Free Radical Reactions
Bond Strengths And Radical Stability
Last updated: April 20th, 2026 |
Bond Dissociation Energies And Radical Stability
- The last article discussed factors which stabilize – and destabilize – free radials.
- So how do we quantify free-radical stability?
- Some caution is required – see this note – but if we keep enough variables constant, the strength of C–H bonds (“Bond Dissociation Energies”, or BDE’s) are a pretty good guide to the stability of carbon radicals, and other radicals besides.

Table of Contents
- Quantifying Free Radical Stability
- Why Does H2O Have A Higher Bond Dissociation Energy Than CH4?
- Bond Dissociation Energy Correlates With Free-Radical Stability
- Factor #1: Stability Increases In The Order Methyl < Primary < Secondary < Tertiary. Bond Dissociation Energies (BDE’s)
- Factor#2: Free Radicals Are Stabilized By Resonance.
- Factor #3: Free Radials Are Stabilized By Adjacent Atoms With Lone Pairs
- Factor #4: Across The Periodic Table, Free-Radical Stability Decreases With Increasing Electronegativity
- Factor #5: Down The Periodic Table, Free-Radical Stability Increases With Increasing Size Of The Atom
- Factor #6: The Stability Of The Free Radical Decreases As The Orbital Is Held Closer To The Nucleus
- Factor #7: Electron Withdrawing Groups Destabilize Free Radicals
- Summary: Quantifying Free-Radical Stability With Bond Dissociation Energies (BDE’s)
- Notes
- (Advanced) References and Further Reading
1. Quantifying Free Radical Stability
In the last article we went through the factors which affect the stability of free radicals (See article – Factors Which Stabilize Free Radicals).
The bottom line is that
- Free radicals are electron-deficient due to their half-filled orbital. Therefore, radicals will be stabilized by any factor which either
- donates electron density to the half-filled orbital, or
- disperses the orbital containing the half filled orbital over a larger volume (a.k.a “delocalize” it) or
- results in the half-filled orbital being further away from the positively-charged nucleus (i.e. feeling less effective nuclear charge)
There were a total of six factors we discussed. You might initially find it hard to keep track of the factors we mentioned. That’s OK, because interestingly, there is one measurement which can help us keep all of these factors straight. [EDIT: provided we confine ourselves to some simple examples provided herein]
It’s called Bond Dissociation Energy [BDE]. You might be familiar with it already!
There’s probably a table of bond dissociation energies in your textbook, usually within the first 100 pages or so.
What many people take some time to realize is that BDE is a measure of the energy required for homolytic bond cleavage, and as we discussed earlier, homolytic bond cleavage leads to the formation of free radicals [heterolytic bond cleavage, which is much more common in organic chemistry, leads to the formation of at least one charged species [ions]].
Therefore, BDE is essentially a measure of free radical stability, at least for C-H bonds. [EDIT: As Prof. Wenthold points out, the stability of the starting molecule is also a factor to consider. In this post we deal the simple cases of bonds to H that are unstrained, but in cases with strained bonds, bonds weakened by significant electron repulsion, or bonds to very electronegative atoms, corrections must be made for these factors in order to ascertain quantitative free radical stabilities]
So let’s use some bond dissociation energy data to help connect these concepts.
2. Why Does Water Have A Higher Bond Dissociation Energy Than Methane?
Take two molecules – methane (CH4) and water (H2O). Which bond is stronger, O-H or C-H ?
Thinking back to some of the chemistry we’ve talked about earlier, such as acid-base reactions, it might be tempting to say that O–H is weaker than C–H, since we can think of many strong bases which will deprotonate water [pKa = 14], but very few that will deprotonate alkanes [pKa = 50].
However when we look at the BDE’s, we see that HO–H is 118 kcal/mol and H3C–H is 104 kcal/mol. So the C–H bond is weaker? What gives?
If you’re thinking about BDE’s and acid-base reactions, you’re using the wrong mental model.
Acid base reactions involve heterolytic cleavage. BDE’s are a measure of homolytic cleavage.
Instead of the stability of the ions (heterolytic cleavage!) we need to look at the stability of the free radicals (homolytic cleavage).
