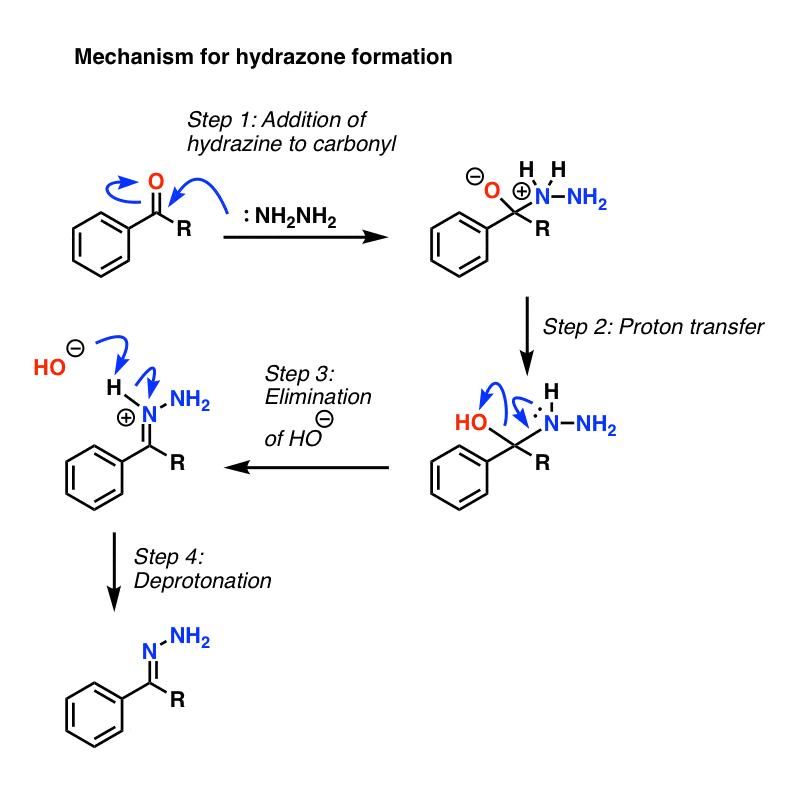{kind=link}
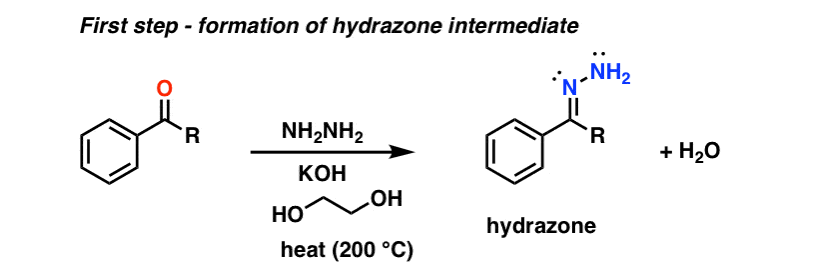
Once the hydrazone is formed, the real action in the Wolff-Kishner begins!
3. Mechanism Of The Wolff-Kishner Reaction
The NH2 of the hydrazone is reasonably acidic (pKa about 21) and can be deprotonated by strong base at a high enough temperature (the base is likely the conjugate base of ethylene glycol, not KOH). This deprotonation appears to be the rate-limiting step.
The next step is the trickiest: protonation on the carbon. With the caveat that resonance forms don’t really exist, it can be helpful to imagine forming the resonance form of this species that has a negative charge on the carbon, and then protonating it with solvent (ethylene glycol).
This gives a species with a nitrogen-nitrogen double bond , which , after deprotonation by base, decomposes irreversibly to give nitrogen gas and a carbanion (i.e. a negatively charged carbon).
Protonation of the carbon completes the process.
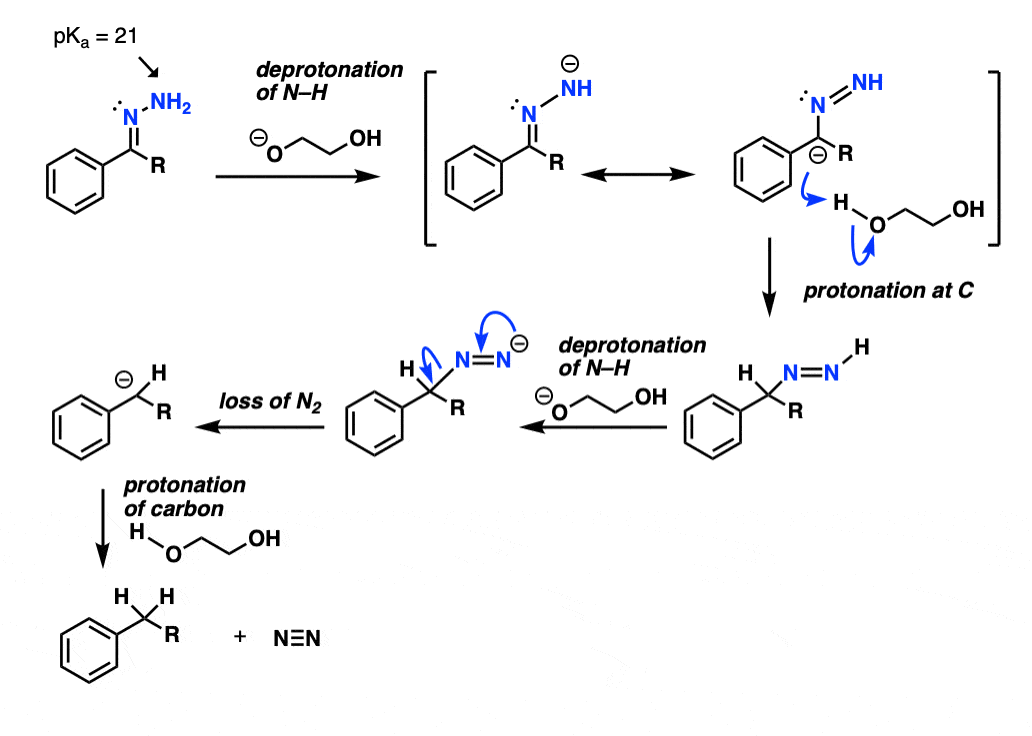
Hover for full arrow-pushing mechanism or click on this link.
