Alkene Reactions
Halogenation of Alkenes and Halohydrin Formation
Last updated: May 29th, 2026 |
Bromination, Chlorination, and Halohydrin Formation from Alkenes
- Alkenes undergo halogenation when treated with Cl2, Br2 and (less commonly) I2 to give vicinal dihalides
- These reactions are stereoselective and give anti-addition products
- The mechanism proceeds through a cyclic halonium ion which undergoes backside attack at carbon by a nucleophile to give the anti-addition product.
- When water or alcohols are used as solvent, halohydrins or haloethers can form. These also give the products of anti addition.
- With unsymmetrical halonium ions, the C-O bond tends to form at the most substituted carbon (“Markovnikov” regioselectivity).
- Dihalides can undergo elimination to give alkynes; halohydrins can be used to form epoxides.
- Overall, these reactions have similar mechanisms to those for oxymercuration and for the opening of epoxides under acidic conditions [See article: Alkene Addition Pattern #2 – The “Three Membered Ring” Pathway]
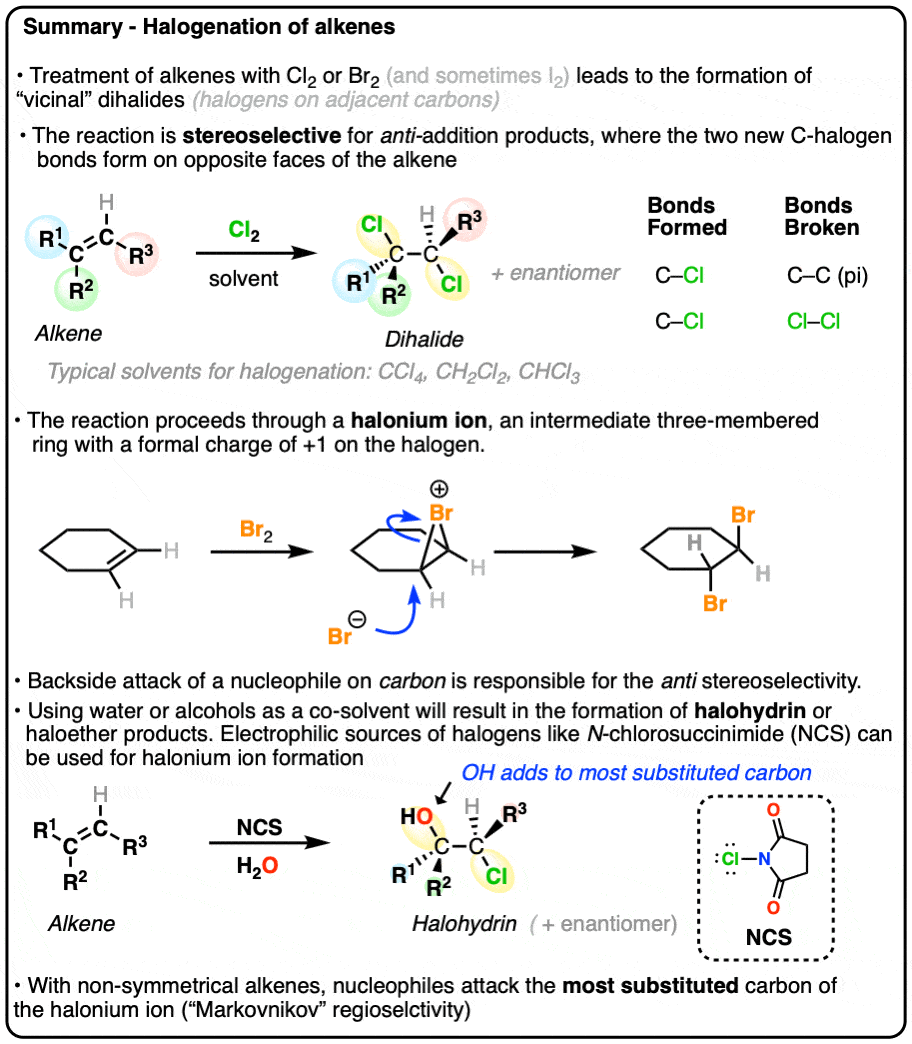
Table of Contents
-
- Bromination and Chlorination of Alkenes With Br2 and Cl2
- Halogenation is Stereoselective for Anti Addition Products
- Halonium Ions
- Halogenation of Alkenes – The Mechanism
- Halohydrin Formation
- Mechanism of Halohydrin Formation
- Haloethers
- Applications of Alkene Halogenation
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Halogenation of Alkenes
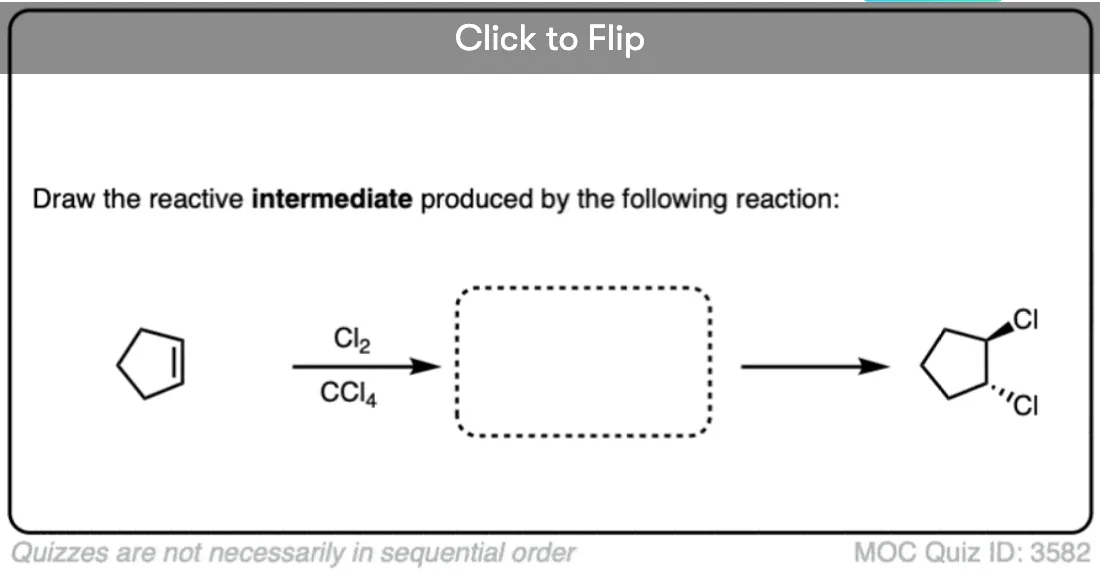
When alkenes (also known as olefins) are treated with bromine (Br2) or chlorine (Cl2) in an inert solvent [Note 1] such as carbon tetrachloride (CCl4) or dichloromethane (CH2Cl2), they are converted into dihalides (specifically, ‘vicinal’ dihalides since the C-halogen bonds are on adjacent carbons).
This reaction results in the formation of two new C-halogen bonds and breaks the C-C pi bond as well as a halogen-halogen bond. The two carbon-halogen bonds add to opposite faces of the alkene.
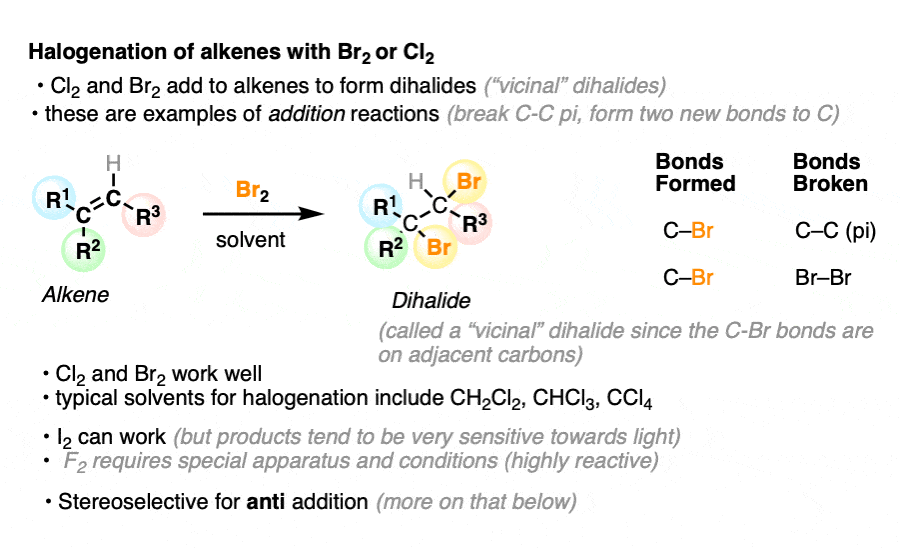
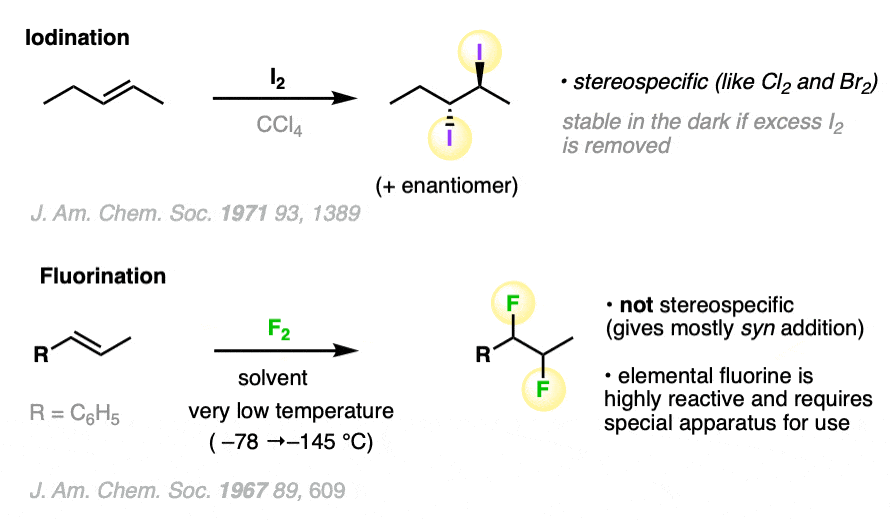
It can also work for iodine (I2) but tends to be reversible, and the di-iodide products tend to be unstable towards light. Fluorine (F2) is such a ravenous beast that it requires special apparatus to work with, which means we won’t discuss it further here. [Note 2]
This reaction is the basis of a common test used in teaching laboratories, the bromine water test.
Molecular bromine (Br2) has a characteristic orange color. In the presence of alkenes, the orange color disappears, indicating that a reaction has occurred.
via GIPHY. Original source: @FranklyChemistry (Youtube)
Note that Br2 does not ordinarily react with benzene (C6H6) or molecules containing an aromatic ring, even though these may appear at first glance to be similar to alkenes. More to say about that in a later chapter. [See article: Halogenation of Benzene]
2. Halogenation Is Stereoselective For anti- Addition Products
Halogenation of alkenes is an example of a stereoselective reaction.
