Substitution Reactions
The SN2 Mechanism
Last updated: June 2nd, 2026 |
The SN2 Reaction Mechanism
Having gone through the two different types of substitution reactions, and talked about nucleophiles and electrophiles, we’re finally in a position to reveal the mechanism for one of the most important reactions in organic chemistry.
It’s called the SN2 reaction, and it’s going to be extremely useful for us going forward.
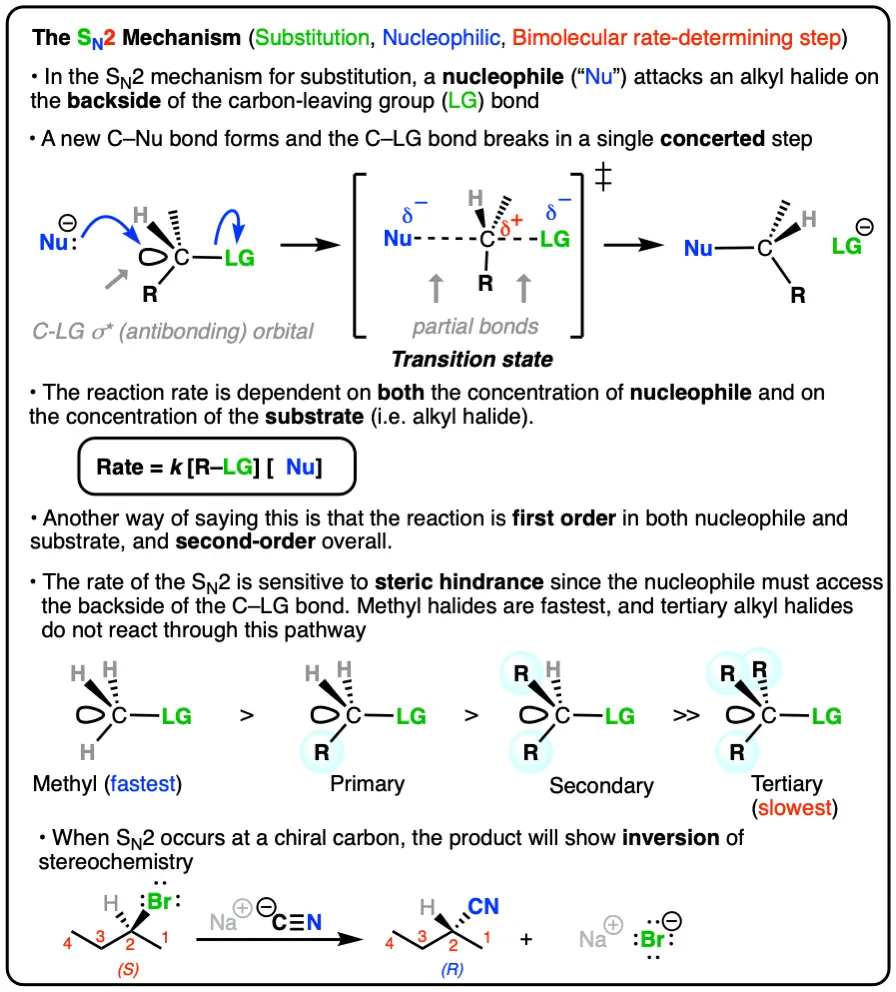
Table of Contents
- The SN2 Reaction Proceeds With Inversion of Configuration
- The Rate Law Of The SN2 Is Second Order Overall
- The Reaction Rate Is Fastest For Small Alkyl Halides (Methyl > Primary > Secondary >> Tertiary)
- The SN2 Mechanism Proceeds Through A Concerted Backside Attack Of The Nucleophile Upon The Alkyl Halide
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
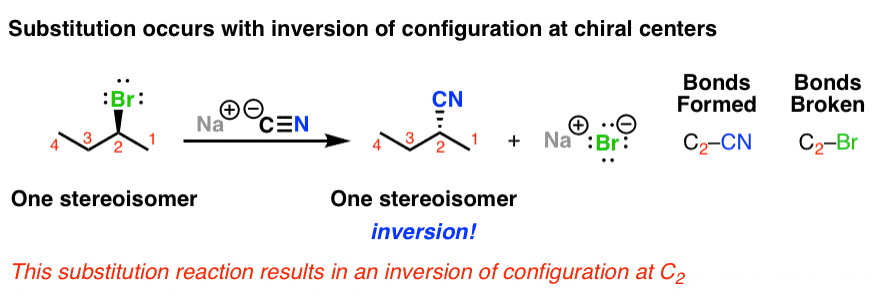
1. The SN2 Reaction Proceeds With Inversion of Configuration
When we start with a molecule with a chiral center, such as (S)-2-bromobutane, this class of reaction results in inversion of stereochemistry. Note how we start with (S)-2-bromobutane and end up with (R)-2-methylbutanenitrile.