Here’s an example of what we need to look at in this case:
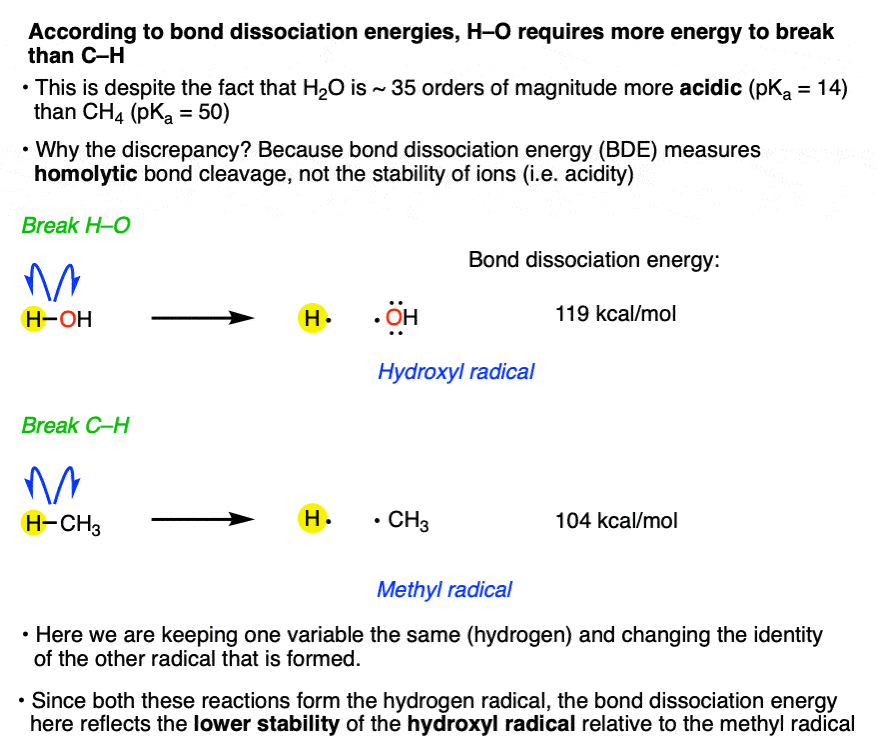
Note how we’re forming a H radical in both cases. What’s different is the identity of the other radical.
That leads us to comparing the stability of H3C• and HO• , and we learned previously that [all else being equal] the stability of free radicals decreases as we go from left to right across the periodic table, since O is more electronegative than C and that partially empty orbital is being held more closely to the positively charged nucleus. More on that below.
3. Bond Dissociation Energy Is Correlated With Free Radical Stability
The bottom line for this post is that bond dissociation energy is correlated to free radical stability.
Low bond dissociation energies reflect the formation of stable free radicals, and high bond dissociation energies reflect the formation of unstable free radicals.
[EDIT: Caveats apply when extending this discussion beyond the scope discussed in this post. See Note 1]
If we keep one variable constant and vary the other variable, we can analyze the influence of structure on free radical stability.
Here we’re going to keep H as the variable which is the same, and by examine the trends which influence free radical stability in a new light.
Let’s look at these seven factors in turn:
- Stability increases in the order methyl (least stable) < primary < secondary < tertiary (most stable)
- Free radicals are stabilized by resonance
- Free radicals are stabilized by adjacent atoms with lone pairs
- Free radicals increase in stability as the electronegativity of the atom decreases
- Free radicals increase in stability as we go down the periodic table (larger size, more polarizable)
- Free radicals decrease in stability as we go from sp3 to sp2 to sp hybridization
- Adjacent electron withdrawing groups decrease the stability of free radicals.
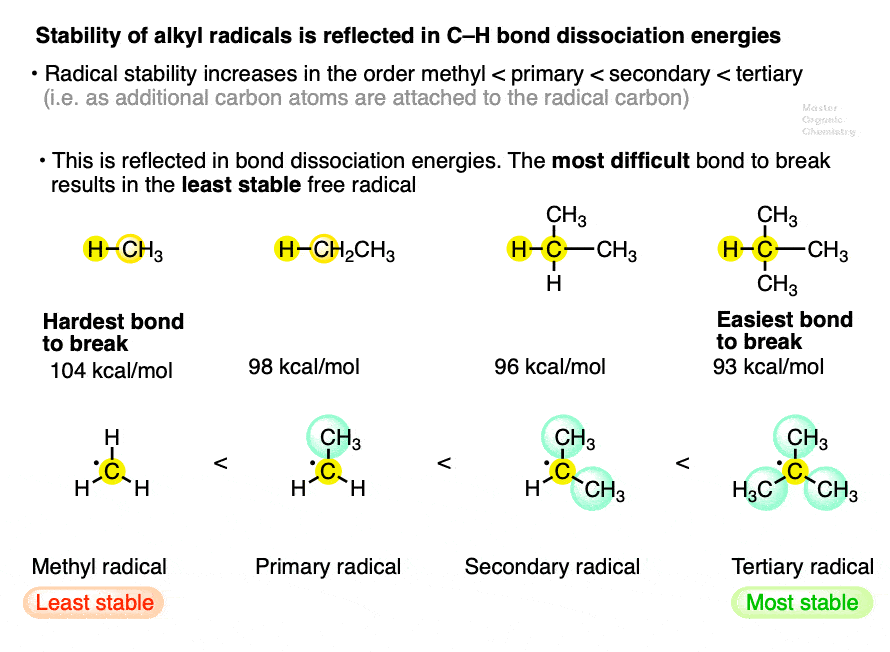
4. Factor #1: Stability increases in the order methyl < primary < secondary < tertiary
Note that the BDE of C-H bonds decreases as we go from methyl to primary to secondary to tertiary. They are easier to break since homolytic bond cleavage results in a more stable radical.
