Alkene Reactions
Oxymercuration Demercuration of Alkenes
Last updated: May 28th, 2026 |
Oxymercuration Demercuration of Alkenes
- Treatment of alkenes with mercury (II) salts and water leads to the addition of water and mercury across the C-C pi bond. This is called oxymercuration.
- Addition to the alkene occurs such that OH forms a bond to the most substituted carbon, making it selective for Markovnikov products.
- The reaction proceeds through attack of the nucleophile on a 3-membered intermediate known as an oxymercurinium ion, which resembles the halonium ion intermediate in reactions such as halogenation of alkenes. As in halogenation, addition occurs to give the products of anti addition.
- In the demercuration step, mercury is removed by treating the product with sodium borohydride (NaBH4), giving the alcohol.
- By using alcohols instead of H2O, ethers can be formed, a reaction known as alkoxymercuration.
- Oxymercuration-demercuration is a nice complement to hydroboration-oxidation, which is regioselective for anti-Markovnikov alcohols.
- Oxymercuration can also be performed on alkynes (see article: Hydroboration and Oxymercuration of Akynes)
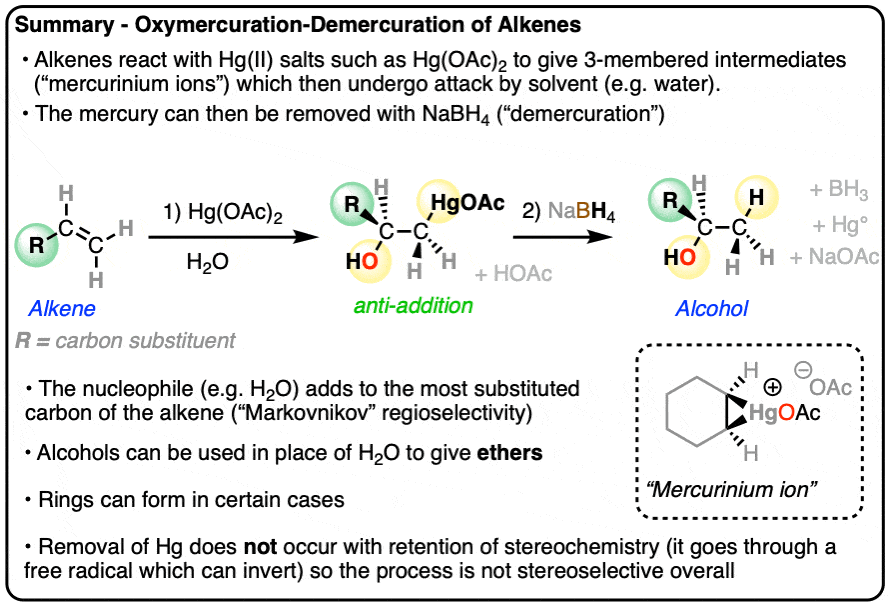
Table of Contents
-
- Oxymercuration-Demercuration of Alkenes
- Oxymercuration is Regioselective for Markovnikov Products
- The Mechanism for Oxymercuration Proceeds Through Attack of a Nucleophile On A Cyclic Mercurinium Ion
- Understanding Markovnikov Selectivity
- The Demercuration Step
- Oxymercuration Provides “Markovnikov” Alcohols Without Rearrangement
- Addition of Alcohols to Give Ethers
- Intramolecular Examples
- Summary
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Oxymercuration Demercuration of Alkenes
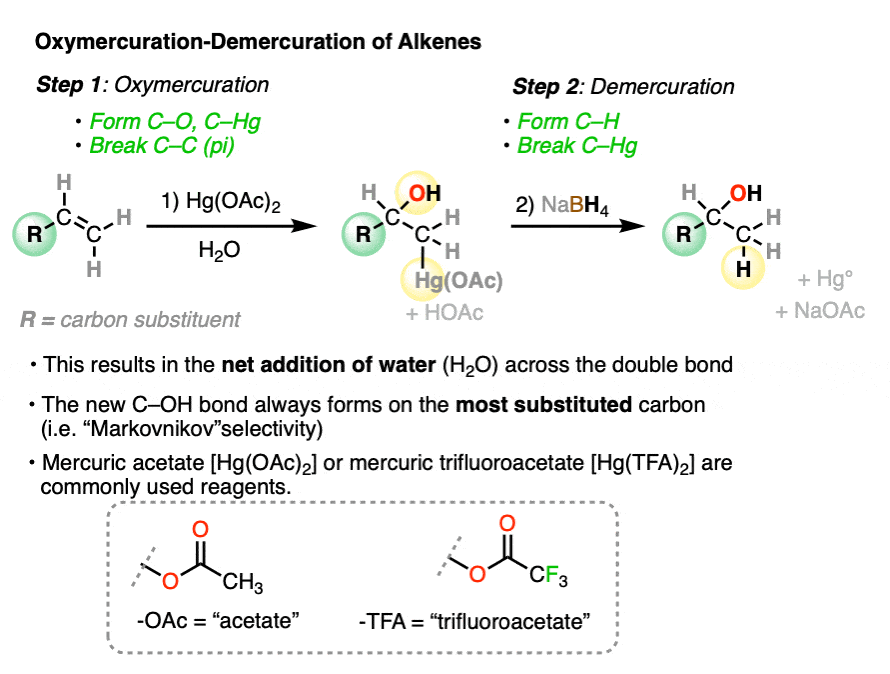
When an alkene is treated with a mercury(II) salt such as mercuric acetate [Hg(OAc)2] or mercuric trifluoroacetate Hg(OCOCF3)2 in the presence of H2O, addition occurs across the C-C pi bond, resulting in a new C-OH bond and a C-Hg bond. This reaction is called oxymercuration.