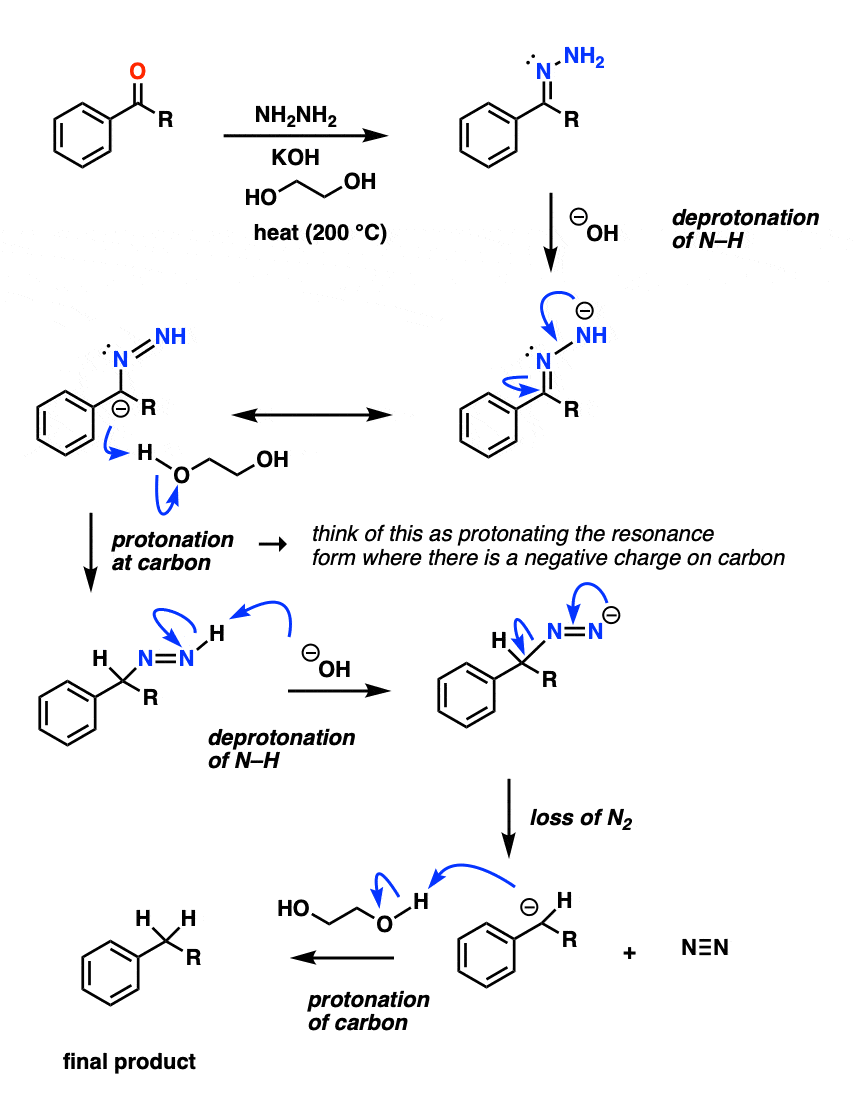{kind=link}
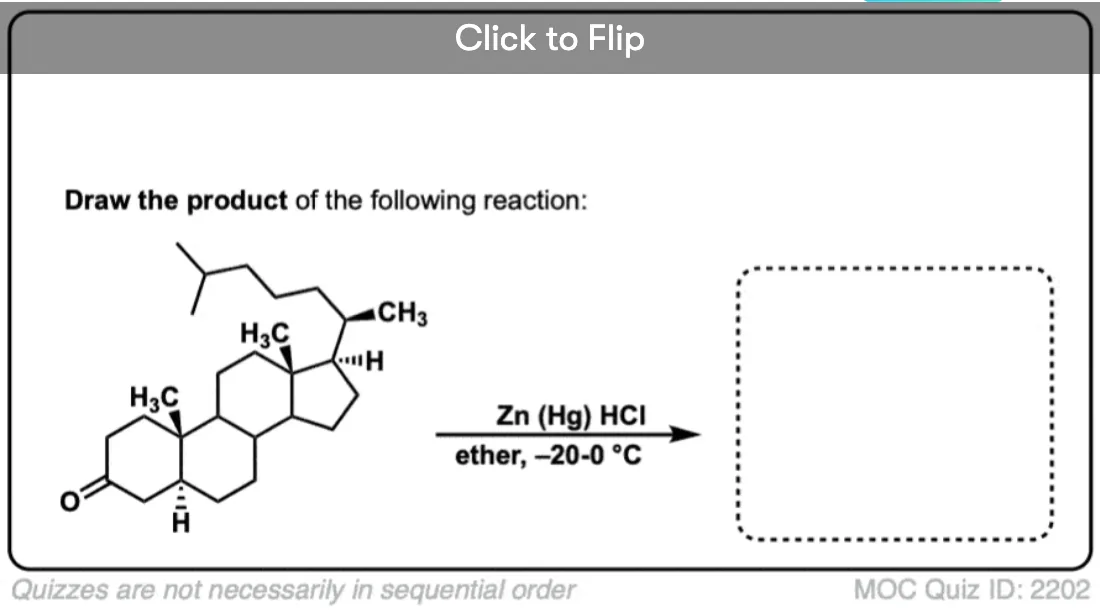
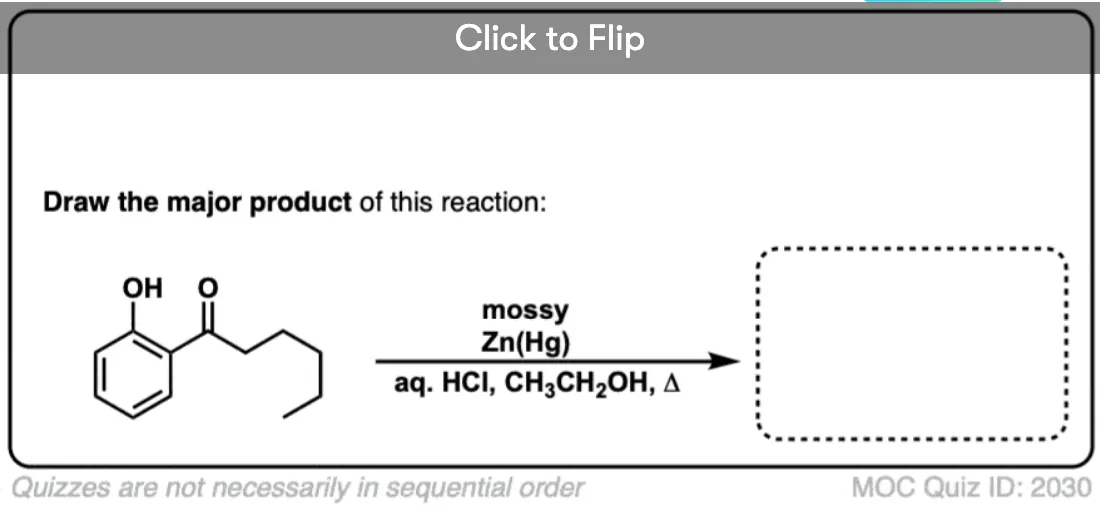
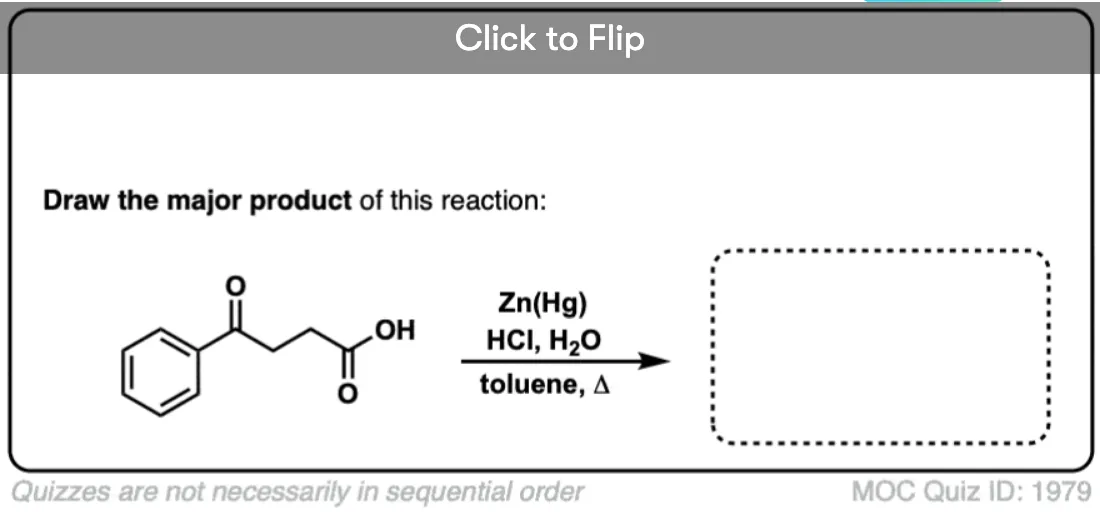
4. The Clemmensen Reduction of Ketones
A second way to go about reducing the carbonyl of an aromatic ketone is to use a reaction known as the Clemmensen Reduction. The reductant here is “zinc amalgam” (Zn-Hg) which is used under acidic conditions; one method calls for the presence of aqueous HCl, for example:
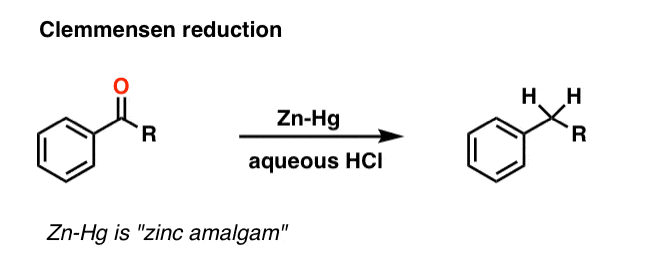
This process works best for aromatic ketones; non-aromatic ketones, not so much. The mechanism has not been thoroughly worked out; it’s thought to occur through a series of one-electron transfers from zinc amalgam.