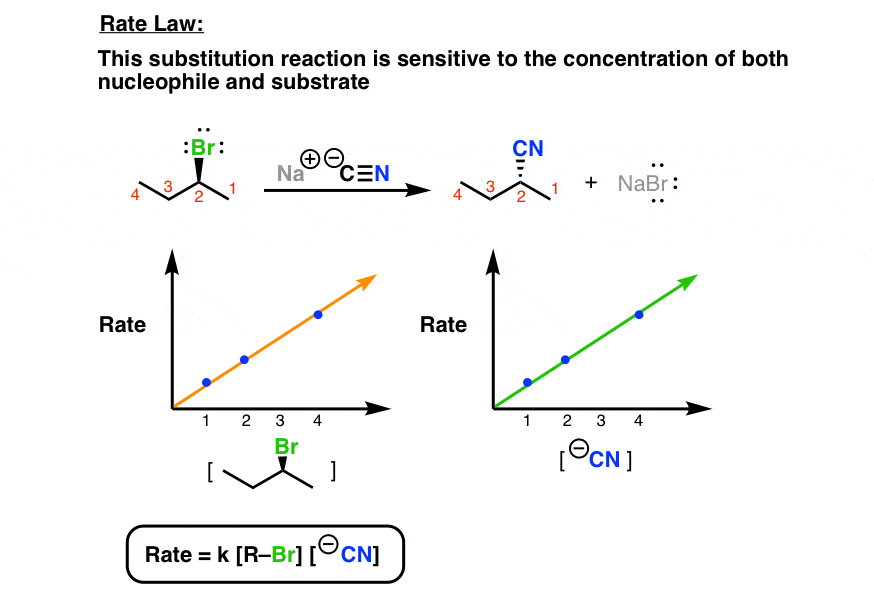
2. The Rate Law Of The SN2 Is Second Order Overall
Note how the rate of the reaction is dependent on both the concentration of the nucleophile and that of the substrate. In other words, it’s a second-order reaction.
3. The Reaction Rate Of The SN2 Reaction Is Fastest For Small Alkyl Halides (Methyl > Primary > Secondary >> Tertiary)
Finally, note how changes in the substitution pattern of the alkyl halide results in dramatic changes in the rate of the reaction.[Note 1] “Smaller” alkyl halides like methyl bromide are fast, while more highly substituted tertiary alkyl bromide doesn’t proceed at all.
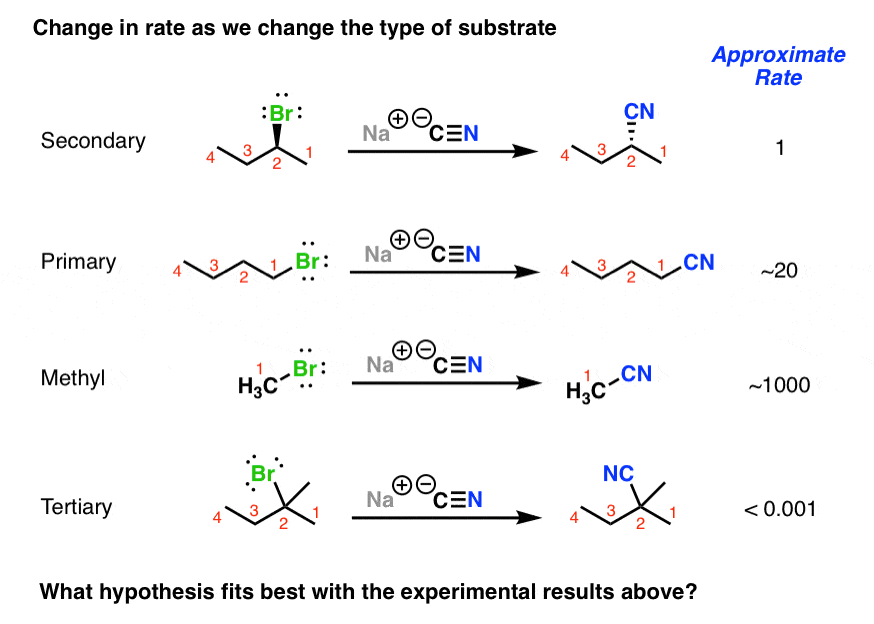
Taking all this data into consideration, we refer to this reaction as the SN2 mechanism. What does SN2 stand for?
- Substitution
- Nucleophilic
- 2 molecules in the rate determining step
So how does it work?
4. The SN2 Mechanism Proceeds Through A Concerted Backside Attack Of The Nucleophile Upon The Alkyl Halide
The best explanation we have for what happens in this reaction is that it proceeds through what organic chemists refer to as a backside attack. The nucleophile approaches the alkyl halide 180° from the C-Br bond, and as the C-(nucleophile) bond forms, the C-(leaving group) bond breaks [Note 2] At the transition state of the reaction, there are partial C-(nucleophile) and C-(leaving group) bonds (denoted by dashed lines). Note the geometry too – instead of tetrahedral, it’s trigonal bipyramidal. This is 5-coordinate carbon – if only for a femtosecond or two.