It is alternatively called “solvomercuration” since addition will generally incorporate any nucleophilic solvent that happens to be around, whether that be H2O, alcohols, carboxylic acids, or others.
Organomercury compounds tend to be highly toxic and it is best to avoid handling them if at all possible. [Note 1] In the demercuration step, treatment with sodium borohydride (NaBH4) replaces C-Hg with C-H, resulting in the alcohol and a little pool of elemental liquid mercury that accumulates at the bottom of the flask.
2. Oxymercuration is Regioselective for Markovnikov Products
Any time two different atoms are added across a C-C pi bond there is the potential for the formation of constitutional isomers. We saw that in the addition of strong acids (such as H-Br) to alkenes, there is high selectivity for formation of the constitutional isomer where the C-halogen bond forms on the more substituted carbon of the alkene, a phenomenon known as “Markovnikov’s Rule” (See article – Markovnikov’s Rule). When a reaction shows a strong preference for the formation of one constitutional isomer over another, we say that reaction is regioselective.
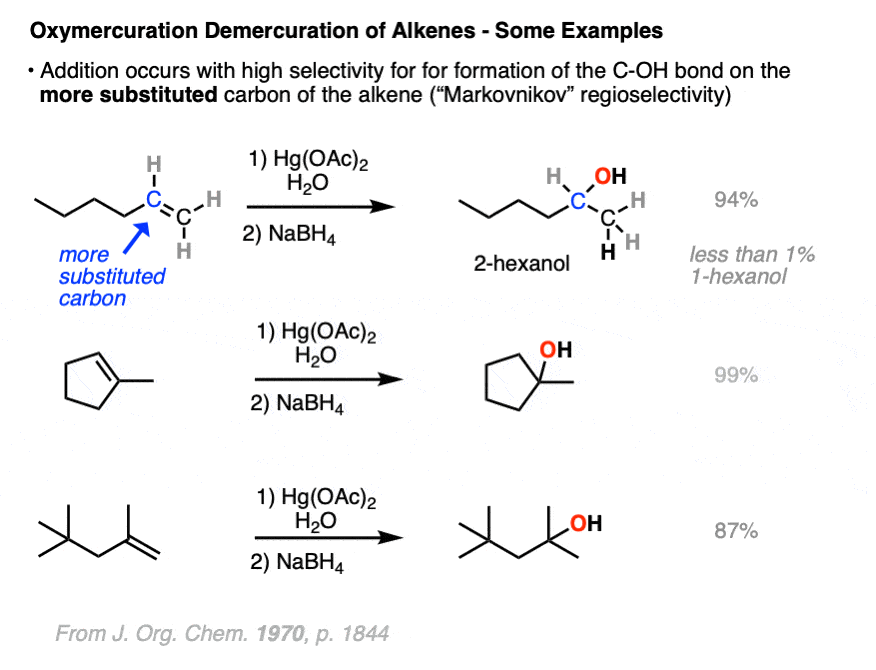
Oxymercuration is also regioselective for the “Markovnikov” alcohol products!
The resulting alcohols that form tend to have the new C-OH bond on the most substituted carbon of the alkene. (The selectivity seems to be very good – one study found only about a 1% yield of the minor anti-Markovnikov alcohol product)
The reaction can be carried out on a variety of alkenes to give the products of “Marknovnikov” hydration after removal of mercury with NaBH4.