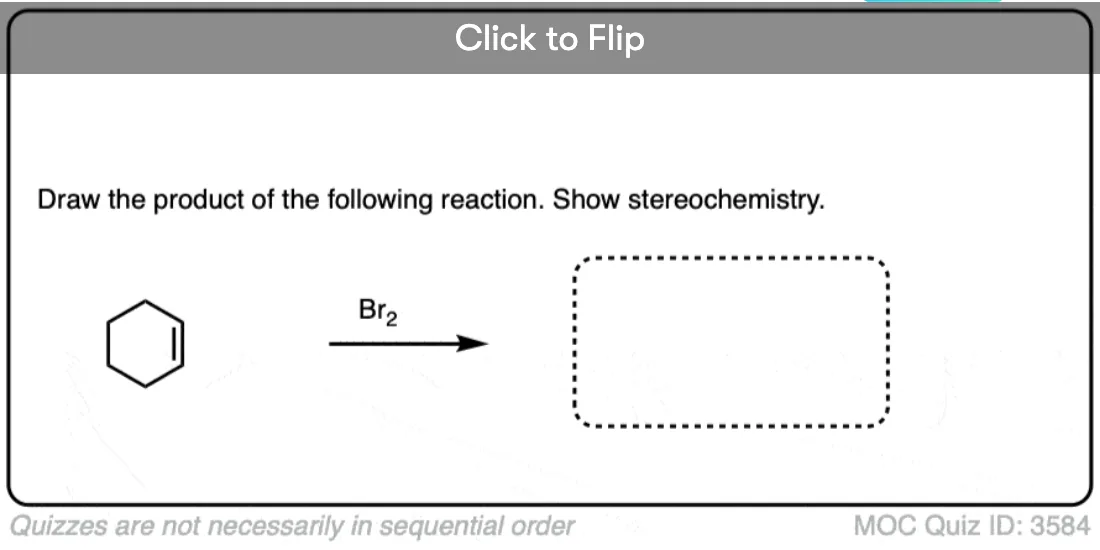
The reaction of Br2, Cl2 and other halogens with alkenes leads to products of anti– addition. A classic example is the bromination of cyclohexene (below), which gives trans-1,2-dibromocyclohexane as a racemic mixture. No cis-1,2-dibromocyclohexane is formed.
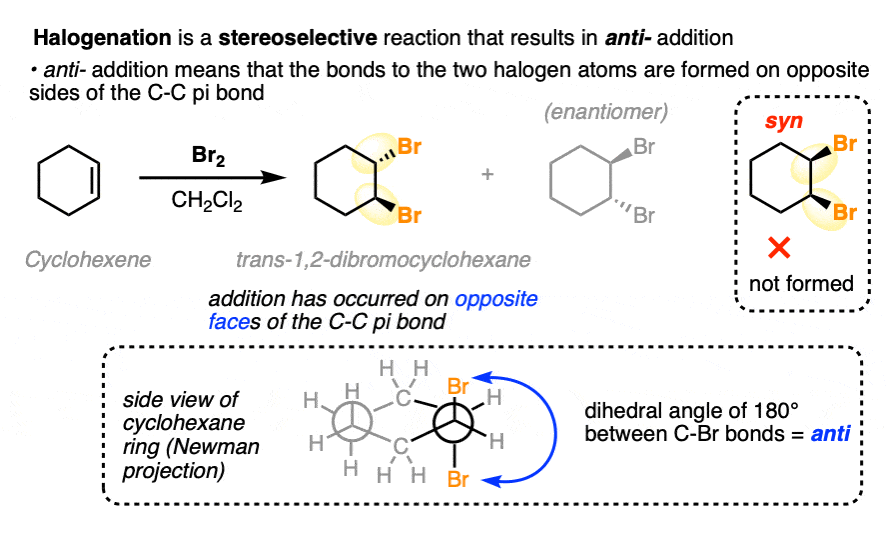
The terms syn– and anti– refer to the dihedral angle observed between the two C-halogen bonds when we look directly along the C-C bond. We say this addition is anti– because the dihedral angle between the two C-halogen bonds is 180°. [See – Staggered vs Eclipsed Conformations of Ethane]
While it’s understandable to use the word “trans” when referring to this orientation (and most people will know what you mean!) we usually reserve “trans” to refer to situations where two groups are on opposite faces of a multiple bond or small ring (i.e. in cis-trans isomerism). The term “anti” is much more broad since it refers to the relative orientation between two groups, as defined by dihedral angle (180° for anti, 0° for syn).
When drawing the products of anti– addition on an alkene, it’s common to draw the two new C-halogen bonds in the plane of the page to clearly show the dihedral angle of 180°.
Pro tip: make sure you keep the relative orientation of the groups on the alkene the same when drawing the product:
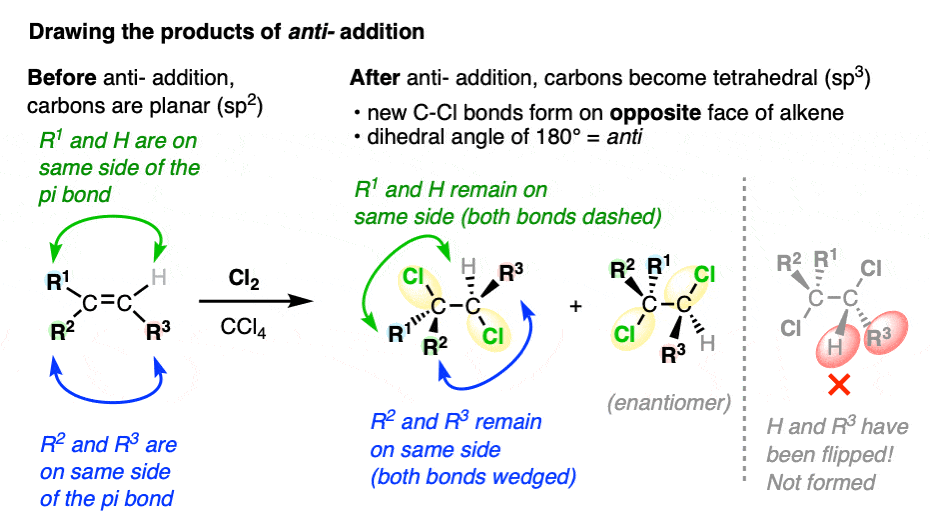
While the products are often drawn with the C-halogen bonds in the plane, be alert that this will not always be the case.
After all, once the C-C pi bond has broken, free rotation may be possible about the C-C single bond. That means it’s possible to draw conformational isomers of an anti– addition product where the two C-halogen bonds are on the same side of the C-C single bond.
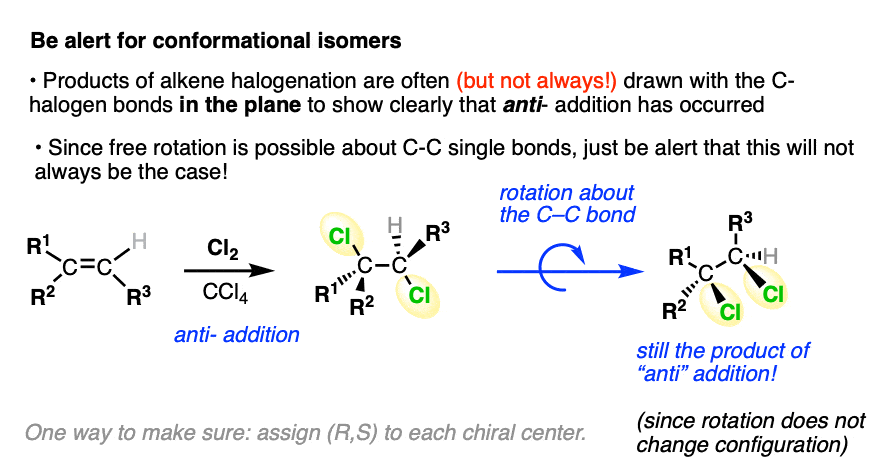
This does not suddenly make it a syn-addition product, however, since rotating bonds does not change their configuration. No amount of bond rotation will convert a chiral center with an (S) configuration into (R) !
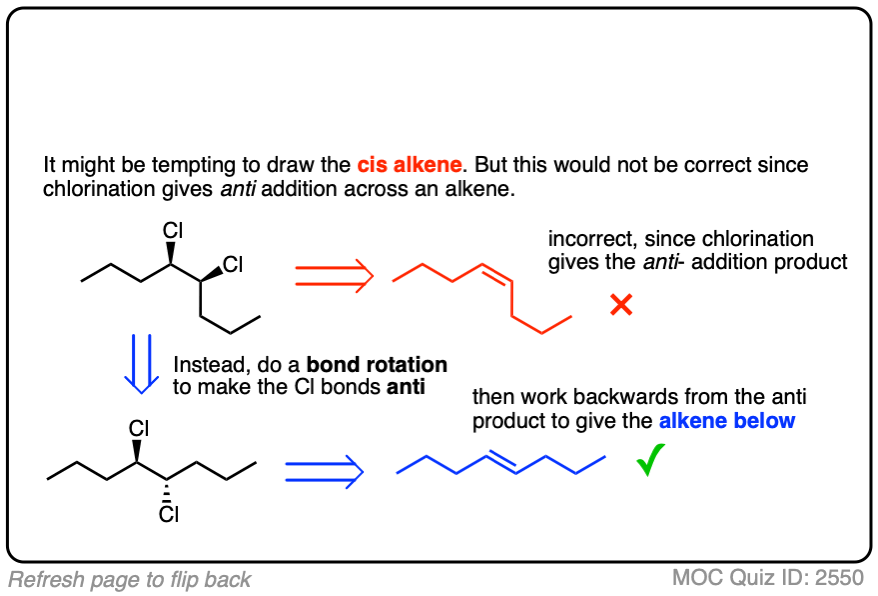
You may have to work backwards sometimes (using bond rotations) from the product to figure out the alkene starting material. This may be a little confusing at first. When in doubt whether you have done a bond rotation correctly, it can be helpful to determine (R)/(S) to make sure you didn’t accidentally flip any stereocenters.
A classic example showing the stereoselectivity of halogenation is cis– and trans-but-2-ene.
- When cis-but-2-ene undergoes bromination, the product is a racemic mixture of (S,S)-2,3-dibromobutane and (R,R)-2,3-dibromobutane.
- When trans-but-2-ene undergoes bromination, the product is (2S, 3R)-2,3-dibromobutane. This molecule has two chiral centers but is an achiral molecule overall due to the presence of an internal mirror plane. This class of molecules are known as meso compounds. (See article: The Meso Trap)
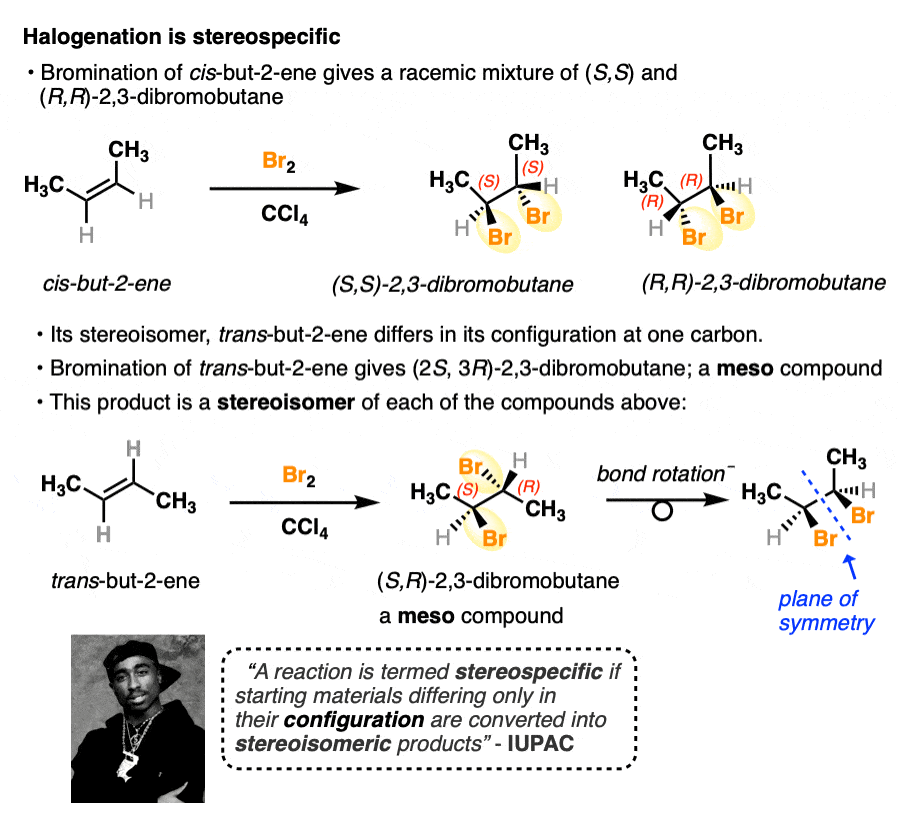
One stereoisomer of but-2-ene produces one set of stereoisomer products; the other stereoisomer of but-2-ene produces a product that is stereoisomeric. There is no crossover between these two reactions.