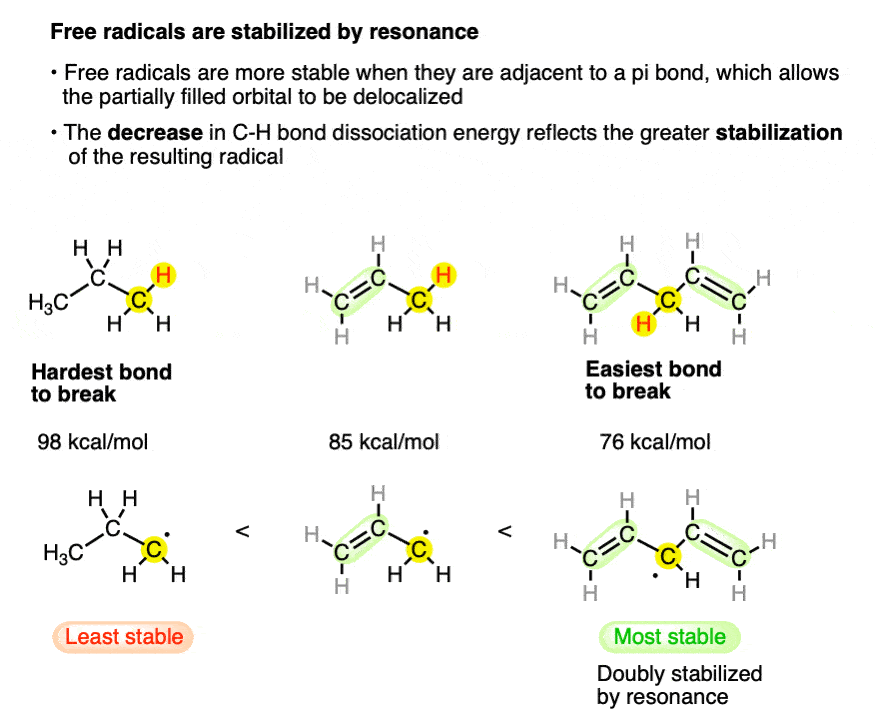
5. Factor #2: Free radicals are stabilized by resonance.
Note the difference in bond strengths between the (primary) C-H bond of propane and of the alkyl C-H bond of propene. The sizeable difference [~13 kcal/mol] is a reflection of the greater stability of the resonance-stabilized “allyl” radical.
Although not directly comparable, look at the C-H bond strength when it is adjacent to two alkenes [76 kcal/mol]. This “doubly allylic” C–H bond is even weaker, reflecting the fact that a greater number of resonance forms are available for the radical species.