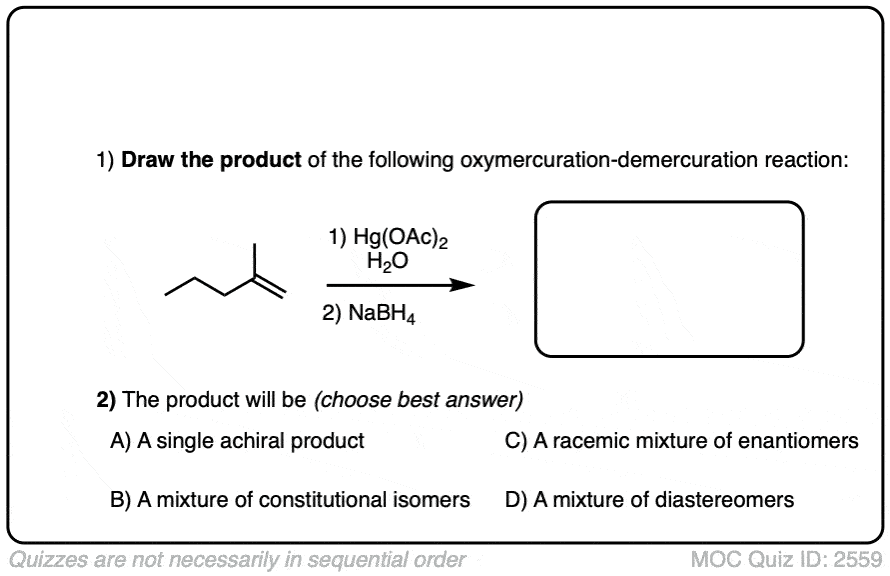
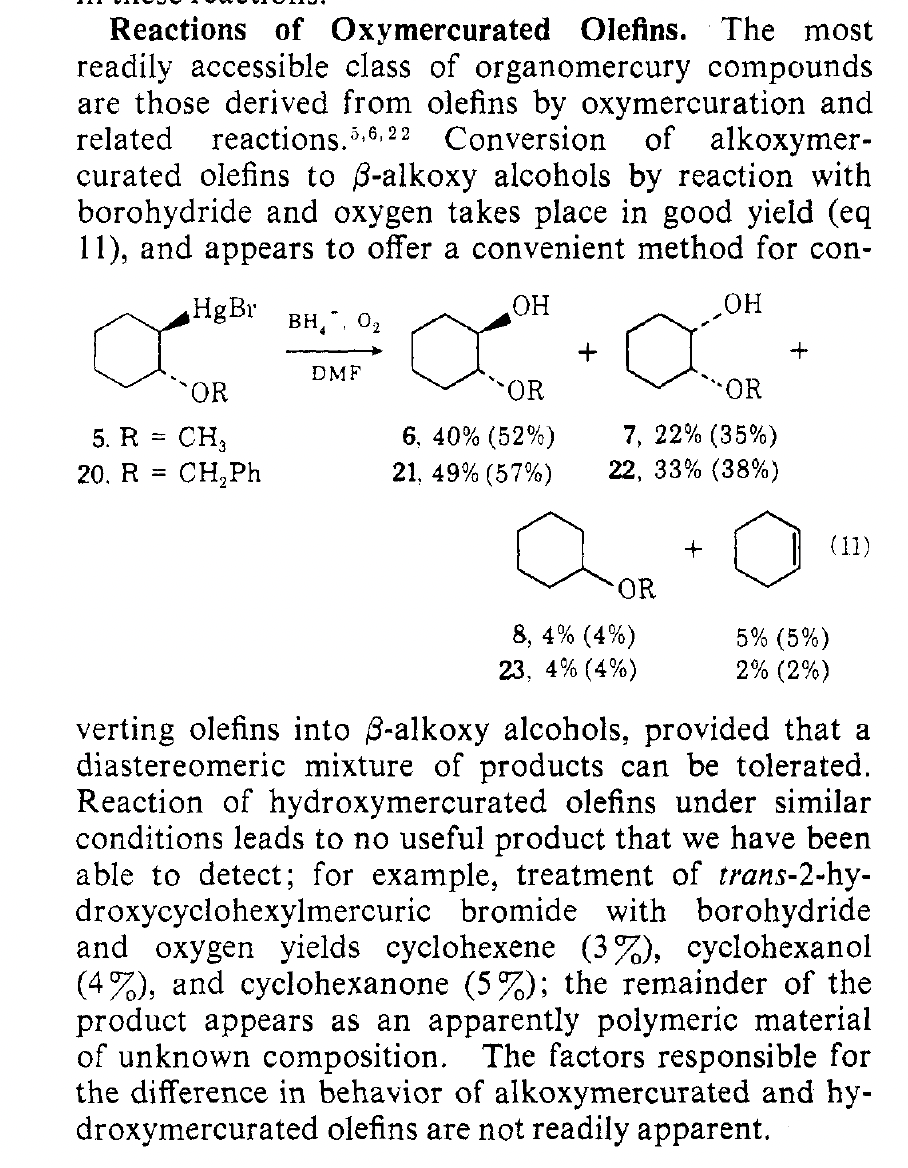
See if you can predict the product of the reaction below:
 Click to Flip
Click to Flip
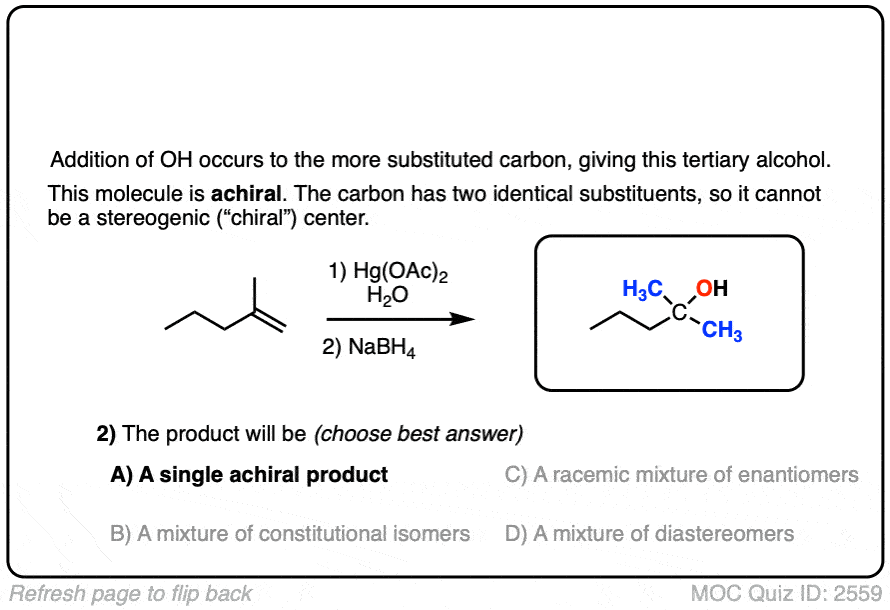
If you are given the product of a reaction, and you know the pattern of bonds that form and break, you should be able to work backwards from the product to the starting material, like in the example below:
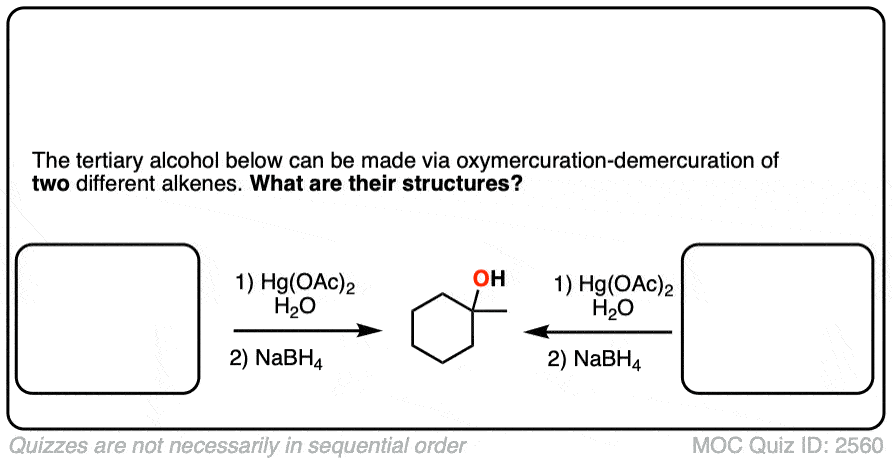 Click to Flip
Click to Flip
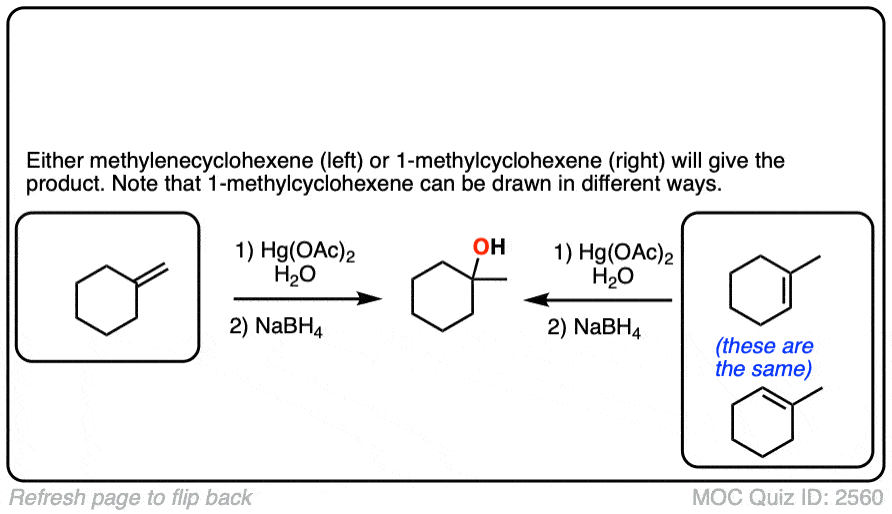
3. The Mechanism of Oxymercuration Goes Through A 3-Membered “Mercurinium Ion”
