Alcohols, Epoxides and Ethers
Demystifying The Mechanisms of Alcohol Oxidations
Last updated: May 28th, 2026 |
Alcohol Oxidation Mechanisms, Demystified
• The mechanisms for the oxidation of alcohols generally involve putting a good leaving group on oxygen, followed by deprotonation of an adjacent C-H bond that results in elimination to give a new C-O pi bond.
• In this sense it greatly resembles an E2 mechanism.
• Oxidation of aldehydes to carboxylic acids usually involves addition of water to the aldehyde first (formation of a hydrate) which then undergoes elimination with base.
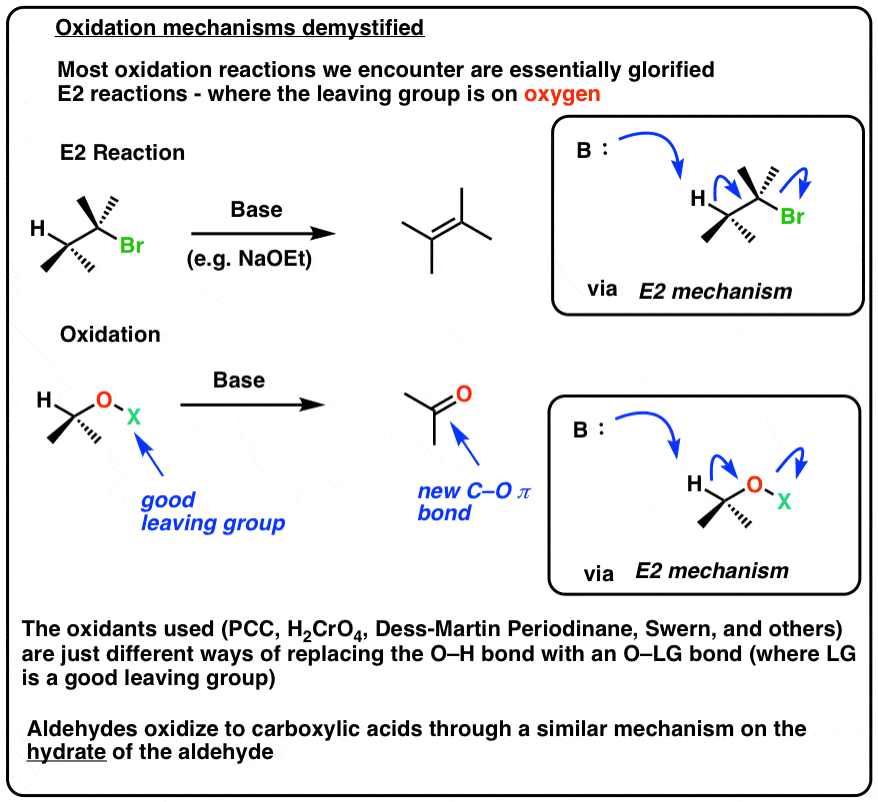
Table of Contents
- E2: The Familiar Key Step At The Heart of (Almost) All Oxidation Reactions
- Oxidants Are Essentially Fancy Reagents For Attaching Leaving Groups To Oxygen
- What About Oxidation of Aldehydes To Carboxylic Acids? (Spoiler: Yes, that too)
- Why Don’t Ketones Oxidize Further?
- Summary: Alcohol Oxidation Mechanisms
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. E2: The Familiar Key Step At The Heart Of (Almost) All Alcohol Oxidation Reactions
When I was learning organic chemistry I remember the reagents for oxidation reactions completely coming out of left field.
KMnO4, K2Cr2O7, PCC, CrO3, Swern, Dess—Martin ? Hold on. Where did these reagents come from? How do they work? Why chromium? What’s the mechanism?
In my course, the details of these reactions were completely glossed over. “ Don’t worry about the mechanism! No time to go through this! “ the instructor said. I was left with the impression that there was something deeply mysterious about alcohol oxidation.
Only later did I learn that it’s not mysterious at all. In fact the key mechanism is very familiar.
Let me show you what I mean.