This fits IUPAC’s definition of a stereospecific reaction. Several other examples of stereospecific reactions of alkenes include hydroboration, dihydroxylation, epoxidation and more.
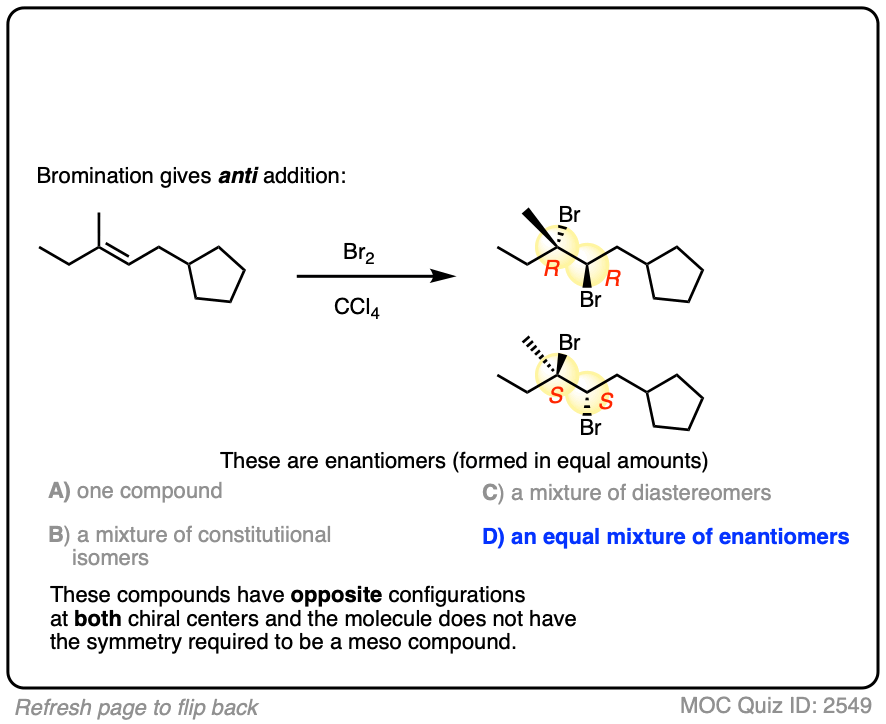
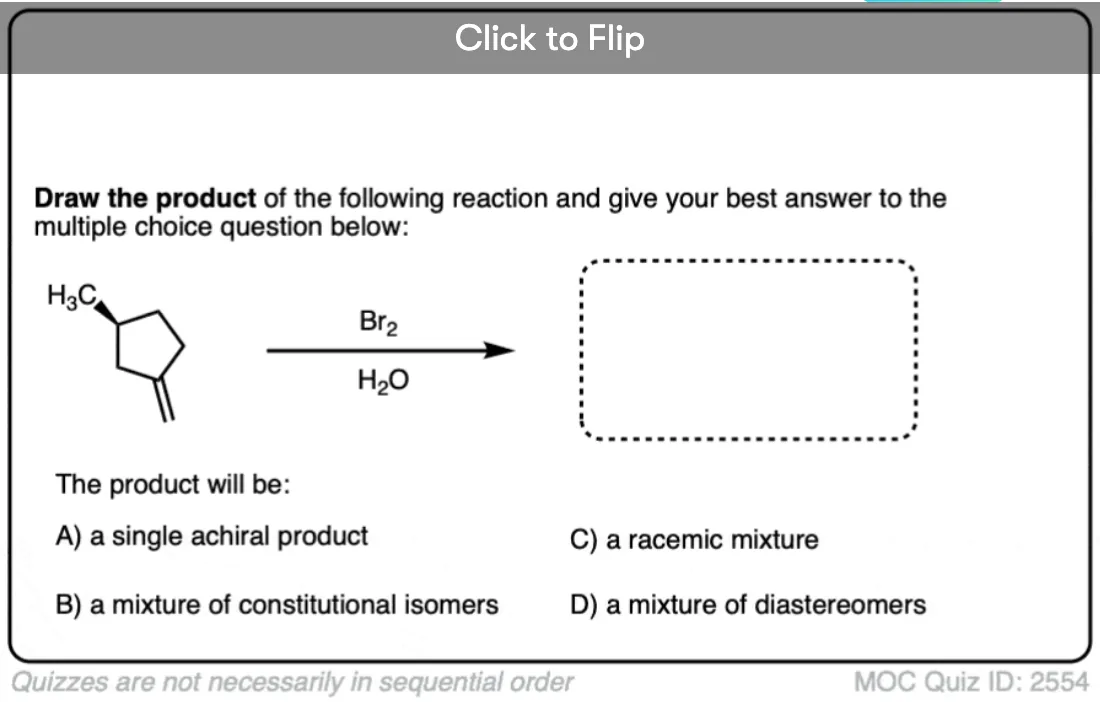
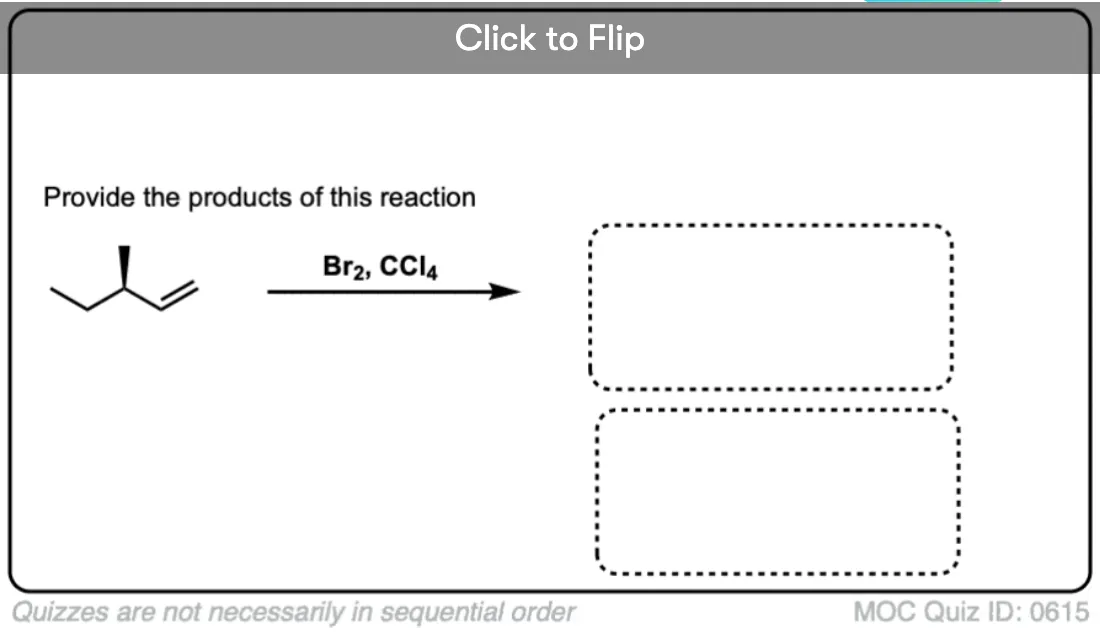
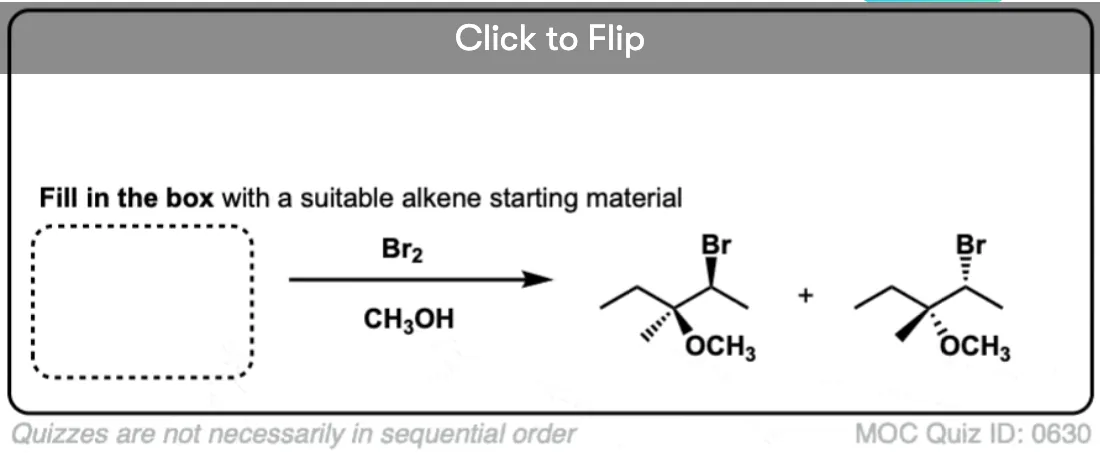
Now for some exercises. See if you can draw the products of the following reaction and determine the products:
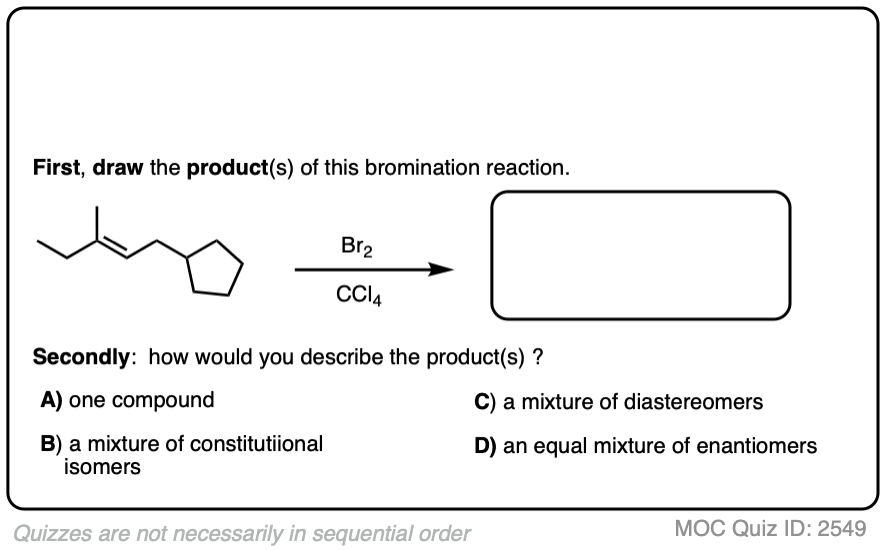 Click to Flip
Click to Flip
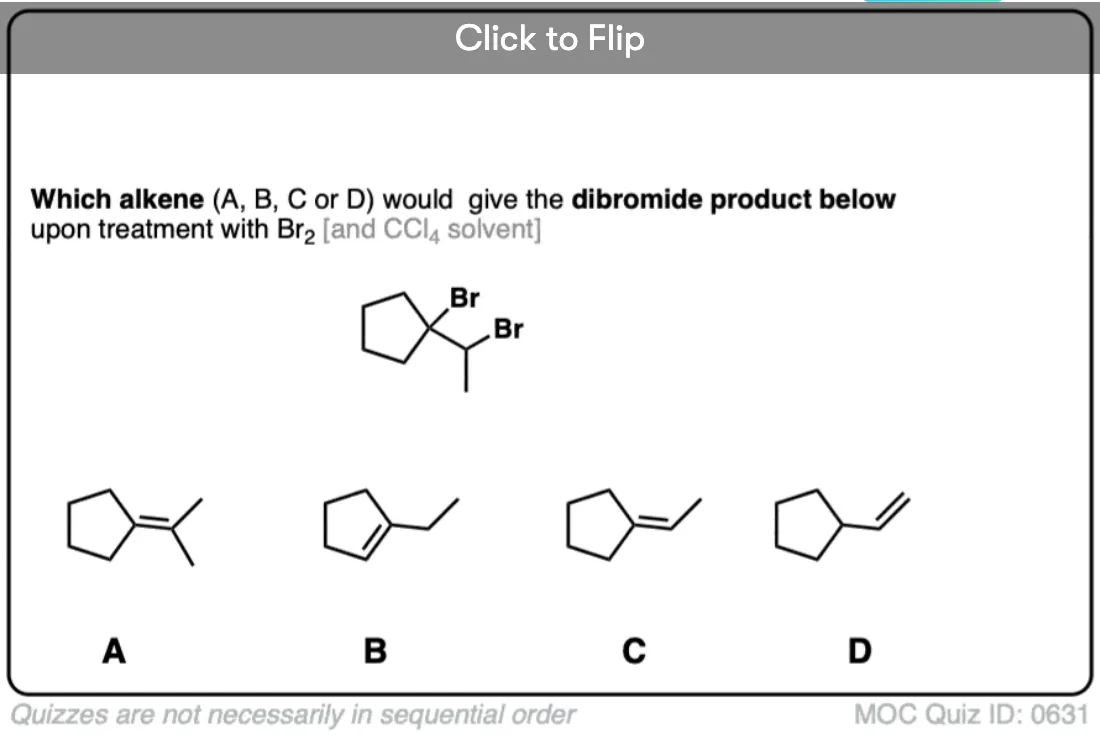
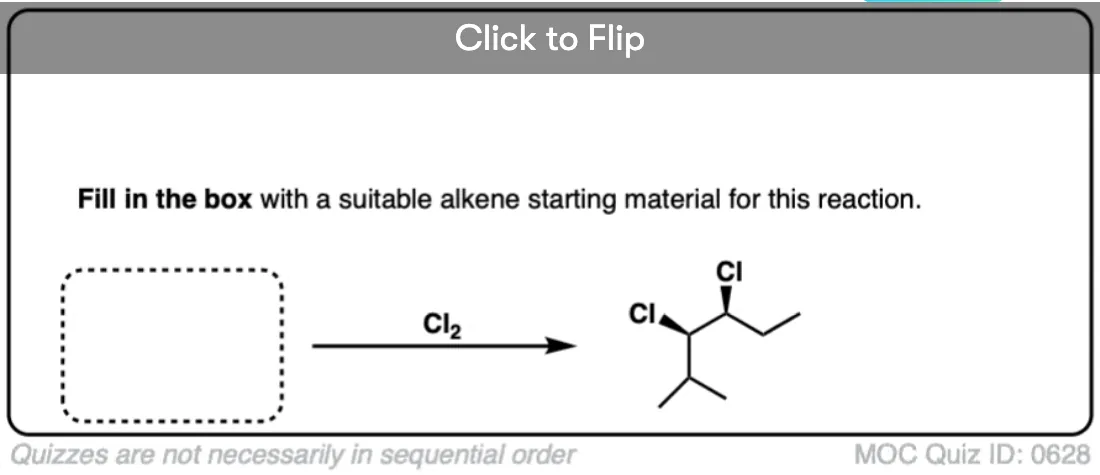
It’s also important to be able to work backwards! Can you identify the starting material of this halogenation reaction?
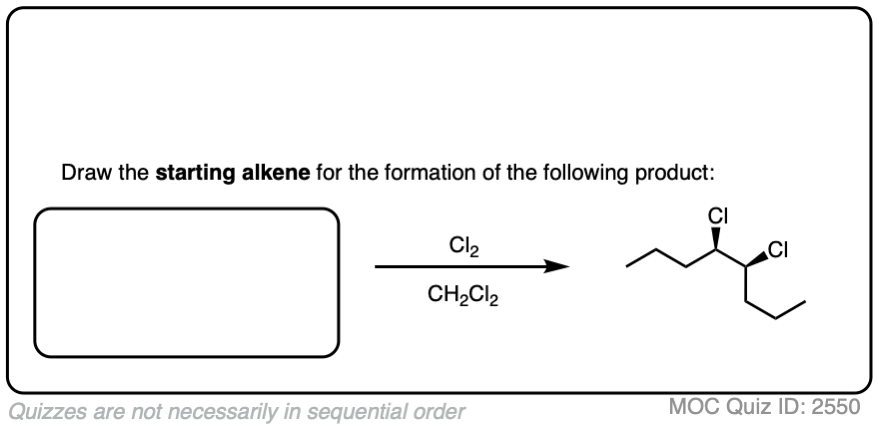 Click to Flip
Click to Flip
3. Halonium Ions
OK. So how does halogenation of alkenes actually work?
One initial idea was that they might proceed through a free carbocation intermediate, like the addition of HCl to alkenes. [See article – Markovnikov Addition of HCl to Alkenes].