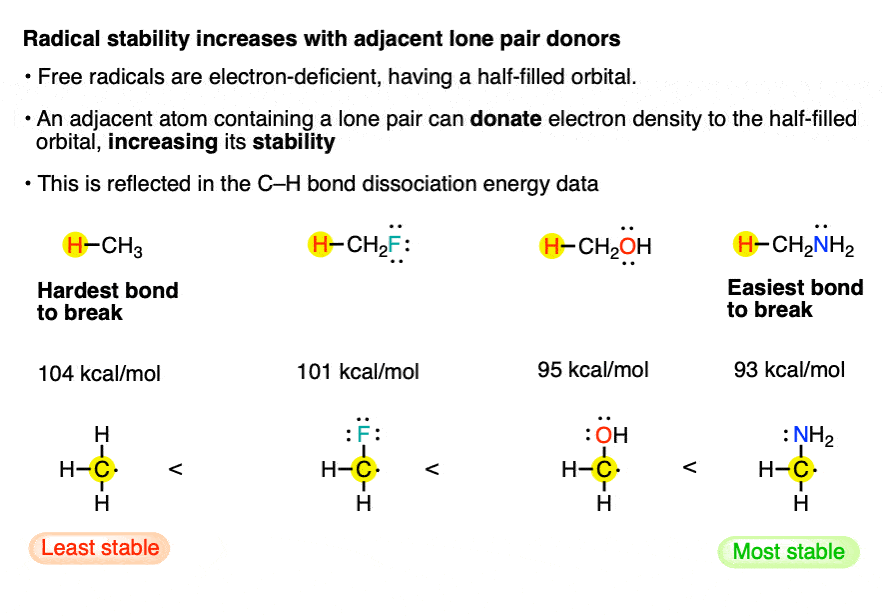
6. Factor #3: Free radicals are stabilized by adjacent atoms with lone pairs.
[This is a subtle point!]. Note the difference in bond strengths between the C-H bond of methane [104 kcal/mol] and that of methanol [95 kcal/mol]. In between we have the C-H bond of fluoromethane [101 kcal/mol].
Note that even though fluorine is more electronegative than H, the presence of the lone pairs on F is actually stabilizing relative to H.
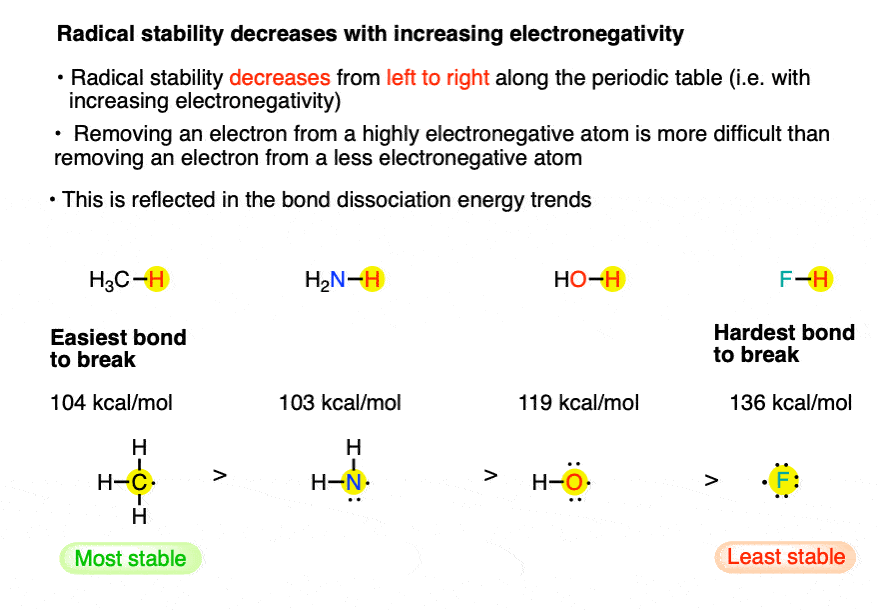
7. Factor #4: Across the periodic table, free radical stability decreases with increasing electronegativity.
Note the difference between H-CH3 [104], H-OH [119] and H-F [136].
The most electronegative element has the least stable free radical and this is reflected in the higher bond strength.