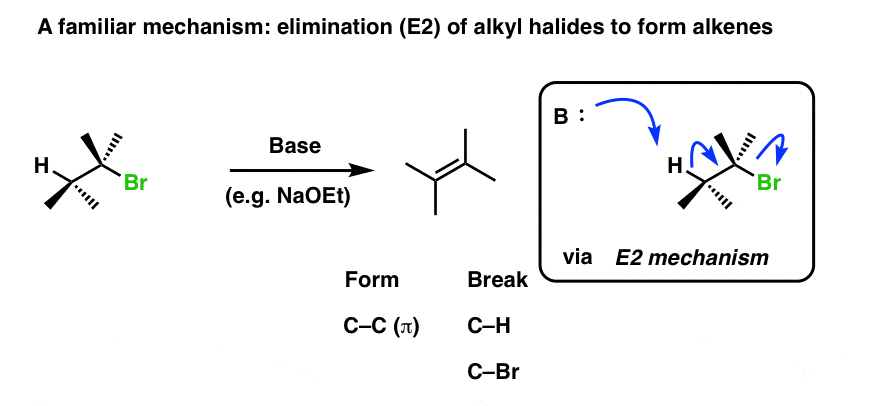
Here’s a reaction we’ve seen before. Elimination of alkyl halides to give alkenes through an E2 mechanism. Base removes hydrogen, we break C-H, form C-C (π) and break C-LG. The result is an alkene.
Now imagine a slightly different E2 reaction, except one where the good leaving group is on oxygen. We’ll leave it vague, as “LG” for now.
Watch!
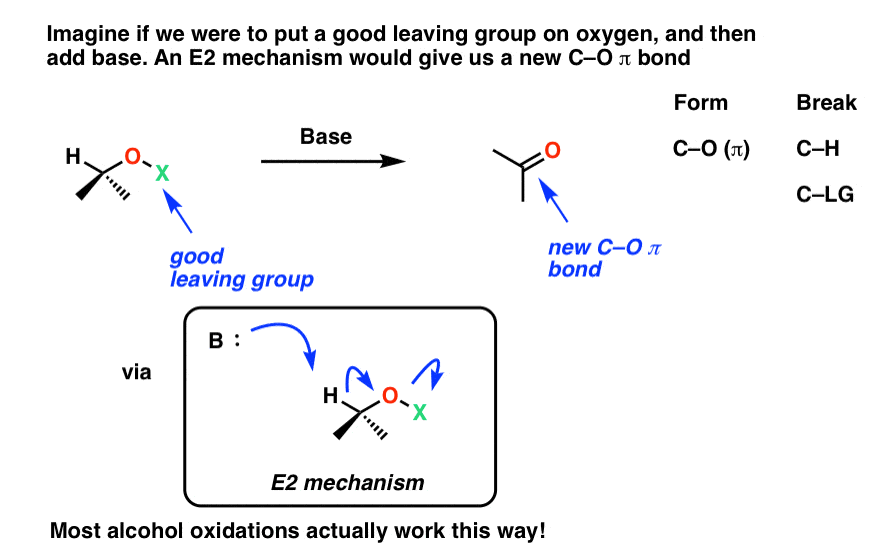
See how we break C-H, form C-O (π), and break O-LG, forming a new C-O π bond in the process. Since we’ve formed a new C-O bond at the expense of a C-H bond, an oxidation has occurred.
Believe it or not, most oxidation reactions of alcohols proceed exactly this way! [Note 1]
I wish I’d known this when I was learning organic chemistry because it would have made alcohol oxidation seem a lot less mysterious.
2. Oxidants Are Essentially Just Fancy Reagents For Attaching Good “Leaving Groups” Directly To Oxygen
Hold on, you might say. It can’t possibly be that simple. Why do we have so many different types of oxidizing agents? And why do the mechanisms (like the Jones oxidation here for example) seem so complicated?
Yes, there are a lot of steps in a typical oxidation reaction. However, most of these steps consist of:
- activating the oxidant (such as in the Swern oxidation, where oxalyl chloride converts DMSO to an electrophilic sulfur species, or in chromate oxidations, where strong acid converts chromate (such as K2Cr2O7) to the active oxidant [H2CrO4]
- coordination of the alcohol to the oxidant, followed by proton transfer(s) (seen in the mechanisms of most chromium oxidants, and Dess-Martin periodinane).
These steps are important, of course, but only in a supporting role. If you’ll excuse the analogy, they’re just foreplay that precedes the main event.
The effect of these beginning steps is simply to install a good leaving group on oxygen. That “good leaving group” can take many forms. It’s illustrated here with each oxidant, in green. There are, of course, many, many more oxidizing agents for alcohols than those depicted, but almost all of them essentially work the same way.