An interesting test of this theory came from halogenation of the alkene below. cis-di-t-butylethylene has a lot of steric strain (9.3 kcal/mol) due to those two bulky t-butyl groups jostling up against each other. (This is technically called, “A-1,2 strain”)
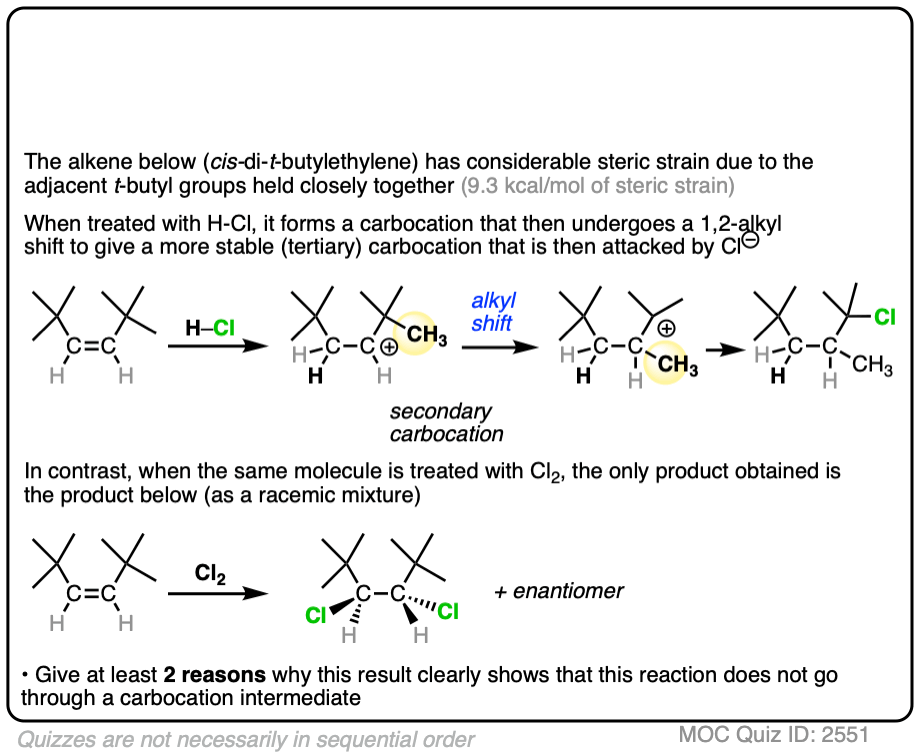 Click to Flip
Click to Flip
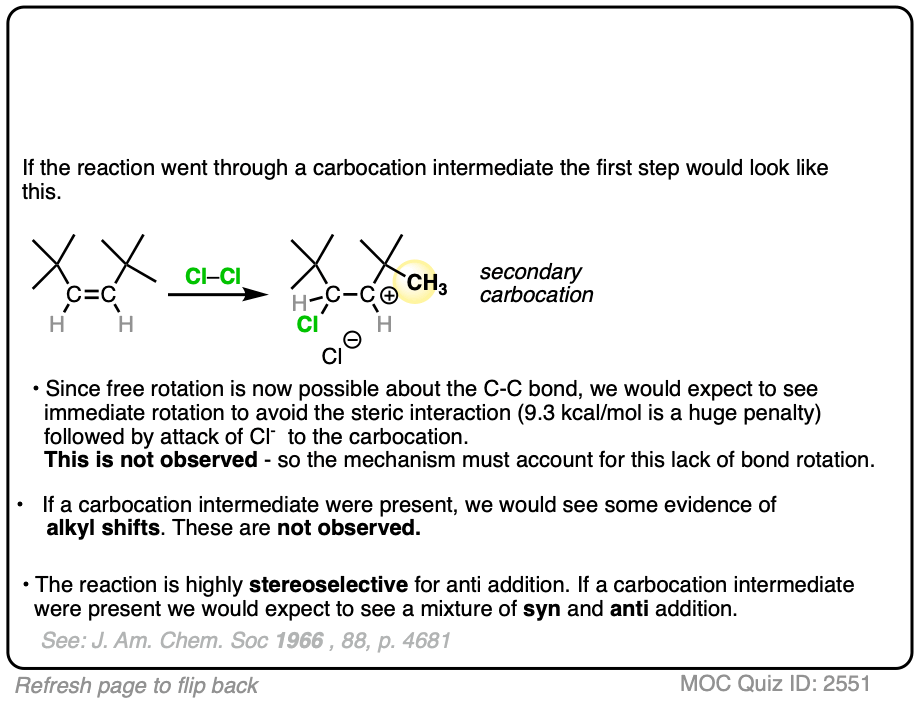
If halogenation went through a free carbocation, rotation would then be possible around the C-C bond. Consequently, we’d expect to see one of the t-butyl groups flip around to relieve the strain.
What was found instead is that this reaction is also highly stereospecific, just like the reaction of cis– 2-butene. Furthermore, there were no alkyl or hydride shifts like we would expect with a free carbocation.
Clearly, something is responsible for the stereoselectivity of this reaction. What is it?
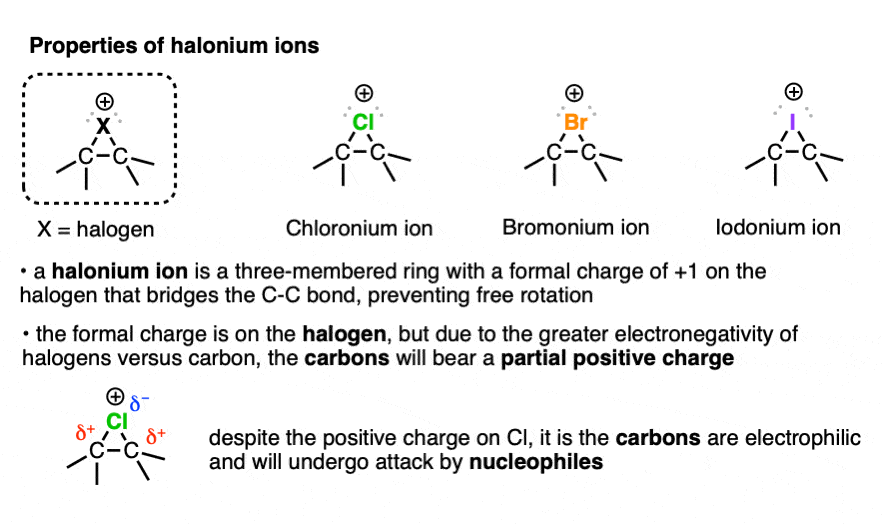
The best proposal we have for what is happening is formation of a cyclic halonium ion, which is a 3-membered ring bearing a positive formal charge on the halogen. “Halonium ion” is the generic term for when a halogen serves as the bridge; we can also use “chloronium”, “bromonium” or “iodonium” when referring to specific halogens.