5. Why Would You Prefer The Wolff-Kishner Over the Clemmensen, Or Vice Versa?
It’s somewhat rare to encounter conditions in an introductory class where a Wolff Kishner would be called for over a Clemmensen, or vice versa, but here are some things to think about.
- The Wolff-Kishner is done under strongly basic conditions using high heat in a polar protic solvent.
- The Clemmensen is performed in strongly acidic conditions. If you have a protecting group somewhere which can be removed with acid, such as an acetal or silyl ether, consider an alternative.
Two other methods deserve mention, although you might not seen them covered until later in the course when ketone chemistry is addressed.
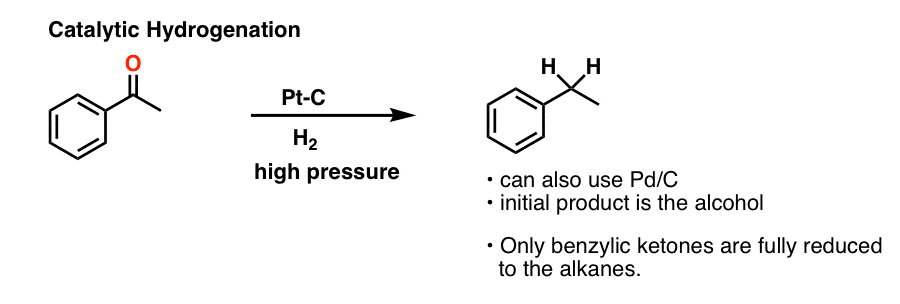
6. A Third Method For Carbonyl Reduction: Catalytic Hydrogenation
The first is catalytic hydrogenation, i.e. using a metal catalyst such as Pd/C or Pt/C with hydrogen gas (H2).
We’ve mostly seen catalytic hydrogenation used for reducing alkenes and alkynes, but it can also be used for ketones if you crank on it enough (i.e. higher temperatures, with higher pressure of H2).
One subtle tweak in conditions, sometimes not mentioned, is that platinum on carbon (Pt-C) or platinum oxide (PtO2) is often used instead of palladium (Pd).
Normally, reduction of ketones usually stops at the alcohol stage. However, in the case where the alcohol is on a benzylic position, (i.e. on a carbon adjacent to an aromatic ring) reduction with can occur further to the alkane (recall that bonds at benzylic positions tend to be easier to break, since the adjacent aromatic ring can donate electron density to them).
7. Reduction of Thioacetals
The second method that sees use is conversion of the ketone to a “thioacetal” with HS-CH2CH2-SH and a Lewis acid such as BF3 . [Note 2] This is followed by treating the thioacetal with a reducing agent known as Raney Nickel: it’s a form of finely divided nickel containing adsorbed hydrogen that cleaves C-S bonds to give C-H bonds, through a somewhat mysterious process also thought to involve free-radicals. [See: Reagents – Raney Nickel]
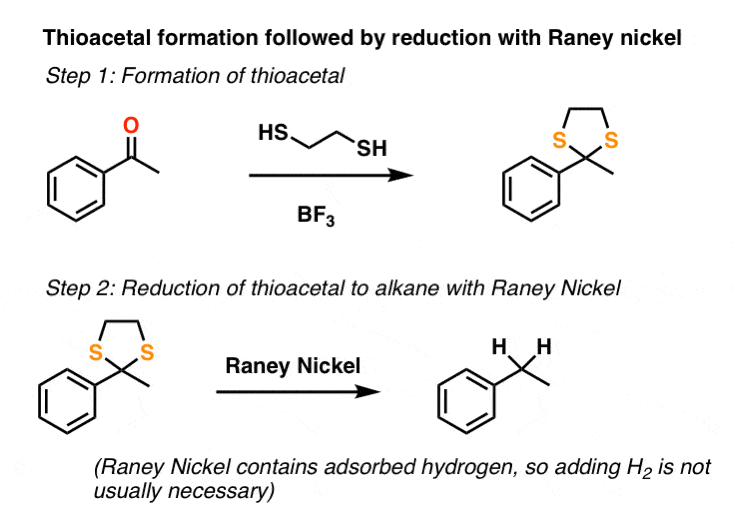
Unlike the Clemmensen and catalytic hydrogenation, thioacetal/Raney nickel method isn’t limited merely to benzylic ketones. It can be used to completely “disappear” an aldehyde or ketone, as was part of the strategy in Woodward’s synthesis of erythromycin.
8. A Workaround Example
With these methods in our toolbox, we can now fill in the vague description “reduction” over the arrow with something a lot more specific.
Here’s a concrete example of a Friedel-Crafts Workaround:
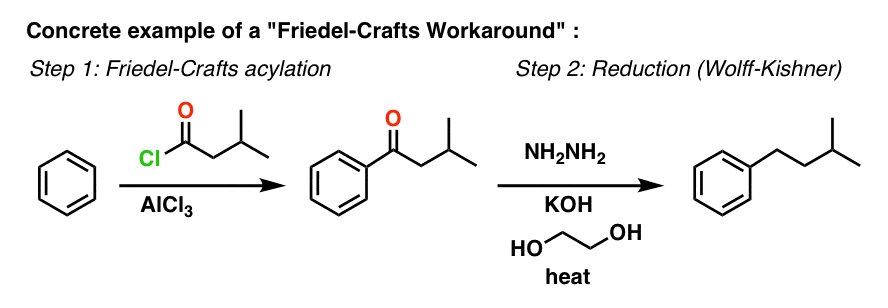
9. A Final Note: Reversing Polarity
A final thing to note here is that reduction of a benzylic ketone to the alkane reverses the polarity of the substituent. It converts an electron-withdrawing meta-director (an acyl group) into an electron-donating ortho-, para- director. We’ll have more to say about this when we address synthesis in aromatic compounds, but just take a gander at these two examples….