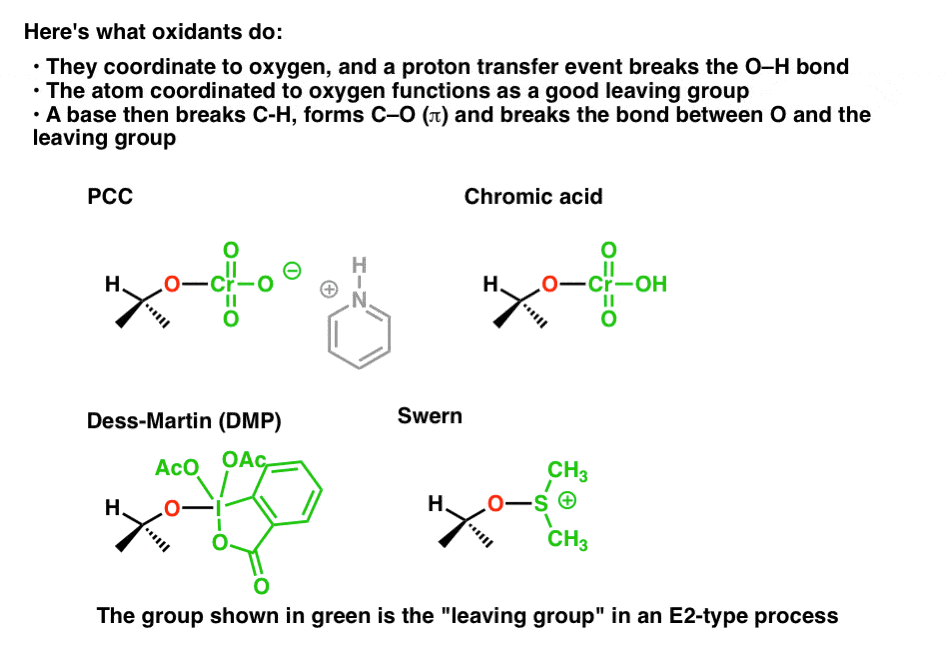
Treatment of each of these substrates with base then results in breakage of C-H, formation of C-O (π) and breakage of O-LG.
Each of these “leaving groups” accepts the pair of electrons from the bond to oxygen, reducing its oxidation state by 2 in the process. [remember – the oxidant is reduced, the substrate is oxidized]
3. What About Oxidation Of Aldehydes To Carboxylic Acids?
So if oxidation of alcohols to aldehydes and ketones is essentially an E2 reaction, how do we explain oxidation of aldehydes to carboxylic acids?
See, given what we’ve just shown, you might initially think it works something like this:
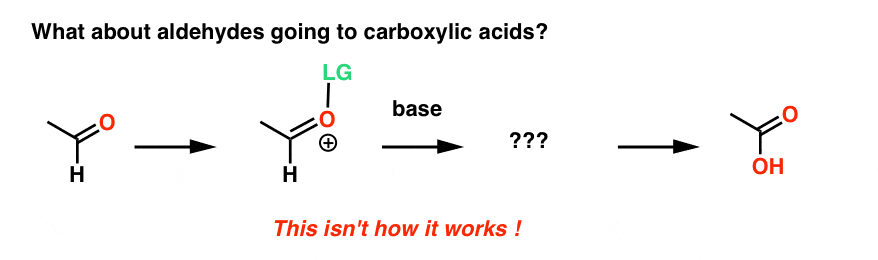
That’s actually not what happens. [Why not? Because the aldehyde carbon is a good electrophile, and any species basic enough to remove the C-H is more likely to add to the aldehyde C ]
It actually follows the same type of process as with alcohols! However, there’s a trick.
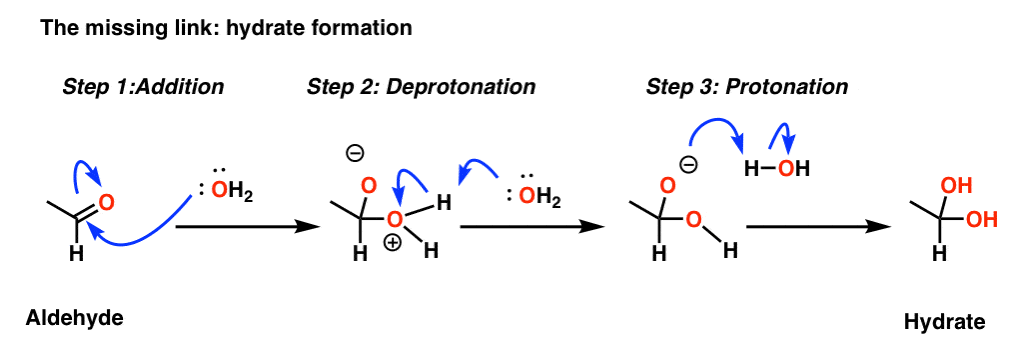
There’s a missing ingredient not mentioned in the diagram above. Water.