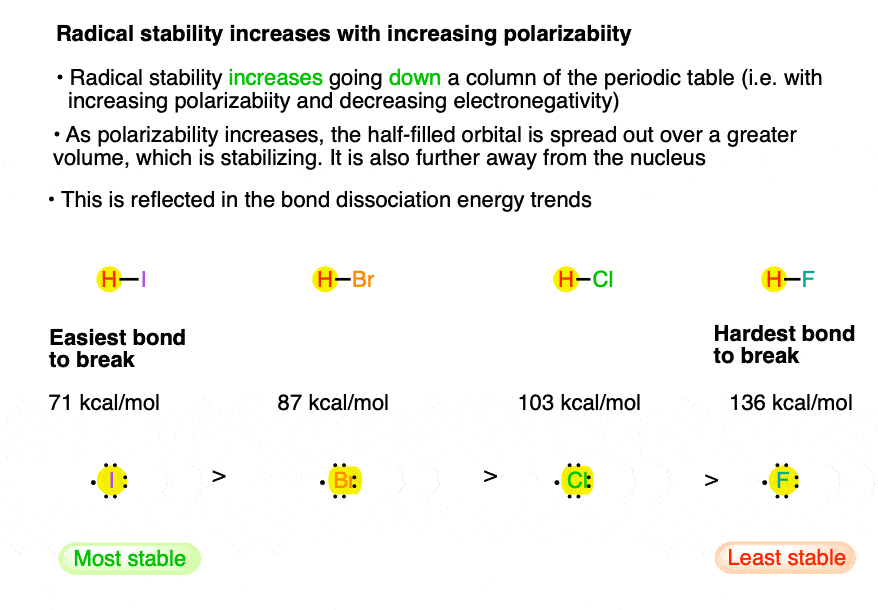
8. Factor #5: Down the periodic table, free radical stability increases with increasing size of the atom.
Look how the BDE decreases as we go from H-F [136] to H-Cl [103] to H-Br [87] to H-I [71]. As we should expect by now, the iodide radical is the most stable, since the orbital is larger in size and is therefore “spread out” over a larger volume. further away from the nucleus, therefore “feeling” less effective nuclear charge than would a smaller atom. [thanks to commenter Xylene for this constructive suggestion].
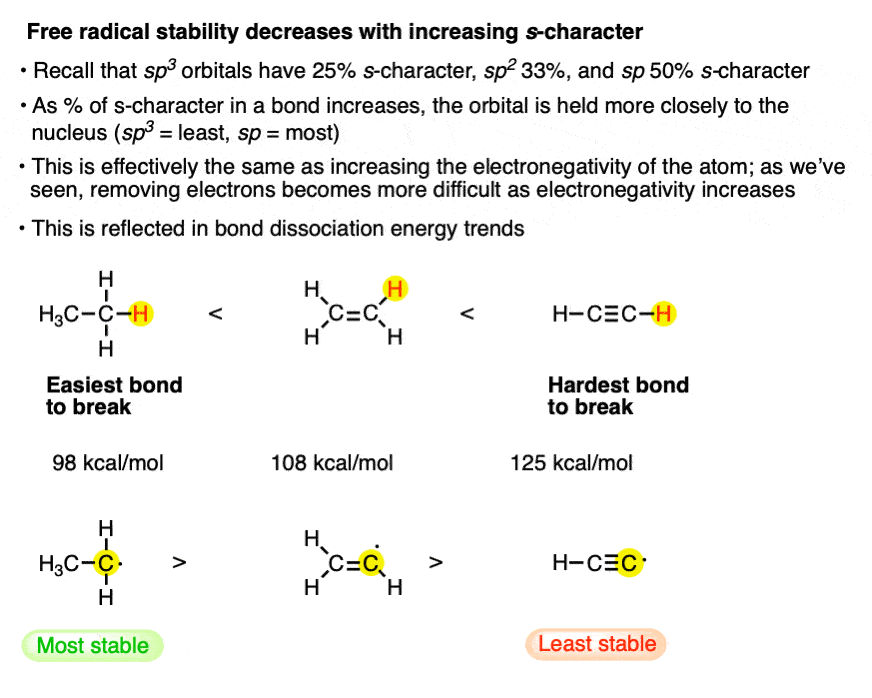
9. Factor #6: The stability of the free radical decreases as the orbital is held closer to the nucleus.
Look what happens to the bond strength as we go from ethane, which is sp3 hybridized [98 kcal/mol] to ethene [sp2, 109 kcal/mol] to acetylene [sp, 125 kcal/mol].
This is largely the same effect as #5 above – the farther away from the nucleus the half-filled orbital is, the more stable it will be.