The first step of oxymercuration results in a new organomercury compound with a C-Hg bond. Unlike many compounds with carbon-metal bonds, organomercury compounds are actually quite stable to handling. [Note 2] They are (reasonably) stable to air, can be purified with column chromatography, and can even be distilled (!)
Oxymercuration is stereoselective for anti– addition products. When cyclohexene is treated with Hg(OAc)2 and water, for example, the new -OH and -Hg bonds always form on the opposite face of the alkene. None of the syn– addition product is formed.
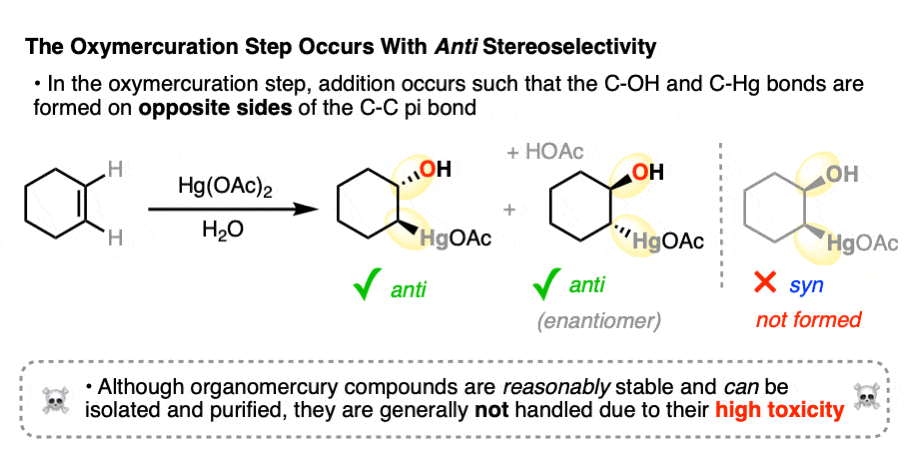