What happens is that water adds to the aldehyde, forming a hydrate. [If this looks unfamiliar, you’ll see MANY variations of this type of mechanism in your upcoming chapter on aldehydes and ketones. This is a sneak preview]
NOW, the oxidant attaches to one of the hydroxyl groups of the hydrate. The E2 from here is much easier to visualize.
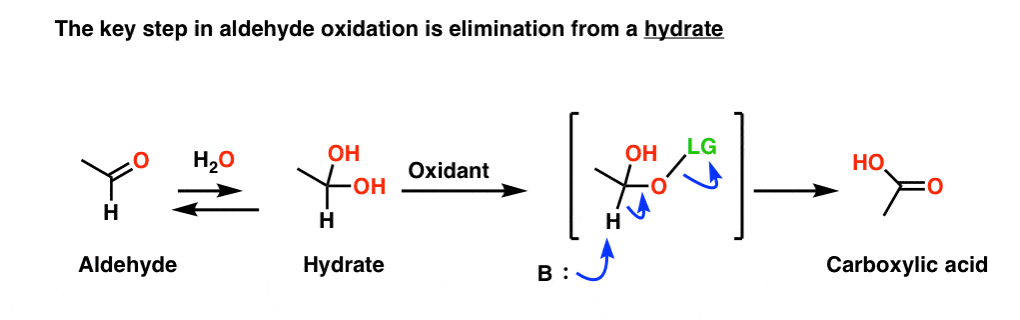
This also helps to explain one key observation made tangentially in the last post. The reagent CrO3/pyridine (Collins’ reagent) will oxidize primary alcohols to aldehydes and stop there.
However, if water is present, this oxidation will go all the way to carboxylic acids. That’s because the water will form a hydrate with the aldehyde, allowing for further oxidation.
No hydrate, no further oxidation.
4. Why Don’t Ketones Oxidize Further?
This also explains why ketones don’t oxidize further. There’s no hydrogen that can be removed in an E2-type process that will lead to a new double bond!
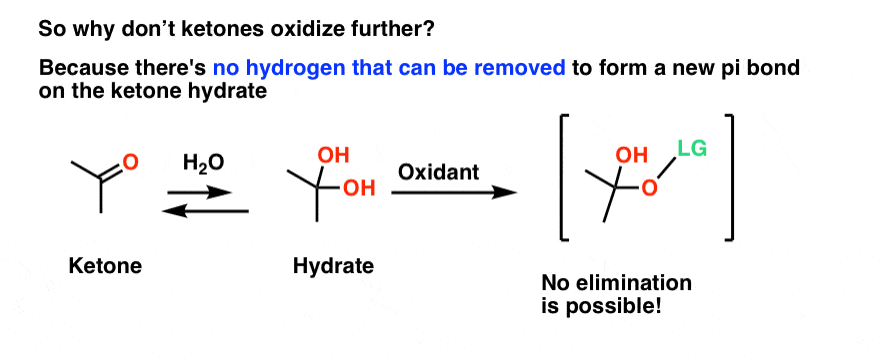
It’s similar to the old question of why this alkyl halide (below) doesn’t undergo elimination. There’s no hydrogen on the “beta” carbon (i.e. on the carbon adjacent to the carbon bearing the good leaving group) that can be removed, so no elimination occurs.
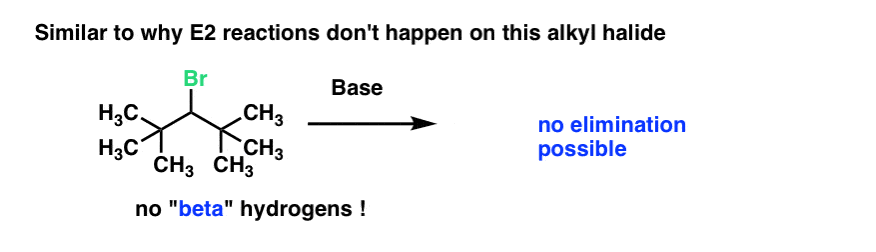
The same could be said for why tertiary alcohols don’t oxidize.