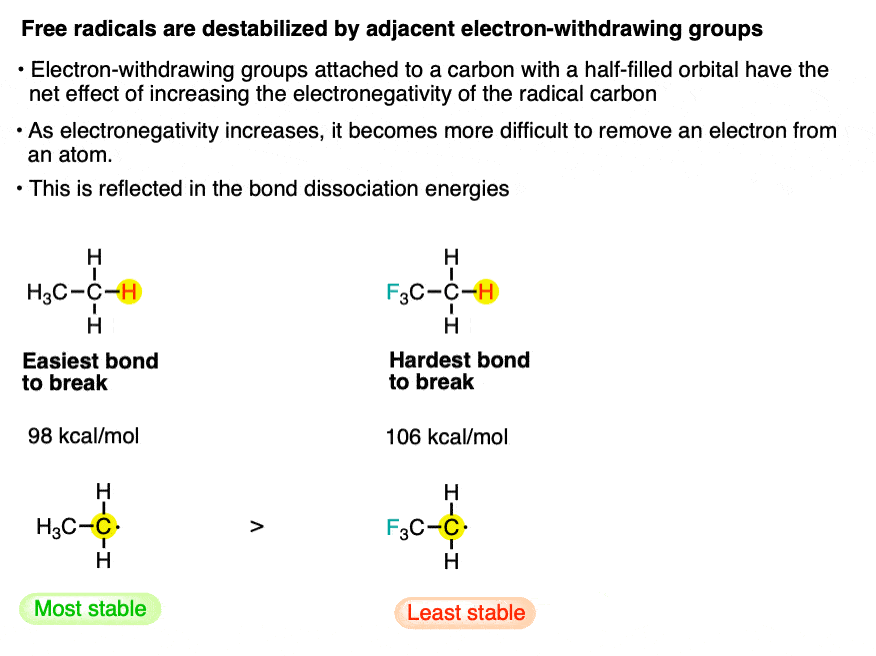
10. Factor #7: electron withdrawing groups destabilize free radicals.
To isolate this effect it’s important to look at examples where the electron-withdrawing group cannot donate a lone pair to the radical (see factor #3). One good example is comparing the C-H strength in ethane vs. trifluoroethane.
11. Summary: Quantifying Free Radical Stability With Bond Dissociation Energies
Hopefully it’s clear by now that where C-H bonds are concerned, by examining bond dissociation energies, we can discern trends in free-radical stabilities.
This will be of prime importance in understanding selectivity in free radical reactions: “which free radical forms?”.
A subtle point is that it is also important in understanding fragmentation patterns in mass spectroscopy, but we’re not there yet.
Next Post: Radical Reactions – Why Is Heat Or Light Required?
Notes
Source of bond dissociation energies in this article (PDF)
Note 1. More caution is required than I had previously indicated regarding the main thesis of this post – that free radical stabilities are solely reflected by bond strengths. They depend on the stability of both reactant and reagent.

Edits are indicated inline. For more discussion see bottom section.[ TL;DR – the general trends in this post are valid because we discuss bonds to H, but use caution when comparing any other type of bond other than hydrogen.] Thanks to Prof. Paul Wenthold (Purdue University) for his input.
Using the bond strengths (BDE’s) of unstrained bonds to hydrogen is a reasonable method for discerning trends in radical stabilities, as discussed in this post.
However, BDE’s in and of themselves are not reliable for discerning absolute radical stabilities in cases where the bond may be weakened by strain, repulsion between lone pairs, or other factors.
For example the BDE for hydrogen peroxide is 51 kcal/mol, which does NOT imply that the HO• radical is stable, but rather that the O–O bond is destabilized by repulsion between the lone pairs.
Note 2. In looking at bond strength data there are often differences in 2-3 kcal between different sources. The important part here is not so much the absolute numbers, but the trends. These two files helped when compiling the numbers for this post:
(Advanced) References and Further Reading:
- Shortcomings of Basing Radical Stabilization Energies on Bond Dissociation Energies of Alkyl Groups to Hydrogen
Andreas A. Zavitsas, Donald W. Rogers, and Nikita Matsunaga
The Journal of Organic Chemistry 2010, 75 (16), 5697-5700
DOI: 1021/jo101127m
Several textbooks, including some advanced ones, provide radical stabilization energies, and this paper discusses why that may not be the best way to quantify the stability of free radicals. - On the Advantages of Hydrocarbon Radical Stabilization Energies Based on R−H Bond Dissociation Energies
Matthew D. Wodrich, W. Chad McKee, and Paul von Ragué Schleyer
The Journal of Organic Chemistry 2011, 76 (8), 2439-2447
DOI: 1021/jo101661c
This paper addresses some of the shortcomings with the approach used in Ref #1 above. The late Prof. Schleyer was a very influential figure in organic chemistry, and was a pioneer in using computational methods to address interesting problems in organic chemistry. - The Radical Stabilization Energy of a Substituted Carbon-Centered Free Radical Depends on Both the Functionality of the Substituent and the Ordinality of the Radical
Marvin L. Poutsma
The Journal of Organic Chemistry 2011, 76 (1), 270-276
DOI: 1021/jo102097n - A Single Universal Scale of Radical Stabilization Energies Does Not Exist: Global Bond Dissociation Energies and Radical Thermochemistries Are Described by Combining Two Universal Scales
Andreas A. Zavitsas
The Journal of Organic Chemistry 2008, 73 (22), 9022-9026
DOI: 1021/jo8018768 - Bond Dissociation Energies by Kinetic Methods
A. Kerr
Chemical Reviews 1966, 66 (5), 465-500
DOI: 10.1021/cr60243a001
This paper describes experimental techniques for measuring homolytic BDEs. - III – Bond energies
Sidney W. Benson
Journal of Chemical Education 1965, 42 (9), 502
DOI: 10.1021/ed042p502