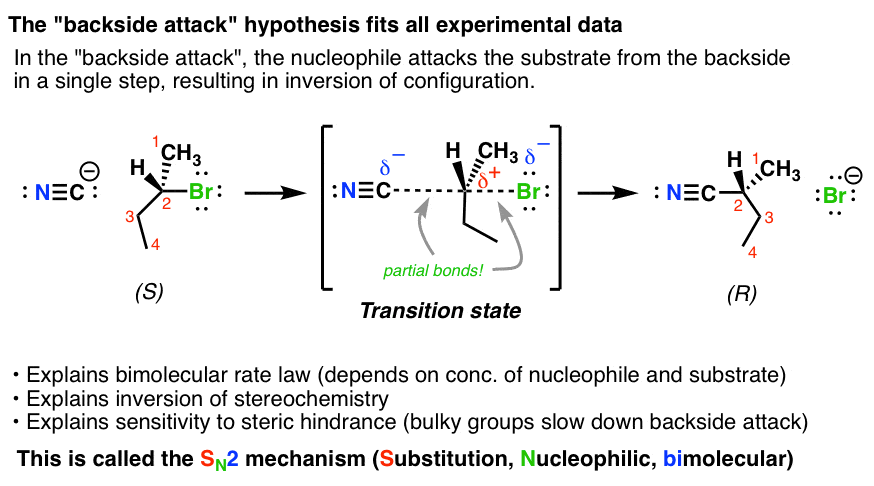
And in an analogy you’ll no doubt hear many times, then, like an umbrella in a strong wind, the three groups flip over as the leaving group leaves, resulting in inversion of configuration. Note that inversion happens at carbons without stereocenters too – it’s just that we can’t observe it because there’s no way to detect the change in configuration.
This umbrella metaphor for the backside attack mechanism is so fundamental and well known in organic chemistry that you can tweet about it and people will know exactly what you mean.
In the next post, we’ll show some more examples of this reaction and explain why it’s one of the most useful reactions in chemistry.
P.S. You may remember that Freda also took this awesome picture of an ozonolysis reaction.
{kind=link}
Next Post: Why The SN2 Is Powerful
Notes
Note 1. Numbers are approximate. Source – Smith, M. and March, J. L. “March’s Advanced Organic Chemistry” 5th ed.
Note 2. backside attack, because the nucleophile donates a pair of electrons into the most accessible empty orbital, which is the antibonding (σ*) orbital of the C-(leaving group) bond, which resides at 180° to the bond. Donation of a pair of electrons into the antibonding orbital results in cleavage of the bond. It’s kind of like an “eject button” in that way.
Quiz Yourself
 Click to Flip
Click to Flip

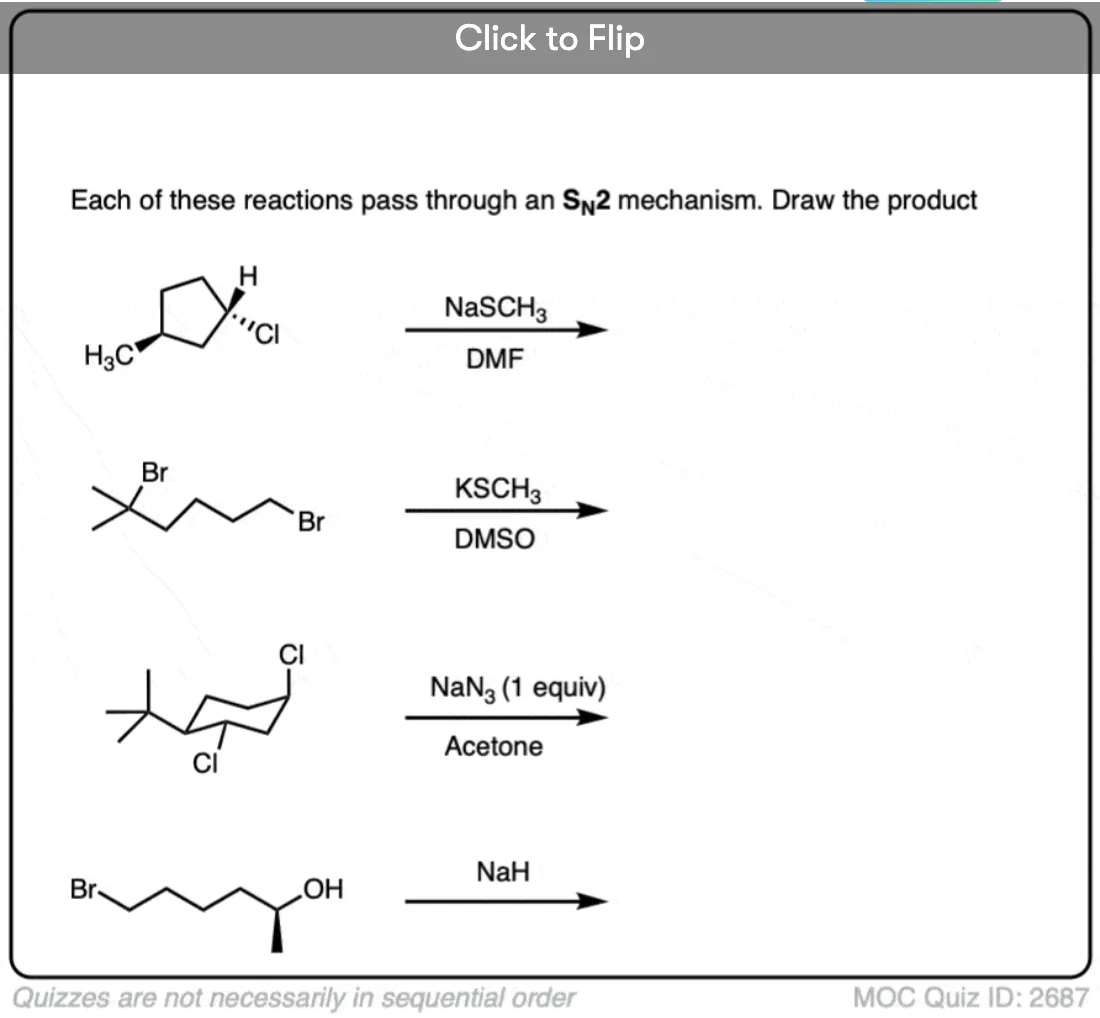
Become a MOC member to see the clickable quiz with answers on the back.