5. Summary: Alcohol Oxidation Mechanisms
So the bottom line for alcohol oxidation is the following.
- Pretty much every alcohol oxidation reaction you’ll encounter has the same key step: an E2-like deprotonation of C-H that results in formation of a new C-O pi bond and breakage of a transient leaving group.
- Aldehydes oxidize to carboxylic acids after formation of a hydrate.
- Ketones don’t oxidize further because there’s no C-H bond that can be broken that would result in a new C-O pi bond.
In the next post we’ll move to something completely different: intramolecular reactions of alcohols, a perennial subject of organic chemistry exams.
Next Post – Intramolecular Reactions Of Alcohols And Ethers
Notes
Note 1. The main exception you’ll encounter is KMnO4, which likely proceeds through a C-H abstraction/internal return type mechanism followed by collapse of the hydrate to give the new carbonyl. That mechanism is mentioned in exactly zero introductory textbooks, so you likely don’t “need” to know this unless you are exceptionally curious about organic chemistry. [back to post]
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
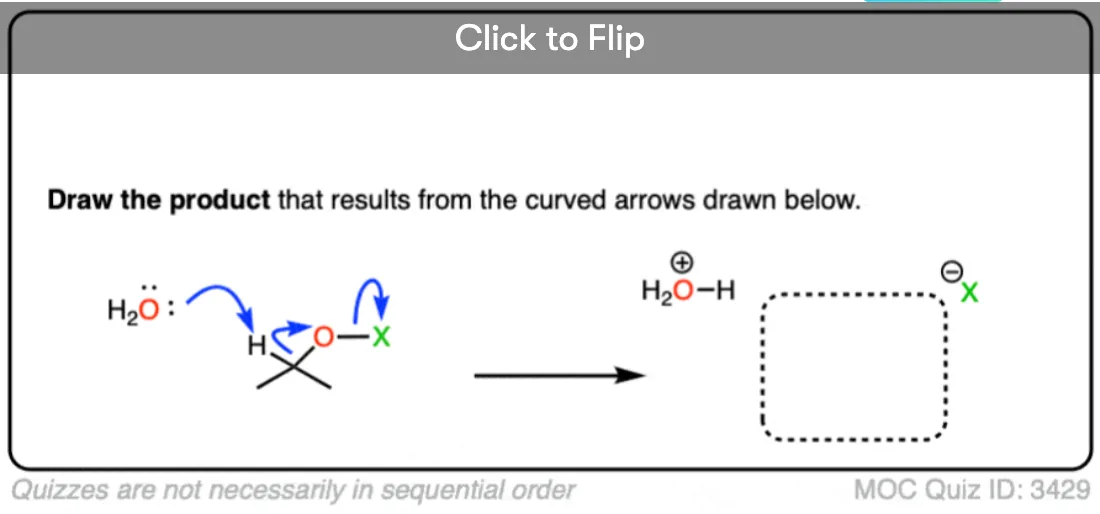
Become a MOC member to see the clickable quiz with answers on the back.