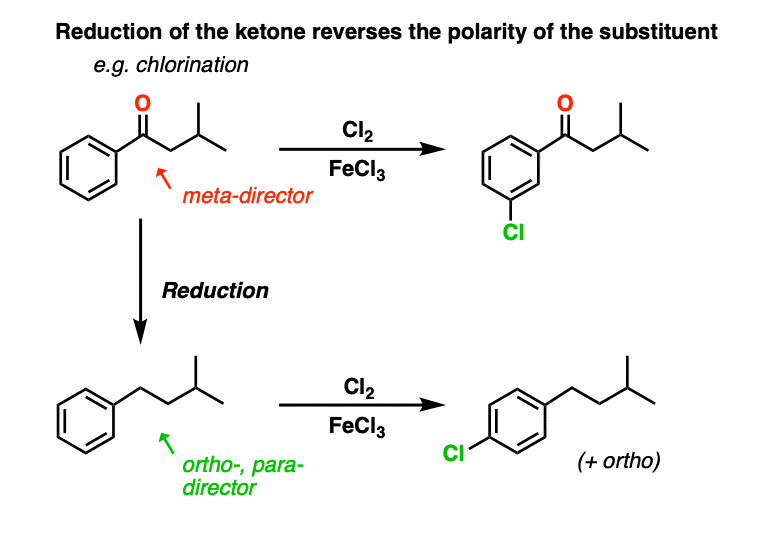
Two more examples of reversing the polarity of a substituent is reduction of the nitro group and Baeyer-Villiger oxidation of a ketone to an ester. We’ll cover those next!
Notes
Note 1. One way of getting around the requirement for high heat in the Wolff-Kishner is to use a strong base like t-BuOK in DMSO, which can be done around room temperature. [Ref: J. Am. Chem. Soc, 1962, 84, 1734-1735.]
Note 2. , Lewis acids such as BF3 or ZnCl2 are commonly used for this reaction [see this Org Syn reference] although in practice, protic acids such as HCl are perfectly fine if the starting material is an aldehyde or ketone [see this Org Syn reference]
In Corey and Seebach’s method for making 1,3-dithiane, the starting materials are 1,3-propanedithiol and dimethoxymethane (CH3OCH2OCH3), an equivalent of formaldehyde. BF3, a strong Lewis acid, gave better yields in this procedure than did anhydrous acid, subsequently BF3 seems to be the Lewis acid of choice in organic chemistry textbooks despite the fact that most thioacetal formation reactions seen in introductory courses are from aldehydes or ketones, not acetals.
Quiz Yourself!
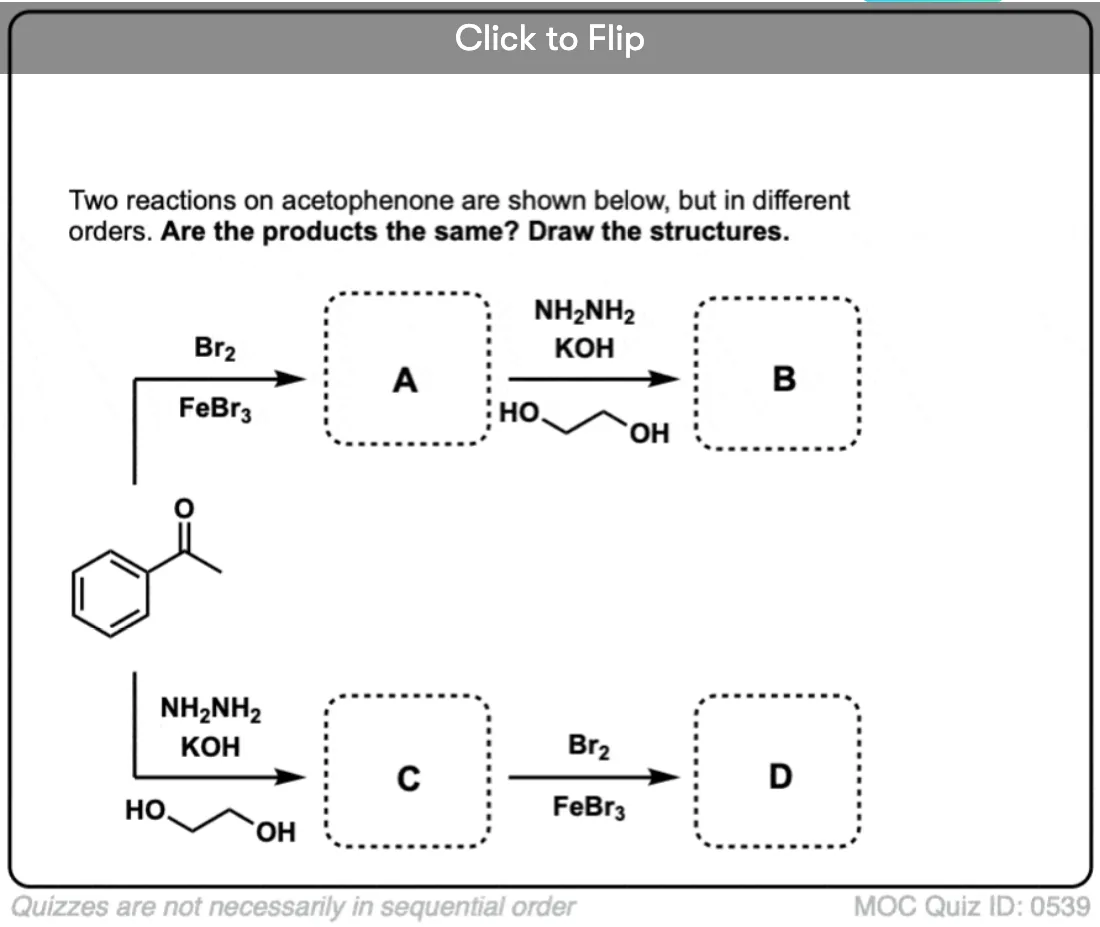
Become a MOC member to see the clickable quiz with answers on the back.