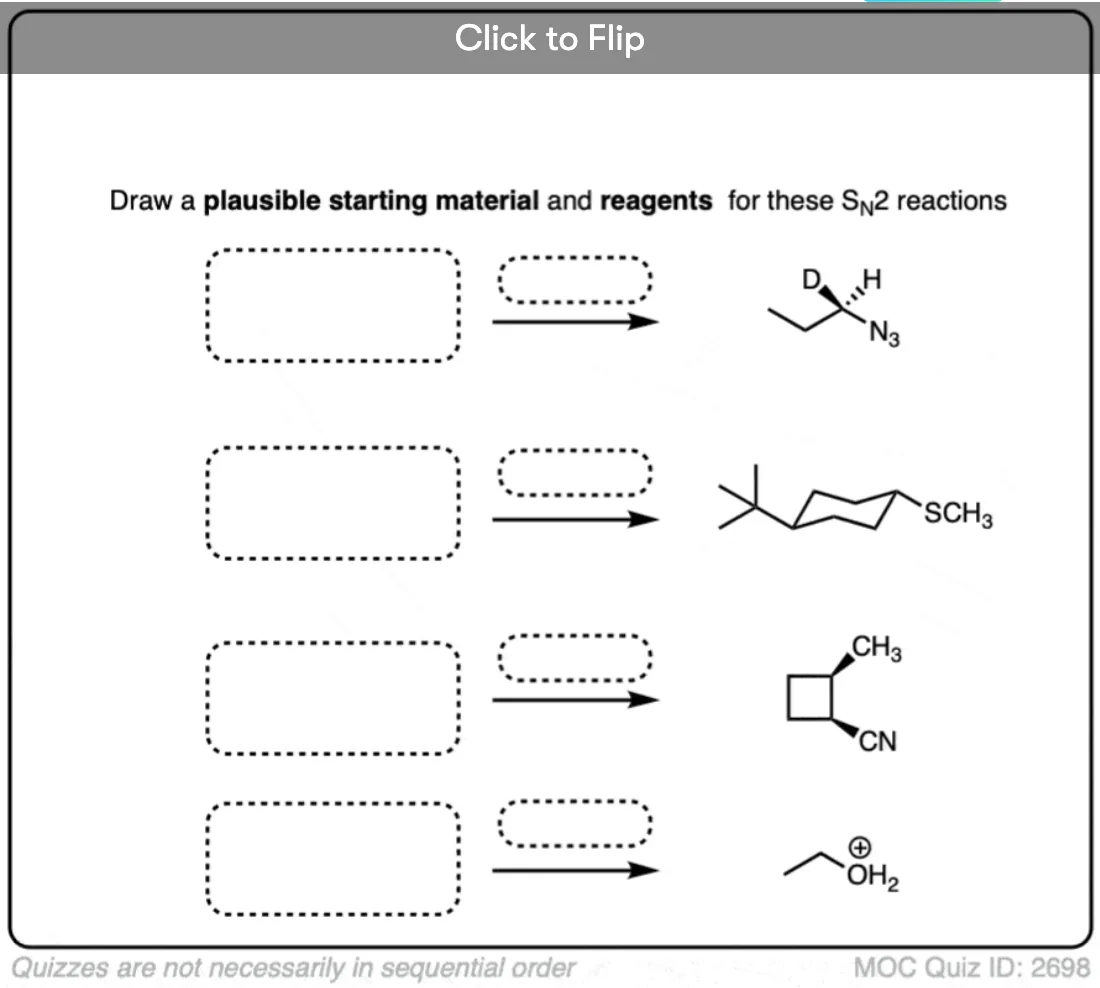
Become a MOC member to see the clickable quiz with answers on the back.