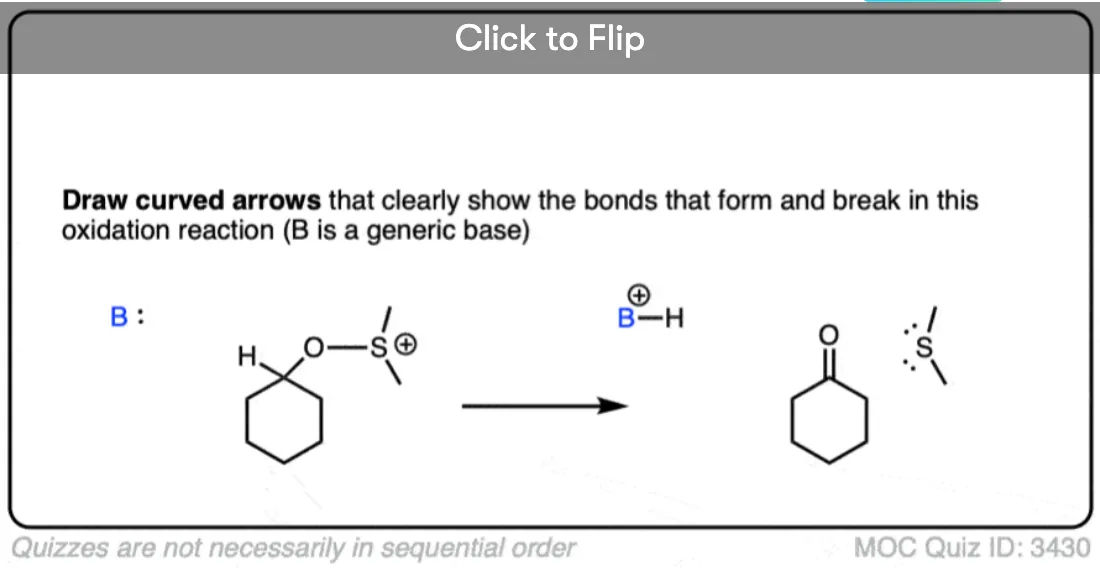
Become a MOC member to see the clickable quiz with answers on the back.
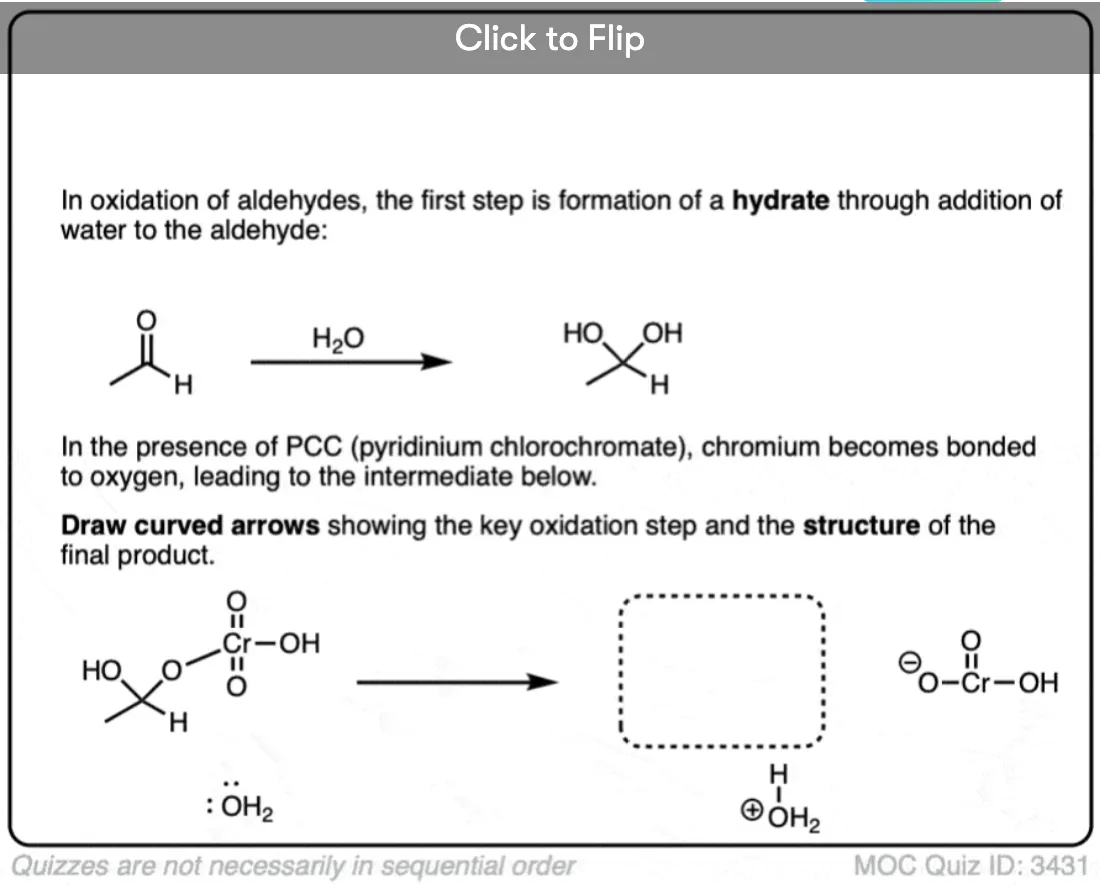
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
Dess-Martin Periodinane:
- A useful 12-I-5 triacetoxyperiodinane (the Dess-Martin periodinane) for the selective oxidation of primary or secondary alcohols and a variety of related 12-I-5 species
Daniel B. Dess and J. C. Martin
Journal of the American Chemical Society 1991, 113 (19), 7277-7287
DOI: 1021/ja00019a027 - Oxidation of fluoroalkyl-substituted carbinols by the Dess-Martin reagent
Russell J. Linderman and David M. Graves
The Journal of Organic Chemistry 1989, 54 (3), 661-668
DOI: 10.1021/jo00264a029
#1 is by the developers of the eponymous ‘Dess-Martin Periodinane’, a hypervalent I(V) compound that has found widespread use as a mild oxidant in organic synthesis. Prof. J. C. Martin spent most of his career at University of Illinois Urbana-Champaign and ended his career at Vanderbilt University. During his career he contributed a lot towards our understanding of hypervalent main-group chemistry, preparing many S(IV), S(VI), Br(III), I(III), I(V), and I(VII) compounds, among others. Ref #2 extends the substrate scope to fluorinated alcohols, and the use of fluorine also enables mechanistic studies of the oxidation via 19F NMR.Swern oxidation: - Structure of the dimethyl sulfoxide-oxalyl chloride reaction product. Oxidation of heteroaromatic and diverse alcohols to carbonyl compounds
Mancuso, A. J.; Brownfain, D. S.; Swern, D.
J. Org. Chem. 1979, 44 (23): 4148–4150
DOI: 10.1021/jo01337a028 - Mechanisms of dimethylsulfoxide oxidations
Kurt Torssell
Tetrahedron Letters 1966 7 (37), 4445-4451
DOI: 1016/S0040-4039(00)70057-8
These papers are on what is now commonly called the “Swern oxidation” after its developer, Daniel Swern. This method is rather mild and uses DMSO, a common solvent, as the oxidant. However, this also results in the formation of dimethyl sulfide (which is notoriously stinky) as the product of the reaction, one of its noteworthy characteristics.Corey-Kim oxidation: - New and highly effective method for the oxidation of primary and secondary alcohols to carbonyl compounds