An example of an arrow-pushing mechanism for halonium formation that shows all the bonds being formed and broken looks like this:
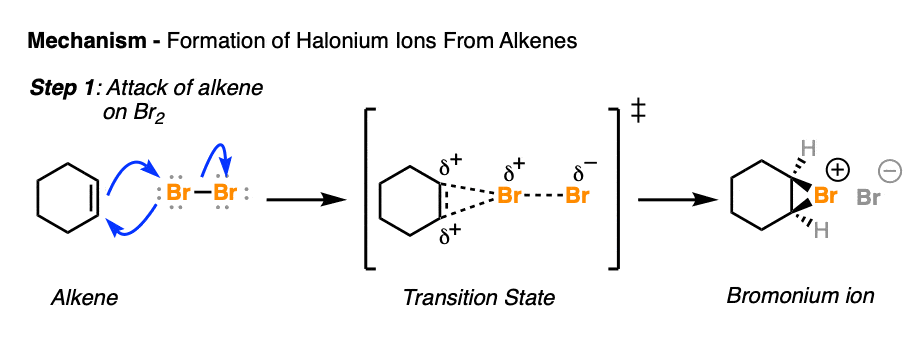
Be aware that many textbooks will only show two arrows, like this (hover here) or click this link.
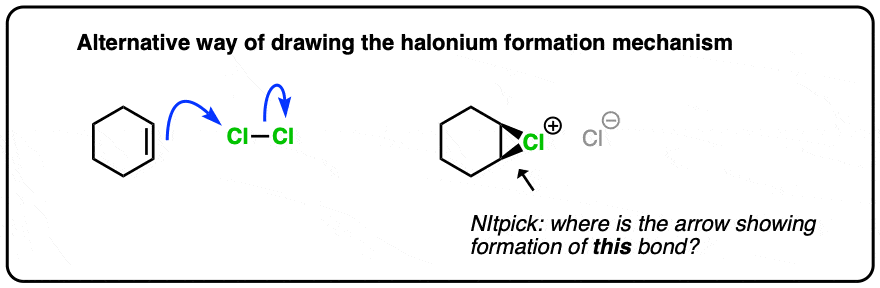{kind=link}
Although a halonium ion has a positive formal charge on the halogen, it’s actually the carbons that are electrophilic. This is easier to recognize if you remember that halogens are more electronegative than carbon, so the carbon-halogen bonds will have partial positive character on carbon. That’s why it’s called, “formal” charge. [See article: How to Calculate Formal Charge]
In a symmetrical halonium ion, the carbon-halogen bond lengths will be equal, as will be the partial positive charges on carbon.
X-ray crystal structures of stable bromonium and iodonium ions were determined by Brown & co-workers a while back [Note 3]
4. Halogenation of Alkenes – The Mechanism
So the first step in halogenation of alkenes is formation of a halonium ion.
In the second step of halogenation, the halide ion attacks the carbon from the backside of the C-halogen bond, resulting in formation of C-halogen and breakage of C-halogen.
This key step accounts for the high stereoselectivity of halogenation for the observed anti products.
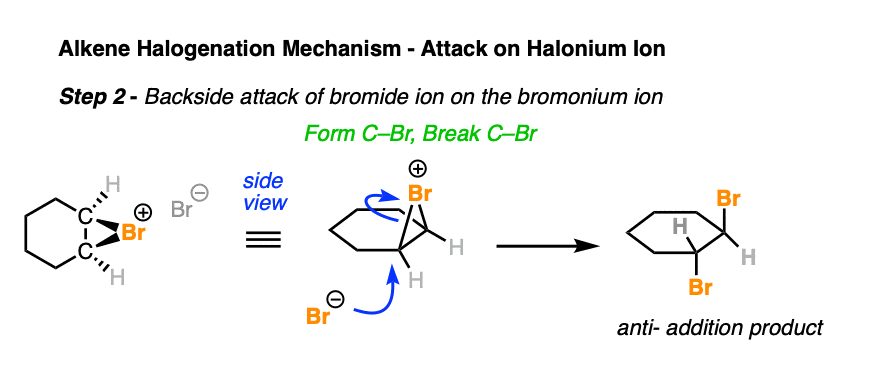
Note that the enantiomer (not shown) is also formed here.
This is similar to the backside attack in the SN2 mechanism [See article – The SN2 mechanism] where a lone pair from the nucleophile overlaps with the sigma* orbital of the leaving group. However, this step does not have the same sensitivity to steric hindrance as the SN2 reaction, as we are about to see.
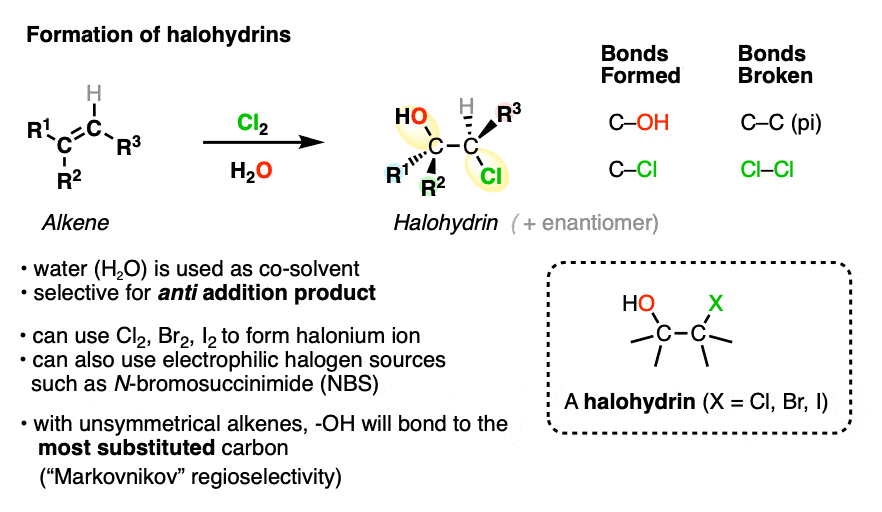
5. Halohydrin Formation – with water and alcohols
When bromination of alkenes is carried out in the presence of water (often as a co-solvent), halohydrin products are formed.
A halohydrin is a molecule containing C-OH and C-halogen bonds on adjacent carbons.
The reaction also proceeds through a halonium ion intermediate. As with halogenation, anti- addition products are formed exclusively.
One difference between halogenation and halohydrin formation is that the two new sigma bonds are formed to non-identical atoms (oxygen and halogen). This gives rise to the possibility of forming a mixture of constitutional isomers. When one constitutional isomer dominates, we say that reaction is regioselective.
Halohydrins are useful for the formation of epoxides. For more, see this article – Formation of Epoxides
It is found that halohydrin formation is regioselective for formation of the isomer where the C-OH bond forms on the more substituted carbon. This is similar to the pattern observed in addition reactions of HX to alkenes, which is known as “Markovnikov” regioselectivity. (See post – Markovnikov’s Rule)
Why might this be? After all, shouldn’t the more substituted carbon be more sensitive to steric hindrance?
Experiments tell us that (in contrast to the SN2) nucleophiles can attack tertiary carbons of halonium ions perfectly well. In other words, steric hindrance is not a significant factor.
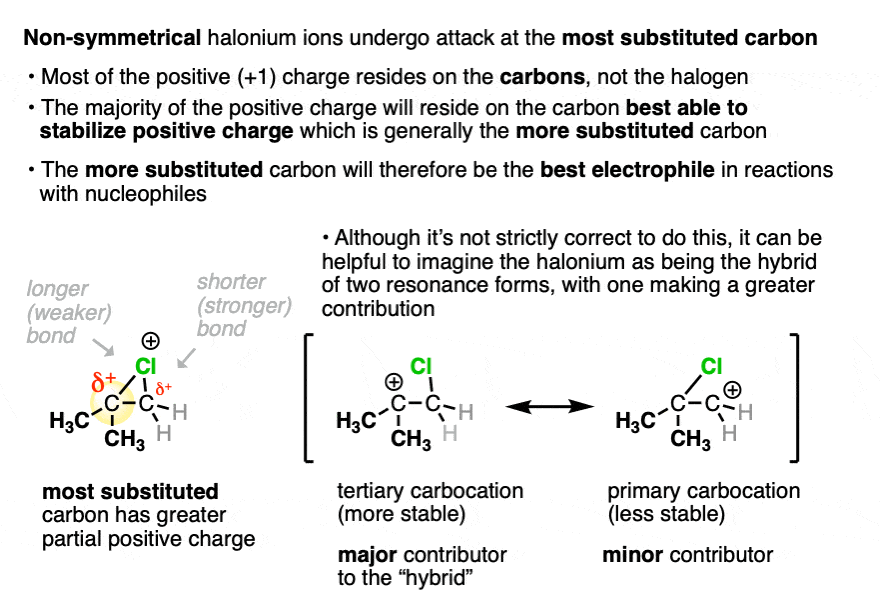
Let’s take a closer look at the structure of a non-symmetrical halonium ion.
As touched on earlier, although there is a formal charge of +1 on the halogen, it’s actually the carbons that bear the positive charge density due to the greater electronegativity of halogens (>3) versus carbon (2.5) . And to the extent that those carbons bear positive charge density, they are electrophilic.
Which of the two carbons is best able to stabilize positive charge? The more substituted carbon, generally. (As well as any carbon capable of delocalizing a positive charge through resonance). [See article – Carbocation Stability]
It’s not really proper to draw resonance forms where single bonds break, so hold your nose for a moment:
It can help to imagine the halonium ion as a “resonance hybrid” of two different carbocations. The resonance form where the carbocation is on the more substituted carbon will make the greater contribution to our “hybrid”.
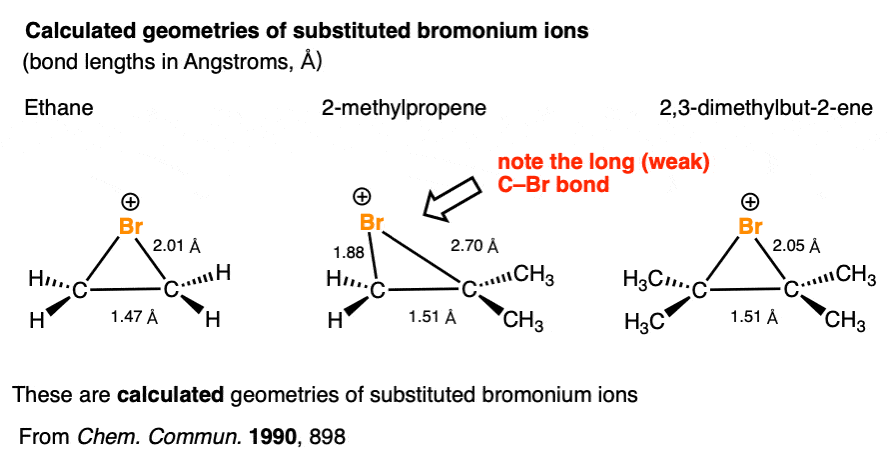
Having greater positive charge density, it will be more electrophilic, and therefore will more readily undergo reaction with nucleophiles. Note – calculations indicate a much weaker C-halogen bond on the more substituted carbon of the halonium ion. See Note
Another question that comes up is, “why does water attack the halonium ion instead of the halide ion? Isn’t the halide ion a better nucleophile?