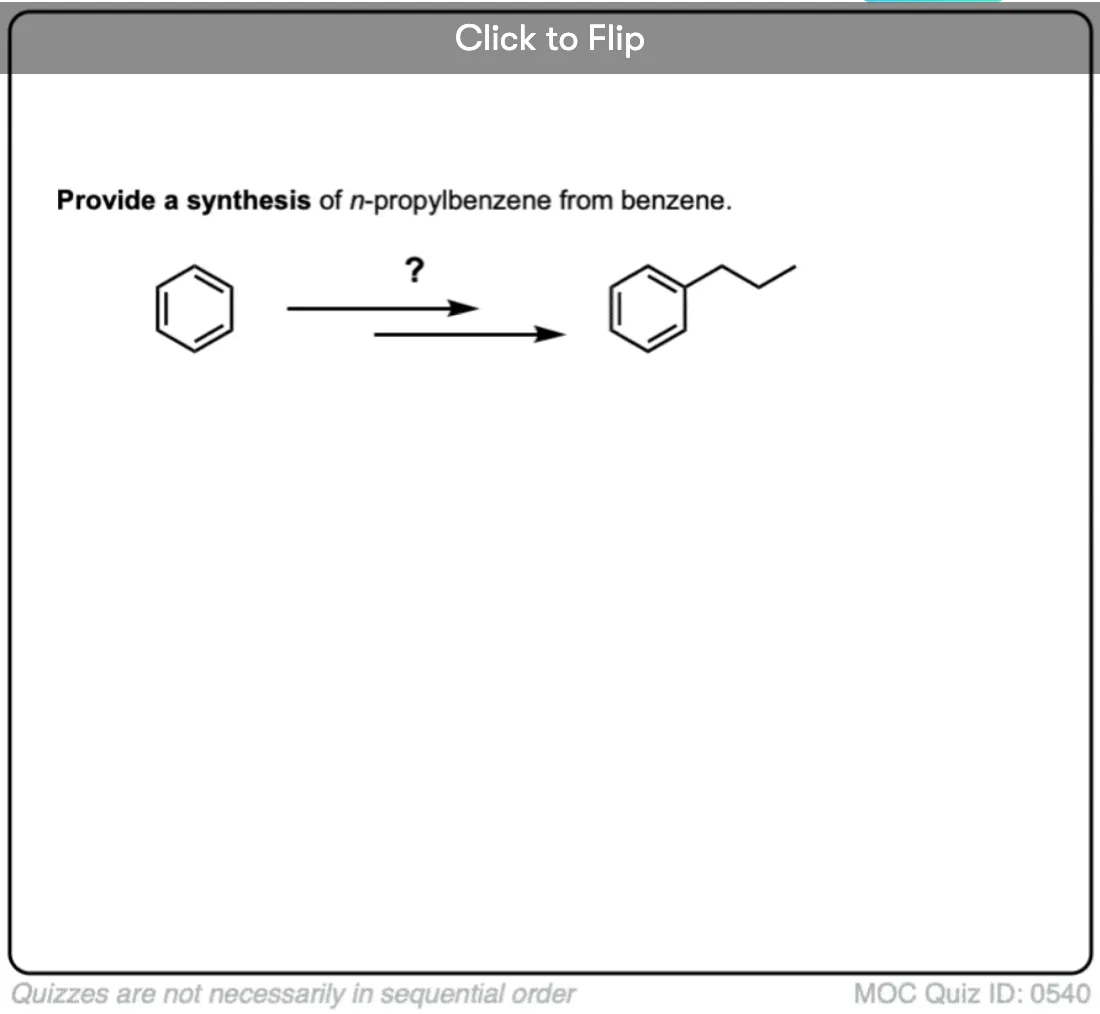
Become a MOC member to see the clickable quiz with answers on the back.