E. J. Corey; C. U. Kim
Journal of the American Chemical Society 1972, 94 (21): 7586–7587
DOI:10.1021/ja00776a056. - A method for the oxidation of sec,tert-1,2-diols to α-hydroxy ketones without carbon-carbon cleavage
E. J. Corey; C. U. Kim
Tetrahedron Letters 1974, 15 (3): 287–290
DOI:10.1016/S0040-4039(01)82195-X
These papers by Nobel Laureate Prof. E. J. Corey (Harvard) are on the development of what is now known as the “Corey-Kim” oxidation. This is very similar to the Swern oxidation in that DMSO is used as the oxidant, except that here NCS (N-chlorosuccinimide) is used instead of oxalyl chloride. The advantage with this procedure is that temperatures above –25 °C can be used, and the disadvantage is that substrates susceptible to chlorination by NCS cannot be used.KMnO4 oxidation: - Oxidations with Manganese Dioxide
P. Papadopoulos, A. Jarrar, and C. H. Issidorides
The Journal of Organic Chemistry 1966, 31 (2), 615-616
DOI: 10.1021/jo01340a520
As this paper shows, MnO2 can also be used for oxidation of secondary alcohols. - Synthesis of a model depsipeptide segment of Luzopeptins (BBM 928), potent antitumor and antiretroviral antibiotics
Marco A. Ciufolini and Shankar Swaminathan
Tetrahedron Letters Volume 30, Issue 23, 1989, Pages 3027-3028
DOI: 1016/S0040-4039(00)99393-6
Step f in the synthesis (Scheme 1) is an oxidation of a primary alcohol to carboxylic acid using KMnO4. - Stereocontrolled addition to a penaldic acid equivalent: an asymmetric of -β-hydroxy-L-glutamic acid
Philip Garner
Tetrahedron Letters Volume 25, Issue 51, 1984, 5855-5858
DOI: 10.1016/S0040-4039(01)81703-2
The final step (g, 6 -> 7) in the synthesis in this paper is an oxidation of a primary alcohol to a carboxylic acid using KMnO4.PCC (pyridinium chlorochromate) oxidation: - Pyridinium Chlorochromate: A Versatile Oxidant in Organic Synthesis
Piancatelli, A. Scettri, M. D’Auria
Synthesis 1982; 1982(4): 245-258
DOI: 10.1055/s-1982-29766
Review on the applications of PCC in organic synthesis. Includes a discussion on the mechanism. - Kinetics and Mechanism of the Oxidation of Alcohols by Pyridinium Chlorochromate
Banerji Kalyan K.