Yes, the halide ion is a better nucleophile than water. However, recall that the reaction rate will depend not just on nucleophilicity but also on concentration. Since water is generally used as the solvent (or co-solvent) here, it will have a vastly higher concentration.
So in a sense we are “overwhelming” the halonium ion with a much higher concentration of a worse nucleophile (the technical term for this is “mass action“).
6. Mechanism of Halohydrin Formation
As with halogenation, the first step in halohydrin formation is creation of a halonium ion.
In addition to Cl2, Br2, and I2, this can also be done with sources of electropositive halogen such as N-bromosuccinimide (NBS). [See article – N-Bromosuccinimide]
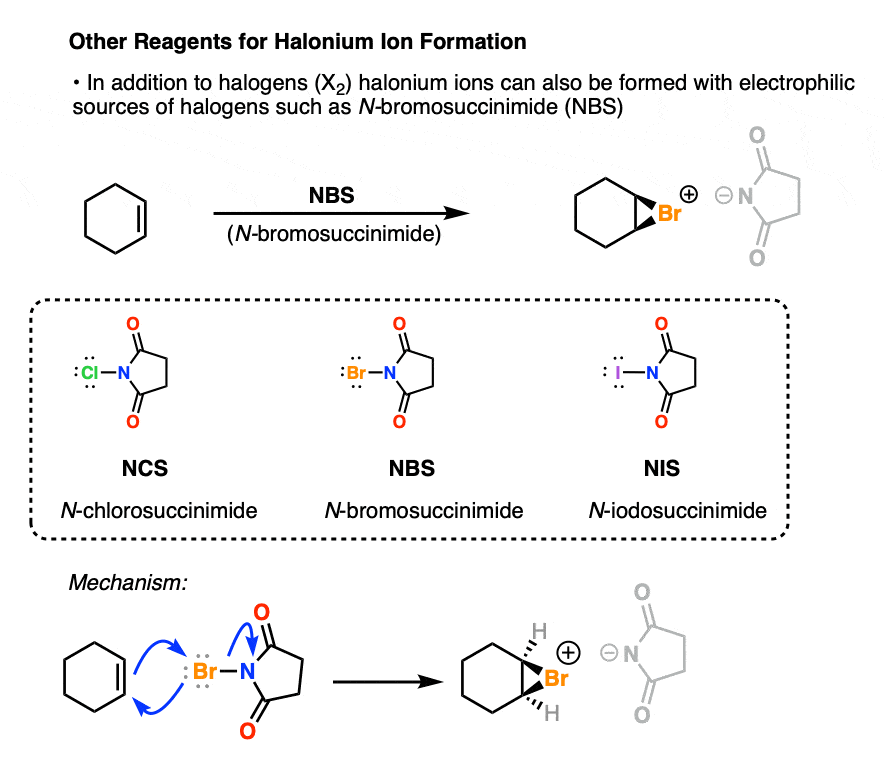
After formation of the halonium ion, the next step is attack on the halonium ion by H2O, at the more substituted carbon.
Why might H2O do this and not the counter-ion? If it’s present as solvent it just outnumbers the concentration of halide ion. [Note 4 ]
This results in the anti product which can then be deprotonated (e.g. with solvent) to give the neutral halohydrin.
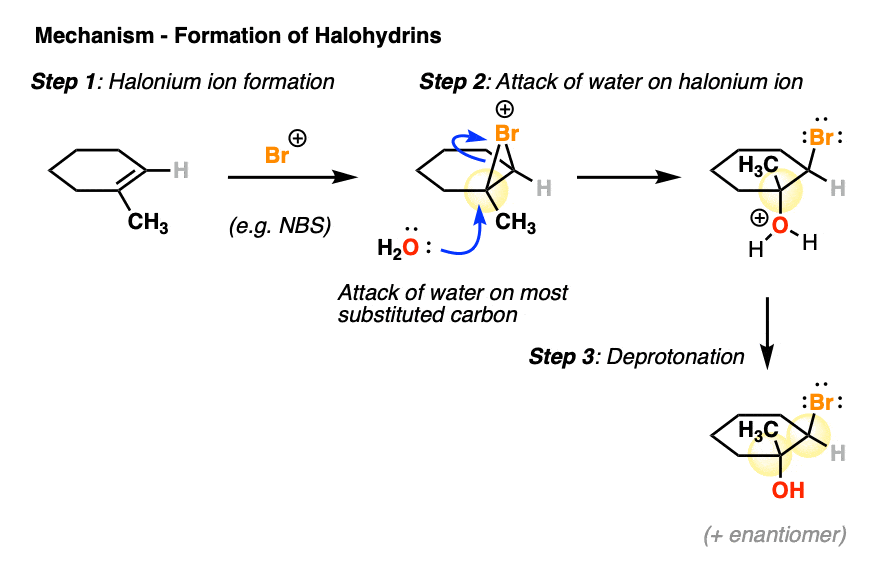
[Note that, like a flat coin that can land on “heads” or “tails” with equal likelihood, the bromine can “land” on either face of the alkene – this will give rise to enantiomers in this case].
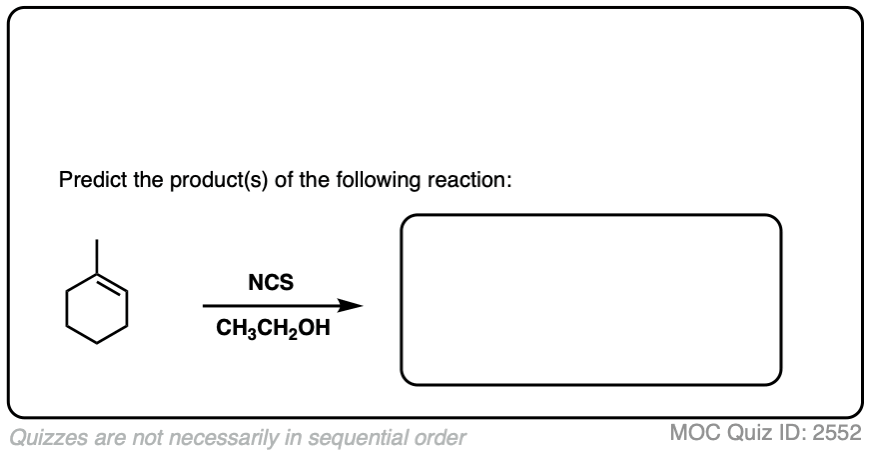
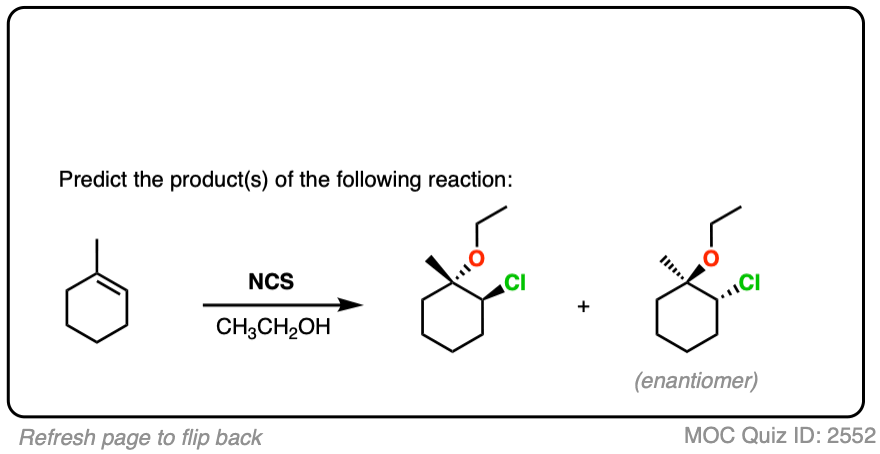
7. Haloethers
Similarly, if alcohols are used as solvent, haloethers may form. The formation of haloethers passes through an identical mechanism to that of halohydrins.
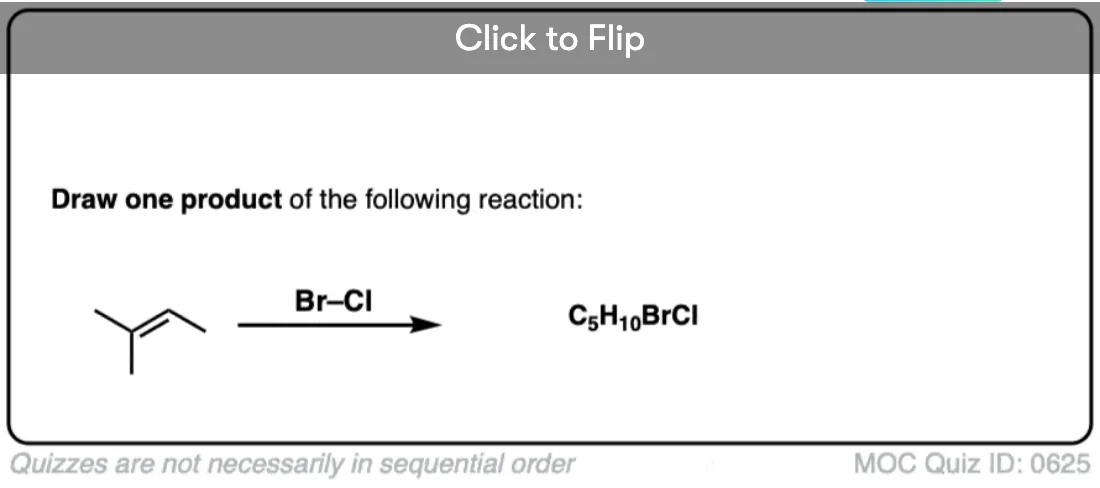
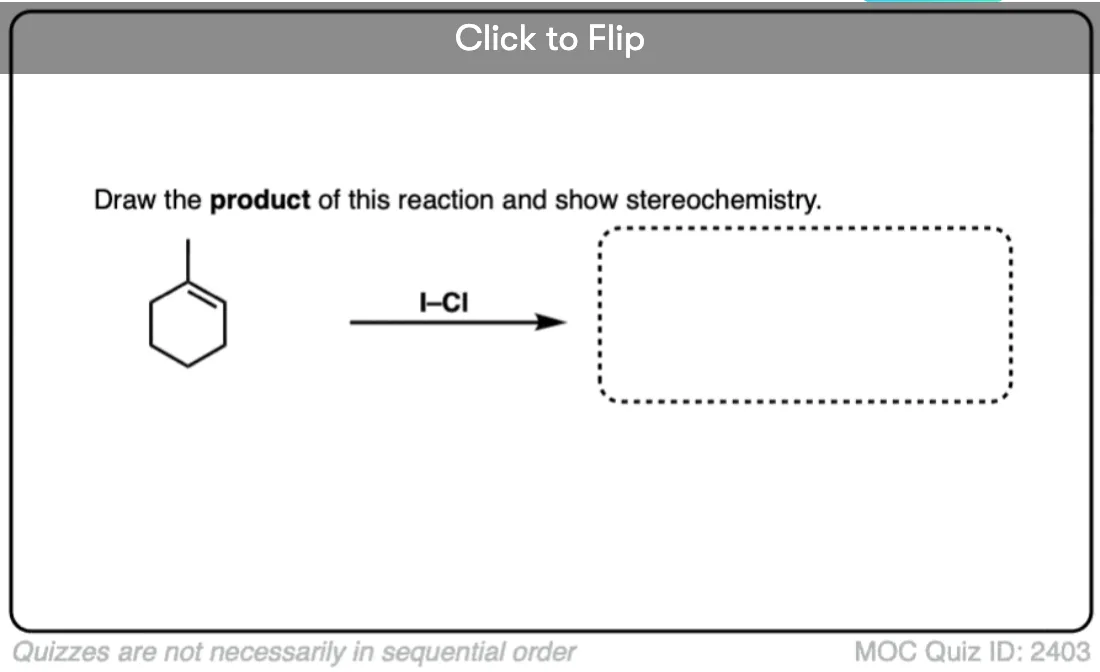
See if you can predict the product of the following reaction.