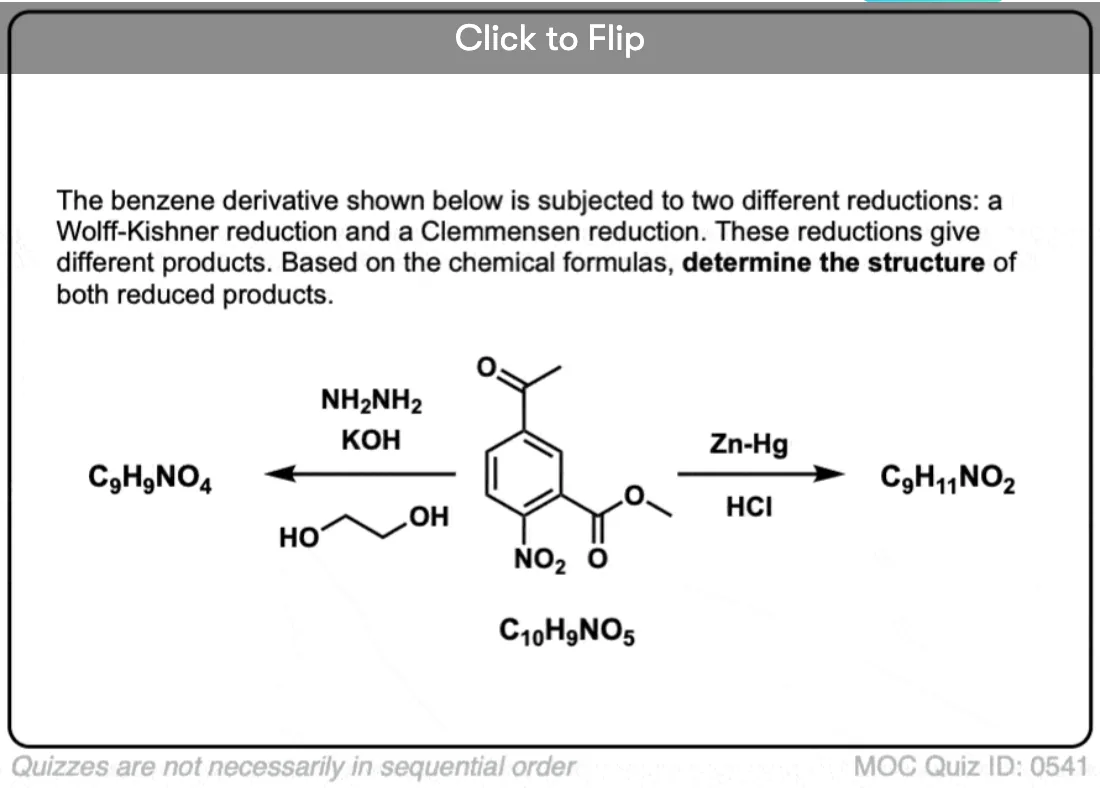
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- Chemischen Institut der Universität Jena: Methode zum Ersatz des Sauerstoffatoms der Ketone und Aldehyde durch Wasserstoff. [Erste Abhandlung.]
Wolff, L.
Lieb. Ann. Chem. 1912 394 (1), 86
DOI: 10.1002/jlac.19123940107
The original paper by Ludwig Wolff on the reduction of aldehydes and ketones with hydrazine. - The Wolff-Kishner Reaction of Hydrazones
Szmant, H. H.; Harmuth, C. H.
J. Am. Chem. Soc.1964, 86 (14), 2909
DOI: 10.1021/ja01068a028
A very nice Physical Organic study on the decomposition of hydrazones, involving Hammett plots (a classic tool in Physical Organic Chemistry), determining the relationship between the electron density of the carbonyl carbon and the mechanism. - Reduction of Steroid Ketones and other Carbonyl Compounds by Modified Wolff-Kishner Method
Huang-Minlon
J. Am. Chem. Soc.1949, 71 (10), 3301
DOI: 10.1021/ja01178a008
A one-pot modification for the Wolff-Kishner reaction that involves distilling off the excess water and hydrazine before heating the hydrazine. This results improved yields and significantly shorter reaction times. - REDUCTION OF KETONES BY USE OF THE TOSYLHYDRAZONE DERIVATIVES: ANDROSTAN-17 β-OL
Caglioti
Org. Synth. 1972,52, 122
DOI: 10.15227/orgsyn.052.0122
Due to the harsh conditions involved in the standard Wolff-Kishner reduction, a number of variations have been developed with milder conditions more amenable for organic synthesis. The use of tosylhydrazide instead of hydrazine allows much gentler reaction conditions – the tosylhydrazone can be reduced with NaBH4 in refluxing methanol (68 °C vs. 200 °C!). - γ-PHENYLBUTYRIC ACID
L. Martin
Org. Synth. 1935, 15, 64
DOI: 10.15227/orgsyn.015.0064
A reliable, tested procedure for a simple Clemmensen Reduction that demonstrates how this reaction can be used to reduce a ketone in the presence of a carboxylic acid. - Elrik Clemmensen: Reduktion von Ketonen und Alahyden su den entspcechenden Kohlenwaaserstoflbn unter Anwendung von amalgamiertem Zink und Salesaure.
Clemmensen, E.
Chem. Ber. 1913 46 (2), 1837-1843
DOI: 10.1002/cber.19130460292
The original paper by Clemmensen on the reduction of aldehydes and ketones with Zn metal in acid. Note that even though this was submitted to a German journal and written in German, Clemmensen was working at Parke-Davis, in the U.S.Two reviews on the Clemmensen Reduction: - THE CLEMMENSEN REDUCTION
Martin, E. L.
Org. React. 1942, 1, 155
DOI: 10.1002/0471264180.or001.07 - CLEMMENSEN REDUCTION OF KETONES IN ANHYDROUS ORGANIC SOLVENTS
Vedejs, E.