This paper describes the empirical measurement of homolytic bond dissociation energies. This paper was written by Prof. Benson while at the Stanford Research Institute (now SRI International), a non-profit research center very close to Stanford University. In 1978, Prof. Benson joined Prof. George Olah at USC and helped established the Loker Hydrocarbon Research Institute there. - From equilibrium acidities to radical stabilization energies
Frederick G. Bordwell and Xian Man Zhang
Accounts of Chemical Research 1993, 26 (9), 510-517
DOI: 10.1021/ar00033a009
This paper attempts to correlate the acidity of a proton with the BDE of the corresponding C-H or X-H bond. - Ab Initio Calculations of the Relative Resonance Stabilization Energies of Allyl and Benzyl Radicals
David A. Hrovat and Weston Thatcher Borden
The Journal of Physical Chemistry 1994, 98 (41), 10460-10464
DOI: 1021/j100092a014
The stabilization energy of a vinyl group (in the allyl radical) and a phenyl group (in the benzyl radical) has been calculated to be 15.7 kcal/mol and 12.5 kcal/mol, respectively. - Effects of adjacent acceptors and donors on the stabilities of carbon-centered radicals
G. Bordwell, Xianman Zhang, and Mikhail S. Alnajjar
Journal of the American Chemical Society 1992, 114 (20), 7623-7629
DOI: 10.1021/ja00046a003
Table I in this paper contains stabilization energies of methyl radicals with various substituents (e.g. ·CH2X).
Hey, as u mentioned in the section (8) ,the Iodine free radical is most stable. But if I’m not wrong alkyl iodides are not prepared using this mechanism as the free radical formed (Of Iodine) forms I2 molecule. Thank You !!
Yes, you are correct that alkyl iodides are not prepared directly through free-radical halogenation because the H-I bond is much weaker than the C-H bond.
Thank you, sir. But what about I–I and F–F, 36 kcal/mole and 38 kcal/mole, BDE’s respectively. Apparently, their radicals have same behaviour but it turns out they don’t. Fluorine free radical is more selective, but Iodine reacts with any Hydrogen. Am I wrong? Why does that happen?
Where did you get that fluorine radical is more selective? Running reactions with fluorine radicals is not for the faint of heart…
Why is methyl radical more stable than NH2 in factor 4
Higher electron affinity of nitrogen versus carbon due to greater electronegativity.
Can someone help me the order of stability of tertairy carbo free radical.
Eg: 3
methyl groups vs three ethyl groups
Probably about the same. Trivial difference.
James, i have 2 questions
Firstly: free radicals decrease in stability as we reduce the amount of hybridization? This seems totally counter intuitive; you’d expect that sp would be much more stable because the radical is closer to the nucleus due to 50% s character… why is this?
Secondly: adjacent lone pairs stabilize a radical? woah, this one totally blew my mind; you’d expect electronic repulsion/increased electron density to destabilize the radical?
Thanks, and glad you liked the mnemonic (i created it for my physical organic test tomorrow :/
1)Stability increases in the order methyl < primary < secondary < tertiary
2) Free radicals are stabilized by resonance
3) Free radicals are stabilized by adjacent atoms with lone pairs
4) Free radicals increase in stability as the electronegativity of the atom decreases
5) Free radicals increases in stability as we go down the periodic table (larger size)
6) Free radicals decrease in stability as we go from sp3 to sp2 to sp hybridization
7)Adjacent electron withdrawing groups decrease the stability of free radicals
4 and 7 say the same thing, so we only need 1 reminding us about electronegativity.
A good mnemonic is S H A R E S:
Substitution
Hybridization
Adjacent lone pairs
Resonance
Electronegativity
Size
Thanks Tim!
Just thought I’d add that in our course it is taught that both EWGs and EDGs stabilise radicals, because of the increased stabilisation of the electron orbitals. It is even more preferred if both groups are present – captodative. Comparing CH3CH2 to F3CH2 is not appropriate as methyl groups are ED.
You’ll notice the EWG I chose is *not* captodative [for exactly that reason : – ) ] . My point was merely that CH3CH2 was more stabilizing than CF3CH2 .