 Click to Flip
Click to Flip
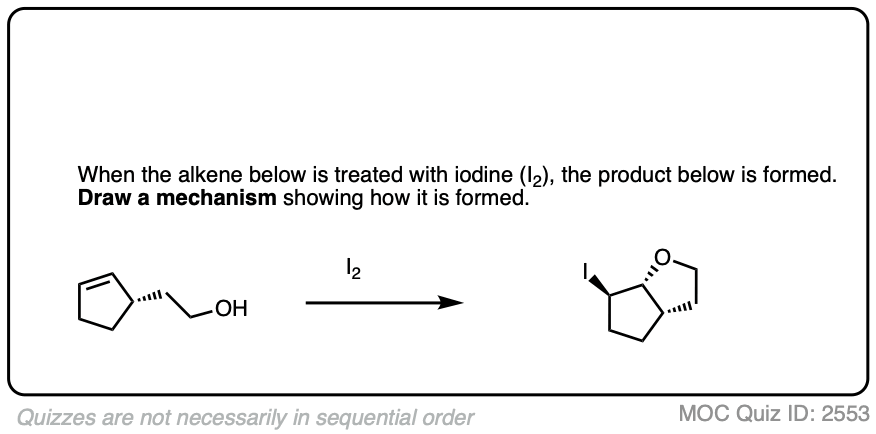
An interesting wrinkle in haloether formation is the possibility for intramolecular reactions.
For example, treating this alkene with NBS (a source of Br+) results in formation of a new ring.
See if you can figure out the mechanism.
 Click to Flip
Click to Flip
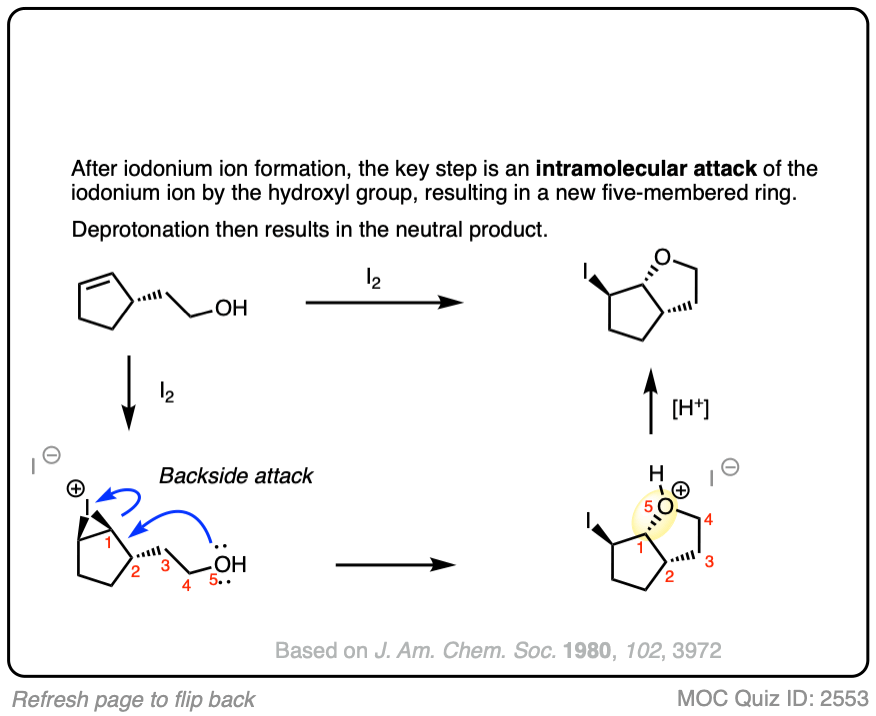
As always, be on the lookout for intramolecular reactions as they make for great exam questions.
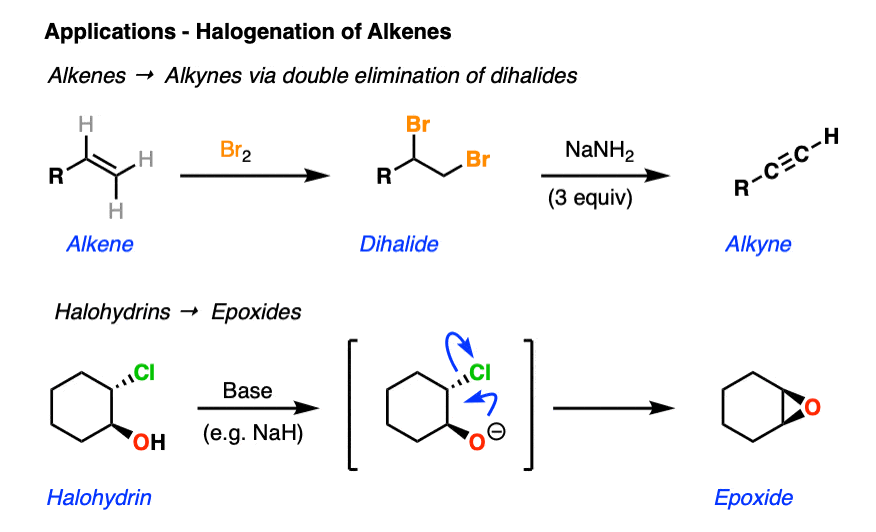
8. Some Applications of Halogenation Reactions
It’s worth knowing how some of these reactions can be applied later on in the course.
Alkynes can be converted into cis-dihaloalkenes through treatment with halogens (See article: Halogenation of Alkynes)
There is no direct way to convert an alkene to an alkyne, but they can be indirectly converted to alkynes through formation of a dihalide followed by double elimination to give the alkyne. (See article: Alkenes to Alkynes via Elimination Reactions)
[Worth noting: bromination of alkenes is technically an oxidation reaction, because each carbon goes from being bound to another carbon (0) to bromine (–1). The oxidation state of each carbon in ethene is +2; the oxidation state of each carbon in dibromoethane is +1. ]
Epoxides can be formed from alkenes in two steps via formation of a halohydrin followed by deprotonation with a strong base (such as NaH). The intermediate alkoxide performs an intramolecular SN2 reaction to give a new 3-membered ring. (See article: Formation of Epoxides)
9. Summary
Remember this reaction and remember this mechanism! It bears a lot of similarity to other mechanisms you will encounter in the chapter on alkenes, such as:
- Oxymercuration (Markovnikov-selective, also goes through a cyclic, 3-membered “mercurinium ion” intermediate)
- Opening of epoxides under acidic conditions (also undergoes addition at the most substituted carbon)
Furthermore, the reaction can also be applied to alkynes.
The stereochemistry of this reaction is very frequently tested. Make sure you can properly draw the products of anti addition and additionally, can work backwards from the dihalide products to the alkene starting materials.
Notes
Note 1. “Inert solvent” here just means a solvent that won’t react with the halonium ion intermediate. CCl4 and CH2Cl2 leave halonium ions alone; H2O, alcohols, and carboxylic acids can potentially undergo reactions with them.
Note 2. Here are some specific examples of iodination and fluorination:
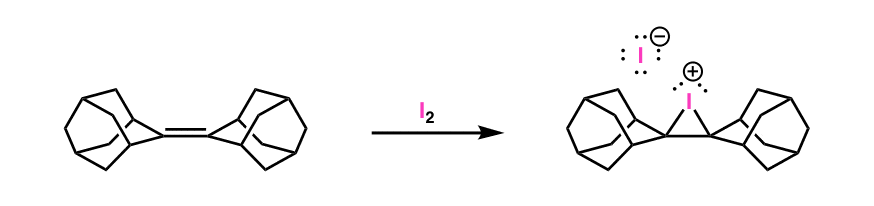
Note 3. Halonium ions are not fictional chemical entities. The halonium ion from the reaction below is particularly stable, since it is very difficult for nucleophiles to attack the very sterically hindered carbon atoms:
R.S. Brown and co-workers succeeded in using X-ray diffraction to obtain a crystal structure of this molecule. Here’s the iodonium ion portion of the structure:
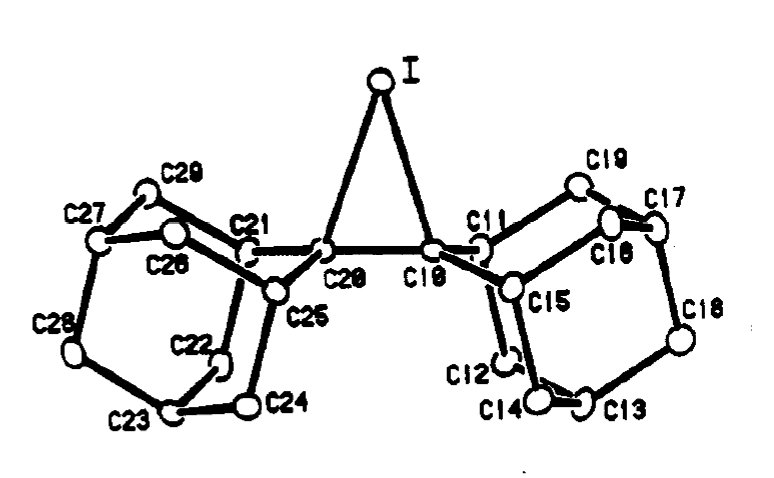
The iodine-carbon bond length is 2.48 Angstrom, significantly longer than the value of 2.13 Angstrom for a typical C-I bond.
The C-C bond length is 1.49 A, which is typical for a C-C single bond.
Note 4. It’s possible to influence the product distribution by adding various salts (e.g. NaCl, sodium acetate, lithium bromide, etc.) that may intercept the halonium ion. See here [Ref] for a study.
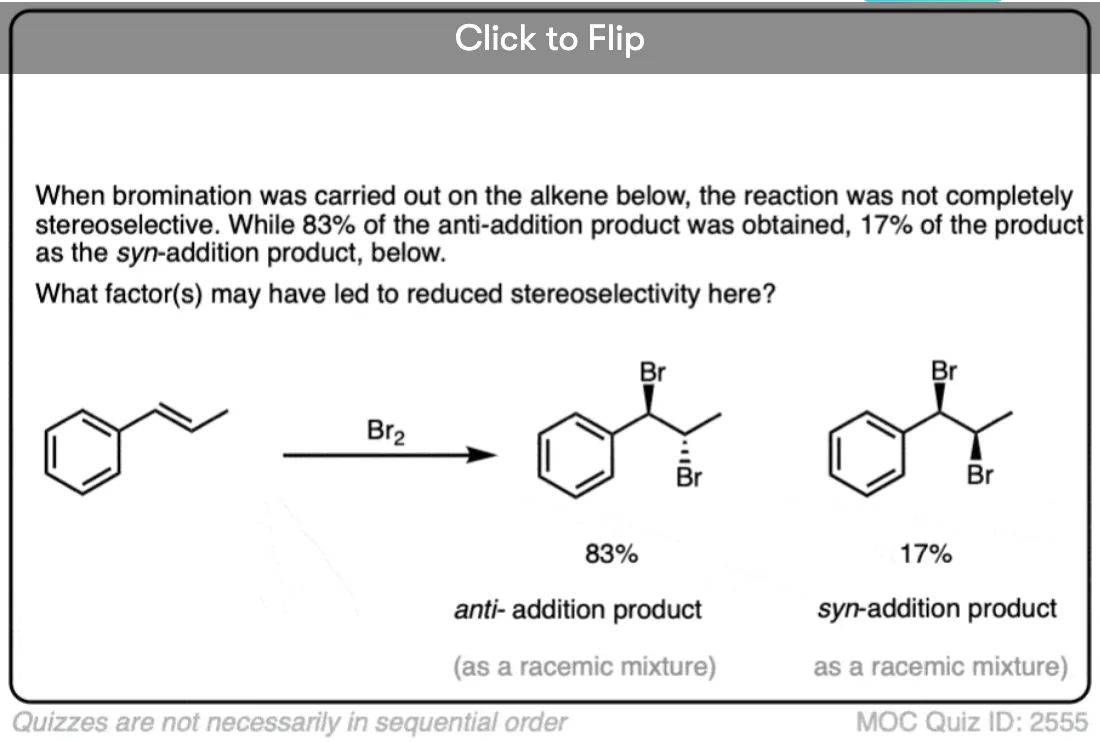
Note 5. In some situations the high selectivity for the anti– product can break down. See [Ref] for more.