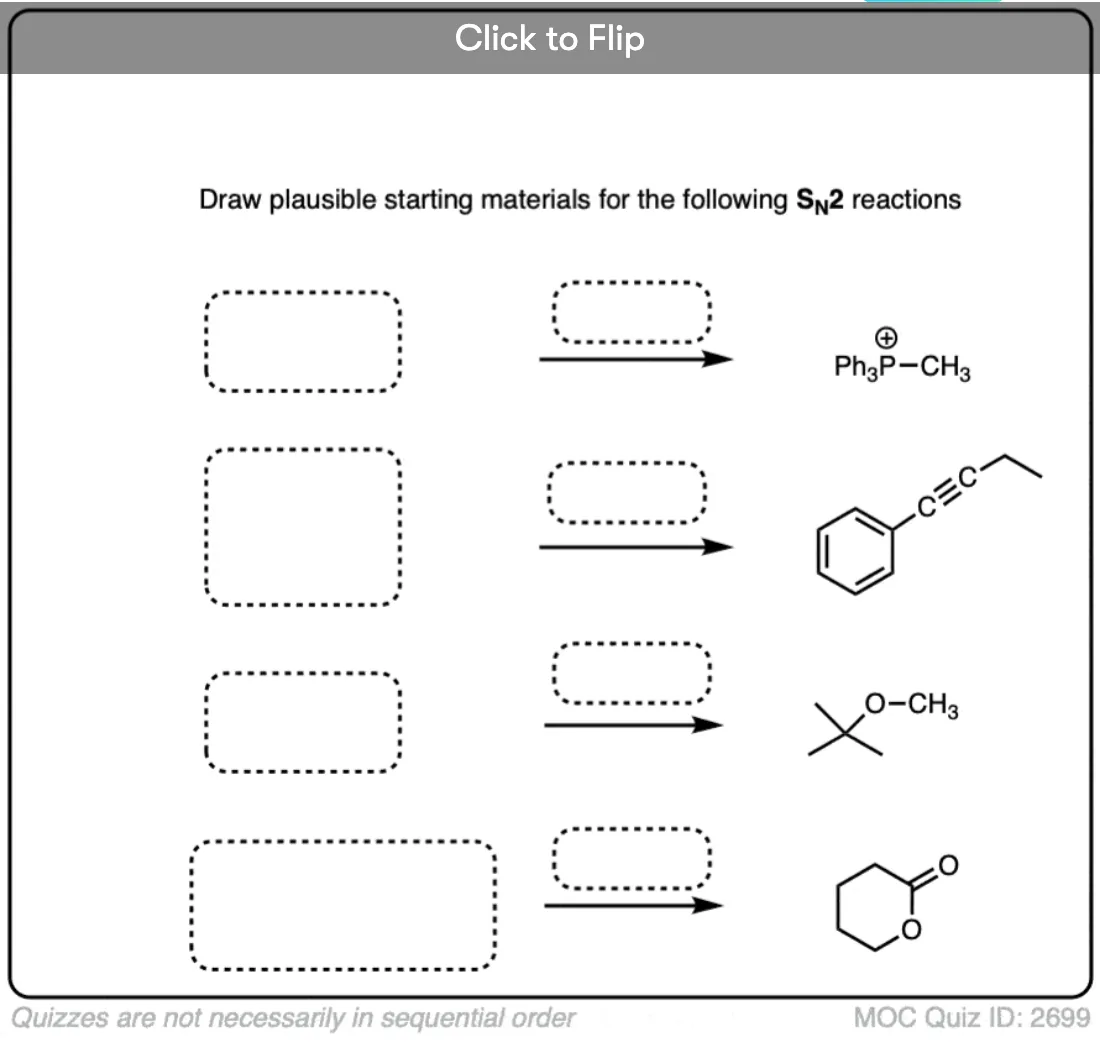
Become a MOC member to see the clickable quiz with answers on the back.
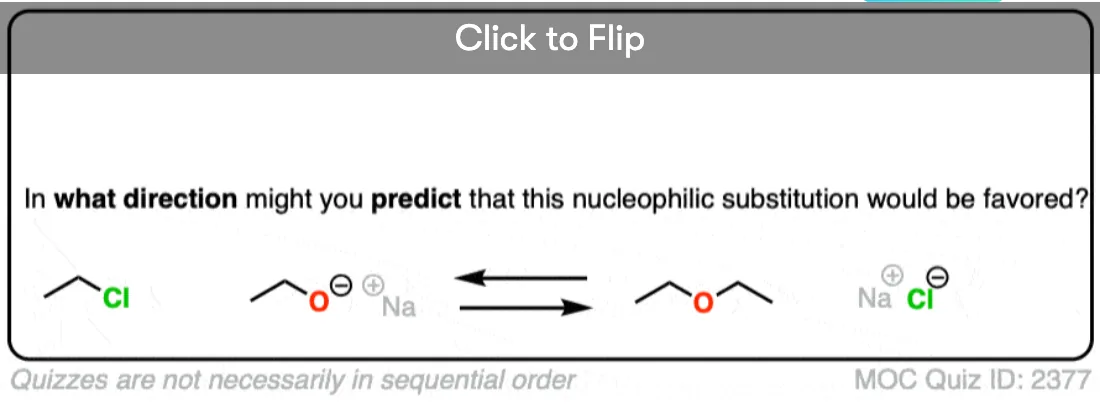
Become a MOC member to see the clickable quiz with answers on the back.