Org. React. 1975, 22, 401
DOI: 10.1002/0471264180.or022.03 - Stereoselective total synthesis of (–)-pumiliotoxin C by an aqueous intramolecular acylnitroso Diels–Alder approach
Masaichi Naruse, Sakae Aoyagi and Chihiro Kibayashi
J. Chem. Soc., Perkin Trans. 1, 1996, 1113-1124
DOI: 10.1039/P19960001113
Clemmensen reductions can be used in total synthesis due to the mild conditions – in this case 34 to 35 is a Clemmensen reduction that selectively reduces a ketone in the presence of an amide.
great website for my jee prep . thanks.
Thanks a lot Sir! This website is saving me in my JEE Prep. Finally found a good website that has everything in a really good way.
Sir, another way we have been taught to convert carbonyl to CH2 is by using red phosphorus(it’s a good reducing agent) along with hydroiodic acid. Can you please also include that reaction.
i came across a question where under clemmensen conditions an amide was changed into COOH in presence of ketone group but how is this possible as it is said that it selectively reduces carbonyl group and please tell its mech.
also please tell that if on beta carbon an alkyl group is present will it undergo reduction via elimination forming a double bond between alpha and beta?please tell the mechanism
Might be the aqueous acidic nature of the Clemmensen resulting in cleavage of the amide? Clemmensen not exactly known for its functional group tolerance.
Why doesnt the carbonyl group in the ester not get reduced?
In general esters and other carboxylic acid derivatives are less reactive towards nucleophiles than ketones. Furthermore the OR group of the ester can act as a leaving group, so in the end what could just happen is nucleophilic acyl substitution at the ester.
Why wolf kishner do not reduce cooh ?
Under basic conditions the carboxylic acid is deprotonated to give a carboxylate, and these do not readily undergo addition to the C=O group (unlike ketones)
Please,I would appreciate it alot if the quizzes solution were dropped aswell,I feel it will go a long way in helping out alot of us.
“Quizzes solution were dropped?” can you explain?
Hey, Could you please clarify why don’t it reduce the -COOH group?
The Wolff Kishner does not reduce the COOH because under the basic conditions, a carboxylate is formed and carboxylates COO(-) do not readily undergo nucleophilic attack at the carbonyl carbon.
https://www.masterorganicchemistry.com/2022/09/09/nucleophilic-addition/
I am not 100% sure about the fate of carboxylic acids in the Clemmensen as it is not used very often (in practice) and the last good review on it was written in like 1944.
Hey ! I am a little confused whether Wolf-Kishner Reduction would substitute a Cl attached to a benzene with an OH . Since the Cl – C bond is partially double bond (due to resonance) , is it still possible for hydrazine to replace it ?
Hi Lucy, no it would not. That would be an example of nucleophilic acyl substitution. While it is possible in some cases to replace a Cl on an aromatic ring with OH, it generally requires very strong base and very high temperatures. See here – https://www.masterorganicchemistry.com/2018/09/17/nucleophilic-aromatic-substitution-2-benzyne/
Oh thank you very much for clarifying! I Highly appreciate your quick reply!
Hey my friend! Great stuff and awesome explanation. I have a question!
Is Zinc amalgam and concentrated HCl (the reagent used in clemmensen reduction) stronger reducing agent than LiAlH4?
I think so because LiAlH4 only reduces carbonyl compounds to Alcohols but Zinc amalgam and concentrated HCl can reduce it directly to Alcohols.
I don’t think so.
The Clemmensen really isn’t all that useful, to be honest. The last really good review on it was written in 1944. It is good for ketones adjacent to benzene rings and that’s about it.
The Clemmensen wont reduce esters or amides, for example, whereas LiAlH4 does.
Why does Clemmenson reduction reduce NO2 to NH2 alongside carbonyl compound, but wolf kishner reaction does not?
Zinc amalgam can reduce many functional groups besides ketones, including nitro groups.
Reduction in the wolff-kishner is through formation of a hydrazone; nitro groups do not undergo any reaction with hydrazine in the absence of any metal catalyst.
Sir, does the HCl used in Clemmensen Reduction also add to double bond in the reactant? (If present)
It’s a possibilty. I don’t personally recommend actually using the Clemmensen in the lab if you have any concerns about functional group compatibility.
Hey, would the aromatic ring itself be reduced in catalytic hydrogenation given that high pressure is used?
Thanks.
It’s very possible to reduce the ketone without touching the aromatic ring. No, the aromatic ring is much more resistant to reduction.
Oh- is no a better leaving group then how oh- will leave in mechanism of Wolff Kishner reaction
Yes, generally HO(-) is a poor leaving group gnerally speaking, but it is a weaker base (and better leaving group) than deprotonated nitrogen.
Hey, just a little doubt, does the Wolff-Kishner reduction work for aldehydes a well?
Also does it work on non-aromatic compounds too?
Yes – it does work for aldehydes, best done under slightly modified conditions (DMSO as solvent, KOtBu as base). https://pubs.acs.org/doi/10.1021/ja00868a048
Hey man, great content. Quick question tho: during the Wolff Kischner reaction, you mention that the hydrazone proton has a pKa of 21, and that the base that deprotanates is likely to be the conjugate base of ethylene glycol. How is it then possible? Using pKa values this doesn’t add up as alcohol groups have pKa around 16 (leading to stronger acid).
A good rule of thumb is that if the pKa difference is 8 or less, then the acid-base reaction is energetically accessible enough for the reaction to proceed. Plus with the Wolff-Kishner there’s enormous heat added.