Here, having what would typically be a electron withdrawing atom (e.g., F, OH) with lone pairs attached to the radical is stabilizing. That is, it is electron donating through induction.
But, when dealing with acidity, these types of atoms connected adjacently to a carbanion in say the conj. base of methyl would stabilize the excess negative charge.
How is it that in this context with radicals, F with lone pairs stabilizes the radical through donation but then withdraws electron density to stabilize a negative charge?
Re: your first paragraph it is NOT electron donating through induction.
Re: your third paragraph – To really discuss this issue, we’d use molecular orbital theory to show how donation of a lone pair into the half-filled orbital is a stabilizing phenomenon, which lowers the energy of the radical. It turns out that the donation of the lone pair is more stabilizing than the removal of electron density via induction.
2nd paragraph last – this phenomenon does not occur in anions, because there’s no empty (or half-empty) orbital to accept the negative charge. That’s why stabilization of negative charge on a carbanion by electron withdrawing atoms such as F is purely an inductive phenomenon.
I think the effect between C and N in factor 4 is due to geometrical relaxation as a consequence of change in hybridisation. Both carbon and nitrogen goes from sp3 in the closed shell molecule to sp2 in the radical. Carbon has three hydrogen atoms to rearrange whereas nitrogen only has two. If the energies of the methyl and amino radicals were to be calculated in fixed geometries (still sp3 hybridisation) I think ammonia would appear to have the higher BDE.
I think I have a reason for why H-NH2 is anomalous. It’s the same explanation why N has near zero electron affinity instead of a positive one. When the .NH2 radical is formed, since radicals are electrophilic, you would expect it to gain an electron to form the NH2- ion. However, the electron does not want to pair up with another electron since it will increase electrostatic repulsion between them, so .NH2 isn’t particularly reactive since there’s that energy barrier. As for O onwards, the added protons increase electron attraction such that it is no longer problematic.
The electron affinity trend of radicals is CH3<NH2<OH<F as can be seen on the NIST Chemistry Webbook. The reason N has a negative electron affinity is because it has a semi closed shell structure.
Regarding factor #5: Is it quite right to say that the radical is more stable because it’s more spread out? After all, the closer an orbital is to the nucleus (lower quantum number or increasing effective nuclear charge), the lower its energy (i.e. the higher its stability). Technically speaking, this would mean that all else being equal, a radical with the lone electron in a large orbital (further away from the nucleus) would be more unstable than a radical with a lone electron in a small orbital (closer to the nucleus).
I believe the real reason at work here has the same basis though. Iodine has much larger orbitals and thus forms weaker bonds to itself simply because overlap isn’t so efficient. The weaker the bonds broken, the more stable the radicals formed (relative to the reactants). On an absolute scale though, I think a chlorine radical would be more stable than an iodine radical – it’s just more unstable relative to chlorine.
A similar point can be made for factor #6: sp and sp2 C-H bonds are just stronger than sp3 C-H bonds. A radical with an electron in an sp or sp2 orbital will probably be more stable than a radical with the electron in an sp3 orbital, on an absolute scale, but relative to the reactants, the order is as you’ve stated.
this is a much better explanation – reduction in potential energy. I will re-edit this. Thank you
Mmm…actually in retrospect I’m not so sure how much “absolute reactivity” matters. After all, chemical equilibria are all based on energy *changes*, i.e. relative energies. So a chlorine radical is still going to be more reactive than an iodine radical (assuming it attacks another Cl atom) because the energy difference between the chlorine radical and dichlorine is higher than that between the iodine radical and diiodine (ignoring kinetic factors, of course).
Inherent in that is the problem that the chlorine radical doesn’t always attack another chlorine atom, and the same goes for the iodine radical. But I suppose you can use the strength of the Cl-Cl or I-I bond as a rough gauge of the bond energy between Cl and another atom (say hydrogen) or between I and another atom.
[In essence what I’m trying to say is that absolute stability is a bit of a red herring here – it’s a valid point but doesn’t have much bearing on “real chemistry”]
Absolute stability is a bit of a red herring, what I’m trying to say is that there is not a 1:1 correlation between the differences in bond enthalpies and differences in radical stability. However, as you say, it is certainly a rough (qualitative) gauge and good enough for our purposes here.
hi I’m feeling a bit confused after reading the article and your comment. So is Cl radical more stable or iodine radical? Or does it depend much on the reacting environment?
I would be grateful if you could explain this in more detail. Thanks!
The H-I bond is weaker, so the iodide radical is more stable. The H-Cl bond is stronger, so the Cl radical is more unstable. Does that make sense?