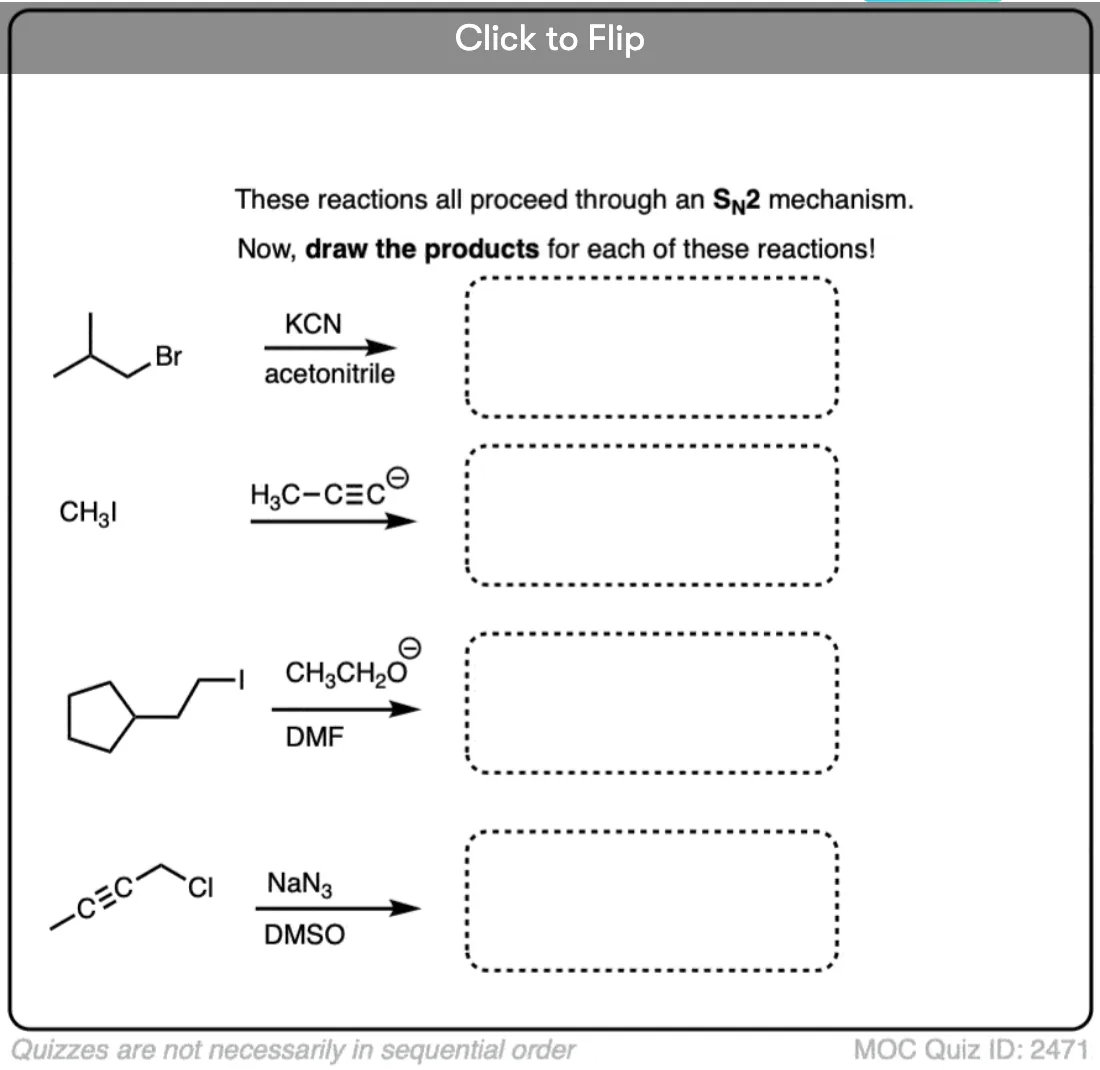
Become a MOC member to see the clickable quiz with answers on the back.
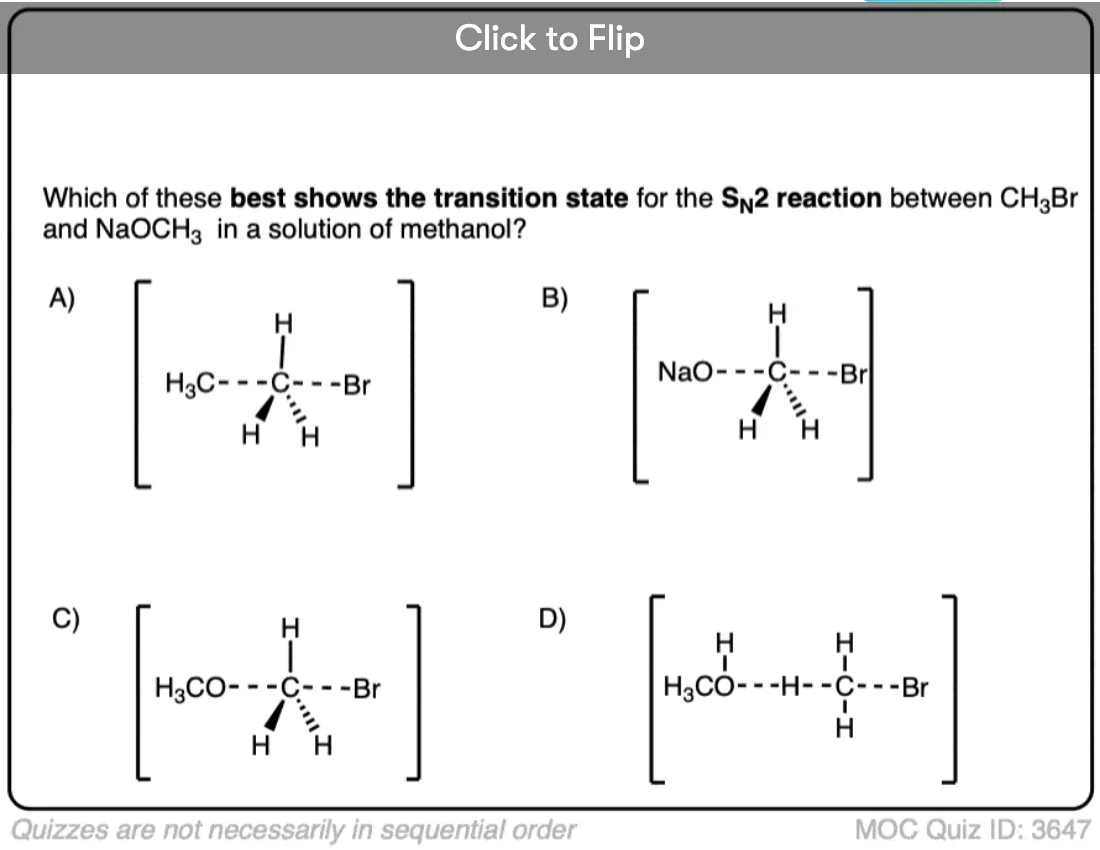
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- Ueber die gegenseitige Umwandlung optischer Antipoden
Walden
Chem. Ber. 1896, 29 (1), 133-138
DOI: 10.1002/cber.18960290127
The inversion of stereochemistry observed in an SN2 reaction is also called Walden inversion, after the chemist Paul Walden. In this paper, he converts (+)-chlorosuccinic acid to the opposite enantiomer and back. This process of double inversion resulting in retention is called a Walden cycle. - Influence of poles and polar linkings on the course pursued by elimination reactions. Part XVI. Mechanism of the thermal decomposition of quaternary ammonium compounds
E. D. Hughes, C. K. Ingold, and C. S. Patel
J. Chem. Soc. 1933, 526-530
DOI: 10.1039/JR9330000526
At the end of this paper, the authors make an important point: “When the various series can be more fully filled in, what has been described as a “ point ” of mechanistic change will probably appear as a region, and thus, just as with reaction (A), we now generalise the original conception of reaction (B) by the contemplation of a range of mechanisms, (Bl)-(B2), both extremes of which have been experimentally exemplified”. Basically, the SN1 and SN2 mechanisms as taught are two extremes of a continuum, and in practice most reactions lie somewhere in between. - Mechanism of substitution at a saturated carbon atom. Part III. Kinetics of the degradations of sulphonium compounds
John L. Gleave, Edward D. Hughes and Christopher K. Ingold
J. Chem. Soc. 1935, 234-244
DOI: 10.1039/JR9350000236
This is a useful paper – in the beginning the terms “SN1” and “SN2” are introduced and defined, and Figs. 1 and 2 depict how the two mechanisms can compete depending on the structure of the substrate. - Aliphatic substitution and the Walden inversion. Part I
E. D. Hughes, F. Juliusburger, S. Masterman, B. Topley, and J. Weiss
J. Chem. Soc. 1935, 1525-1529
DOI: 10.1039/JR9350001525
Classic study correlating the rate of racemization with rate of uptake of radioactive iodide ion. Because this experiment was conducted in the early 20th century when nuclear chemistry was in its infancy, the iodide ion was made radioactive by exposure to a neutron source (Ra/Be) and the exact isotope was not determined. - Mechanism of substitution at a saturated carbon atom. Part XLII. Introductory remarks, and kinetics of the interaction of chloride ions with simple alkyl chlorides in acetone
B. D. de la Mare
J. Chem. Soc. 1955, 3169-3173
DOI: 10.1039/JR9550003169
Table 2 in this paper demonstrates the rate decrease of a simple SN2 reaction (Finkelstein displacement of Cl with 36Cl– in this case) upon going from methyl -> ethyl -> isopropyl. The rate decreases 50-80 fold upon going from methyl -> ethyl or ethyl -> isopropyl. - Mechanism of substitution at a saturated carbon atom. Part XXVI. The rôle of steric hindrance. (Section A) introductory remarks, and a kinetic study of the reactions of methyl, ethyl, n-propyl, isobutyl, and neopentyl bromides with sodium ethoxide in dry ethyl alcohol