Note 6. How might we resolve the role of “nucleophiles” and “electrophiles”in formation of the bromonium ion? This is not easy, because it entails making further assumptions about the reaction mechanism that might not be based on solid evidence. With that hedge out of the way, here’s a proposal. : instead of thinking of C-C π and Br monolithically, break down each component into molecular orbitals. The C-C π orbital could act as a nucleophile while the C-C π* acts as an electrophile; the Br-Br σ* orbital could act as an electrophile while the Br lone pair could act as a nucleophile.
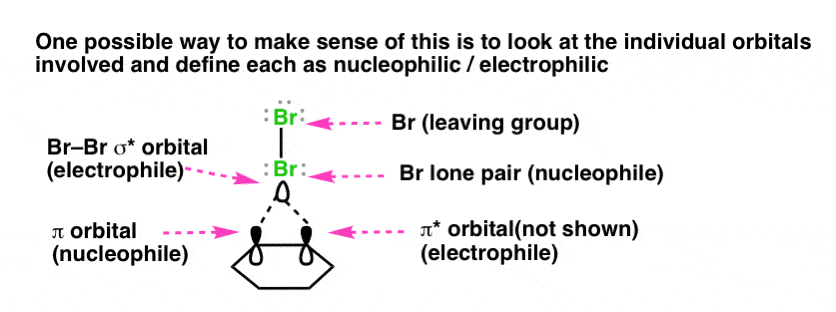
As it turns out, calculations indicate the bromination of alkenes with Br2 to be more complex than we might initially suppose. The initial step is coordination of Br2 to the alkene in a loosely bonded structure known as a “π complex”. The π complex then breaks down to give the bromonium ion. A proper treatment of the orbitals would therefore not strictly be of the alkene and Br2, but of the orbitals in the π complex itself.
It gets more complicated. In some solvents it turns out to be energetically favorable for a second molecule of Br2 to be involved in bonding to the Br- that is expelled in the process (yes, a “termolecular” mechanism). For more details see here (J. Phys Chem A, 2007, 111, 13218)
Note 7. Calculations on several bromonium ions indicate a much weaker C-Br bond on the more substituted carbon of the halonium ion [Ref]
Here’s an interactive model, courtesy of Rowan
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- THE RELATIVE RATES OF BROMINATION OF THE OLEFINS
Harold S. Davis
Journal of the American Chemical Society 1928, 50 (10), 2769-2780
DOI: 1021/ja01397a031
An early paper studying the kinetics of alkene bromination under a variety of conditions. - The Halogenation of Ethylenes
Irving Roberts and George E. Kimball
Journal of the American Chemical Society 1937, 59 (5), 947-948
DOI: 1021/ja01284a507
One of the earliest descriptions in the literature of a three-membered bromonium ion, accounting for the anti stereochemistry of this reaction. - The question of reversible formation of bromonium ions during the course of electrophilic bromination of olefins. 2. The crystal and molecular structure of the bromonium ion of adamantylideneadamantane
H. Slebocka-Tilk, R. G. Ball, and R. Stan Brown
Journal of the American Chemical Society 1985, 107 (15), 4504-4508
DOI: 10.1021/ja00301a021
This paper describes the X-ray crystal structure of an isolated, stable bromonium ion. This is significant because it proves the intermediacy of these three-membered cyclic bromonium ions in the electrophilic addition of bromine to alkenes. - Stable Bromonium and Iodonium Ions of the Hindered Olefins Adamantylideneadamantane and Bicyclo[3.3.1]nonylidenebicyclo[3.3.1]nonane. X-Ray Structure, Transfer of Positive Halogens to Acceptor Olefins, and ab Initio Studies
R. S. Brown, R. W. Nagorski, A. J. Bennet, R. E. D. McClung, G. H. M. Aarts, M. Klobukowski, R. McDonald, and B. D. Santarsiero
Journal of the American Chemical Society 1994, 116 (6), 2448-2456
DOI: 10.1021/ja00085a027
Cyclic iodonium ions, analogous to the bromonium ions, can also be isolated and characterized. The parent alkene in these studies, adamantylideneadamantane, is prepared using a McMurry reaction with 2-adamantanone. - ALKYNE via SOLID-LIQUID PHASE-TRANSFER CATALYZED DEHYDROHALOGENATION: ACETYLENE DICARBOXALDEHYDE TETRAMETHYL ACETAL AND ACETYLENE DICARBOXALDEHYDE DIMETHYL ACETAL
Rufine Akué-Gédu and Benoît Rigo
Org. Synth. 2005, 82, 179
DOI: 10.15227/orgsyn.082.0179
Formation of alkynes via double elimination of a dihalide. - Principles of an Electronic Theory of Organic Reactions.
Christopher K. Ingold
Chemical Reviews 1934, 15 (2), 225-274
DOI: 1021/cr60051a003 - —The relative directive powers of groups of the forms RO and RR′N in aromatic substitution. Part IV. A discussion of the observations recorded in parts I, II, and III
James Allan, Albert Edward Oxford, Robert Robinson, and John Charles Smith
J. Chem. Soc. (Res.) 1926, 129, 401-411
DOI: 10.1039/JR9262900401
Nobel Prize winner Robert Robinson was wrong about the mechanism of halogenation reactions. - Polar Additions to Olefins. II. The Chlorination of Di-t-butylethylene
Robert C. Fahey
Journal of the American Chemical Society 1966 88 (20), 4681-4684
DOI: 10.1021/ja00972a030
Study on the halogenation of cis– and trans- di-t–butylethylene, showing the stereospecific nature of the halogenation reaction. - Polar additions to the styrene and 2-butene systems. II. Medium dependence of bromination products
John H. Rolston and Keith Yates
Journal of the American Chemical Society 1969 91 (6), 1477-1483
DOI: 10.1021/ja01034a034
In some situations the stereoselectivity of halogenation reactions can be significantly eroded by using highly polar solvents or electron-releasing substituents on the alkene that will result in more stabilized carbocation intermediates.
What would this reaction between ethene and KBr
(i) without water, give
(ii) with water, give
No real reaction between KBr and alkenes. KBr is a salt of the bromide ion, very similar to table salt, NaCl.
I think you mean something else, but I will let you think about it….
First you say that halogenation is stereoselective, then you say it is stereospecific. So which one is it?
Stereospecific is a subset of stereoselective.
It’s like saying two people are siblings, and then later on saying that they are brothers. They are both siblings and brothers, but brothers is a subset of siblings.
Draw out the mechanism of the reaction between bromine water and propen giving the bromohydrin and predict which of the two possible bromohydrins CH3CH or CH2OH or CH3CH(OH)CH2Br will be formed.
Good morning! You may wish to double-check your oxidation numbers in this note:
[Worth noting: bromination of alkenes is technically an oxidation reaction, because each carbon goes from being bound to another carbon (0) to bromine (–1). The oxidation state of each carbon in ethene is +2; the oxidation state of each carbon in dibromoethane is +1. ]
This is in Section 8: Some Applications of Halogenation Reactions. Oxidation number should become more positive / less negative if carbon is being oxidized (which it is here). Looks like the numbers are right but the sign is incorrect. Thank you :)
Hello,
I’m studying for a quiz and the problem I came across was an alkene addition with Br-Br and NaCl. I know that the Cl should add first but I can’t seem to figure out the mechanism for it.
No, what happens is that you form the bromonium ion with Br2 and then Cl(-) is the active nucleophile which attacks the resulting bromonium ion at the most substituted position.
Kind of a strange question though, because they probably don’t mention solvent. NaCl is not going to dissolve in an organic solvent. Better choice would have been Bu4N+ Cl- .
How can we invoke an sn2 type ring opening on a tertiary carbon to give the most substituted alcohol? Why is the regioselectivity of the ring opening opposite from that observed for epoxide opening under basic conditions?
The leaving group is excellent, first of all. The C-halogen bond length is much longer than a normal C-leaving group bond length. (e.g. 2.48 A for C-I in the iodonium vs 2.13 A for normal C-I).
Secondly, because of the 3-membered ring, the geometry on the carbons is not the same as in your typical tetrahedral carbon, so there is more “room” for the nucleophile to approach.
Good day Sir. I understand that halogenation reactions of alkenes involve the bridge ion as intermediate, and that no rearrangement occurs. But I don’t get how a 1,4 addition product is produced when conjugated dienes are involved as starting material. Since the intermediate is a bridge ion, how can resonance be formed? Thank you in advance for your reply.
See this post: https://www.masterorganicchemistry.com/2017/04/11/more-on-12-and-14-additions-to-dienes/
How can i synthesis only cis dibromo alkane from an alkene ?
Something I don’t understand (my professor couldn’t explain it well either): logically I would assume that the second attack be on the less substituted carbon as there would be less steric hindrance. Also, being more substituted (more neighboring carbons) means that these carbons would donate more of their electron density to that same carbon, which would make it more negative (hence less electrophilic and less reactive).
I noticed you didn’t go into more detail than “[…] the reaction will proceed at the carbon best able to stabilize positive charge.” Is there a deeper explanation to this?
Hi Idan. The reason the second attack occurs on the more substituted carbon, instead of the less substituted, is explained by the following. In the transition state leading to the product, one C-Br bond in the bromonium ion is already partially broken. Therefore, one of the two C atoms will bear a partial positive charge–it will be cation-like, in other words. Now, think about the relative stability of carbocations. Since more substituted carbocations are more stable, the more substituted carbon will undergo partial C-Br bond cleavage, and therefore nucleophilic attack will preferentially occur at the tertiary carbon in the above example.
While you’re right that the more substituted carbon is more sterically hindered, this effect is less important than the electronic effect of carbocation stability.
This is complicated by the fact that the major product isn’t 1,2-dibromoethane. The water also gets involved in the reaction, and most of the product is 2-bromoethanol.
Is there any reason why in case of water, H2O reacts before Br- does? H2O should be a weaker nucleophile than Br-.
Water is the solvent, so it is present in tremendously high concentration (pure water has a concentration of 55 M !). This explains why water attacks preferentially over Br-, which otherwise is a much better nucleophile.
Hey Prof., I get everything and you explain everything in a simpler way, but I don’t understand the part about “the reaction will proceed at the carbon best able to stabilize positive charge?” Could you perhaps show me that it is that C with greater partial charge in the corresponding example by using partial charge symbols?
Thanks,
Soji
It would be the most substituted carbon – think about carbocation stability, which follows the trend tertiary (most stable) > secondary > primary (least stable)
I was wondering about this, too. Thanks for taking time to explain it.