Bull. Chem. Soc. Jpn. 1978, 51 (9), 2732
DOI: 10.1246/bcsj.51.2732
A nice mechanistic study of PCC oxidation, and includes a probable mechanism of the reaction. - Stoichiometry of the oxidation of primary alcohols with pyridinium chlorochromate. Evidence for a two-electron change
Herbert C. Brown, C. Gundu Rao, and Surendra U. Kulkarni
The Journal of Organic Chemistry 1979 44 (15), 2809-2810
DOI: 1021/jo01329a051
In this paper, Nobel Laureate H. C. Brown proves that PCC oxidations involve a transfer of 2 electrons from the Cr to the substrate. Therefore, one does not need to use an excess of PCC – 1 equivalent works fine.
The Jones oxidation, which uses chromic acid (CrO3 in H2SO4) is a common method for the oxidation of primary alcohols to carboxylic acids. The drawback is of course the production of stoichiometric amounts of chromium waste. - Researches on acetylenic compounds. Part XIV. A study of the reactions of the readily available ethynyl-ethylenic alchohol, pent-2-en-4-yn-1-ol
Sir Ian Heilbron, E. R. H. Jones and F. Sondheimer
J. Chem. Soc., 1947, 1586-1590
DOI: 10.1039/JR9470001586 - An Improved Procedure for the Oxidation of Alkynols to Alkynoic Acids
C. Holland and N. W. Gilman
Synth. Commun. 1974, 4, 203-210
DOI: 10.1080/00397917408062073Oxidation with PDC (pyridinium dichromate): - Useful procedures for the oxidation of alcohols involving pyridinium dichromate in aprotic media
E. J. Corey, Greg Schmidt
Tetrahedron Letters Volume 20, Issue 5, 1979, 399-402
DOI: 10.1016/S0040-4039(01)93515-4
Nobel Laureate Prof. E. J. Corey (Harvard) shows that PDC (pyridinium dichromate) in DMF can be used for the oxidation of primary alcohols to carboxylic acids.
Why do some reactant like Collins reagent stop their oxidation at the aldehyde level?
Is it just the absence of water?
Question:
If the hydrate happens BEFORE the actual attachment of the leaving group, why do we have different outcome with different leaving group?
Paragraph 2:
“2. Oxidants Are Essentially Just Fancy Reagents For Attaching Good “Leaving Groups” Directly To Oxygen”
Just fabulous
Hi, will there be a major product if a molecule with both primary and secondary alcohol is oxidized? I mean, is there a preference for oxidizing primary or secondary?
There are some reagents that will preferentially oxidize primary over secondary, and vice-versa. You usually don’t learn about such details in introductory organic, but an example of the first type (primary over secondary) is TEMPO, and an example of the second type is Bobbitt’s reagent (among others)
See this super useful handout. http://hwpi.harvard.edu/files/myers/files/6-oxidation.pdf
I was wondering if the prof could give a quick answer on NAD+ as a leaving group. Nicotinamide Adenine Dinucleotide acts as a leaving group in the same way? Probably a weak oxidant?
Thank you.
Hi!
I loved your site! You explain very well!
I have a doubt…
How can I convert secondary alcohols into aldehydes? it’s possible?
Not without breaking a C-C bond somehow.
Which book covers the KMnO4 oxidation mechanism correctly?
Good luck. It’s complicated. Start with March’s advanced organic chemistry and dig in there.
You’ve mislabeled the ketone as aldehyde in the second last diagram.
Yes – thank you, finally fixed!
Hey, I have a doubt.
Can ketones be oxidised to carboxylic acids in the presence of H2O?
Not under any conventional conditions we cover, because a C-C bond would need to break.
Non-conventionally though, and by a completely novel mechanism, methyl ketones can be oxidized to carboxylic acids in the haloform reaction. :)
Excellent post, makes a lot of things clearer even for me.
One minor observation: in the hydrate formation image, in the first reaction it looks like the water molecules attacks the C=O bond; please shift the arrow tip a bit to the left.
Will fix. Thanks for your suggestions, as always. James