I. Dostrovsky and E. D. Hughes
J. Chem. Soc. 1946, 157-161
DOI: 10.1039/JR9460000157
Table I in this paper shows the reduction in reaction rate for the SN2 reaction of R-Br with OEt- when R goes from methyl -> ethyl -> n-propyl -> isobutyl -> t-amyl. This can be attributed to sterics, as backside attack of the substituted carbon becomes increasingly challenging. - Bicyclic Structures Prohibiting the Walden Inversion. Further Studies on Triptycene and its Derivatives, Including 1-Bromotriptycene
Paul D. Bartlett and Edward S. Lewis
Journal of the American Chemical Society 1950, 72 (2), 1005-1009
DOI: 1021/ja01158a094
1-bromotriptycene is inert under a wide variety of conditions due to the structure – backside access to the C-Br bond is basically impossible, and due to the bridgehead geometry, forming a sp2 carbon at that position is strongly disfavored, precluding the formation of a cation or radical there. In the first paragraph, Prof. Bartlett mentions, “Work which was then under way to extend this study to the carbonium ion and the free radical was interrupted by World War II and is here reported.” - Reaction kinetics and the Walden inversion. Part VI. Relation of steric orientation to mechanism in substitutions involving halogen atoms and simple or substituted hydroxyl groups
W. A. Cowdrey, E. D. Hughes, C. K. Ingold, S. Masterman, and A. D. Scott
J. Chem. Soc. 1937, 1252-1271
DOI: 10.1039/JR9370001252
The points listed in the summary are worth reading for understanding what influences the SN1 and SN2 pathways. - Conformational Analysis. IV. Bimolecular Displacement Rates of Cyclohexyl p-Toluenesulfonates and the Conformational Equilibrium Constant of the p-Toluenesulfonate Group
Ernest L. Eliel and Rolland S. Ro
Journal of the American Chemical Society 1957, 7i9 (22), 5995-6000
DOI: 1021/ja01579a040
This paper describes a classic SN2 experiment on cis- and trans-4-t-butylcyclohexyltosylate. The trans ester reacts 19 times faster than cis because of the chair flip. Due to the t-butyl group, the barrier for ring inversion is extremely high (t-butyl is basically ‘locked’ in the equatorial position), and this is described further in the section on A-values. - Nucleophilic Substitution (SN2): Dependence on Nucleophile, Leaving Group, Central Atom, Substituents, and Solvent
Trevor A. Hamlin, Marcel Swart, F. Matthias Bickelhaupt
ChemPhysChem 2018, 19 (11), 1315-1330
DOI: 1002/cphc.201701363
A more recent paper computationally examining the effects of various variables on SN2 reaction energetics. - Solvolytic Displacement Reactions At Saturated Carbon Atoms
Andrew Streitwieser, Jr.
Chemical Reviews 1956 56 (4), 571-75
DOI: 10.1021/cr50010a001
This early review by Prof. Streitwieser (U.C. Berkeley) is a comprehensive review of the early literature of the SN1 and SN2 reactions.
What would the end Product be named in tue example?
In the backside attack example, why does the ethyl invert, but not the wedge and dash of the H and CH3
Hi! To show inversion, all you need to do is to swap the places of any two groups, while keeping the rest the same. So in this example I showed the ethyl group inverting with the nucleophile, but I could have kept the position of the ethyl and LG the same and swapped H and CH3 instead.
(This is the single swap rule – quick video right here – https://www.youtube.com/watch?v=6qKbyrF2tXw )
Thanks for this post! How can we compare the rate of SN2 for chloroethane and benzyl chloride?
Published rates indicate that the rate of reaction for methyl halides is roughly 30 times the rate for primary halides (e.g. benzyl chloride) assuming the nucleophile is the same. That’s the rough difference in rates.
According to the organic chemistry textbook by Clayden, Warren and Greeves,
Alpha halo ketones undergoes the fastest SN2 in the world!!
James, can you please pour some light over this?
James , you are just awesome and the best.
The most accurate ,easy to understand and concise explanation of each and every topic.
Thanks a ton.
OK, thank you for visiting the side Madhav and I am glad you find it useful. Please let me know if you see any mistakes / typos. James
“chiral center” is a completely wrong expression. No center could ever be chiral. Chirality owns only to object/ molecules,not to atoms. That carbon is a stereocenter.
“Chirality center” is an approved IUPAC term. https://goldbook.iupac.org/terms/view/C01060 .
The reason I dislike stereocenter in this context is that it also applies to the carbon atoms on double bonds capable of cis/trans isomerism. For that reason I choose not to use it here.
For the audience of this website I personally see no problem with shortening “chirality center” to “chiral center” since pretty much everyone knows that it means a carbon with four different substituents.
What will be the hybridization of the transition state in biomolecular substitution reaction (SN2)? Is it sp3 or sp2?
Thank you!
The transition state contains a partial bond and it is not really appropriate to use hybridization terminology to describe it.
My query is regarding point number 4
Should not the inversion product have H connected with dashed line and CH3 group connected with wedge line?
Thank you for the wonderful work you are doing.
Hello
I want to know that when CH3 (CH2)15CH2CH2Br reacts with methoxide in the presence of methanol at 65°C , why the major product will form according to SN2 Mechanism and not E2 Mechanism.
Please answer, I’ll thankful to you.
Primary alkyl halide.
Hi James,
Great post. Just want to clarify, would there be any positive charge on the carbon in the transition state as shown in the mechanism ?
Balaji.
Yes, there should be a partial positive charge, which is shown as “delta plus” (in red)
Thank u so much for your website you make everything so easy to understand and this website is literally saving my life rn for my exam
Primary halides which have an unsaturated group attached to the carbon react
much faster than bromomethane in SN2 reactions.
What is SN2′ (sn2 prime) and NGP(neighbouring group participation )
I think diagram 2 is incorrect possibly? Should Br be replaced by CN after the arrow?
Yes, fixed!
awesome explaination is given and was every much useful for me…:)
i am taking an exam in organic chemistry tommorrow and i am wishing i had started going through this website earlier this is awsome stuff comprehensive too