This high selectivity would rule out an intermediate carbocation, which would be expected to result in a mixture of syn– and anti- addition products. [See article – Alkene Addition Pattern #1 – The Carbocation Pathway].
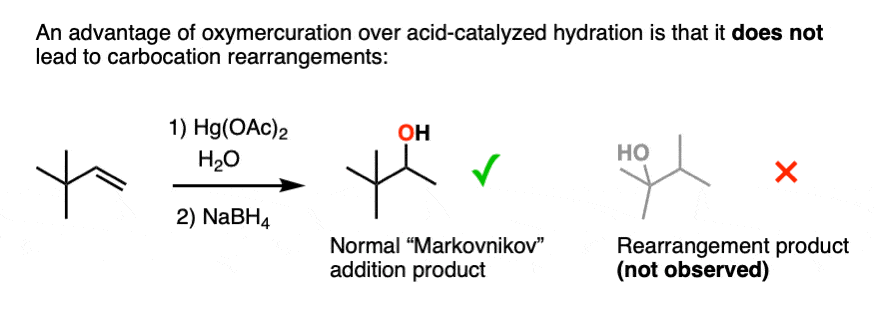
Furthermore, oxymercuration reactions do not give products arising from carbocation rearrangements. [See section 6, below]
The best proposal we have for explaining the stereoselectivity of oxymercuration is that it goes through an intermediate 3-membered “mercurinium” ion. (Note – it likely isn’t crucial for you to remember the name “mercurinium” but we still need to call it something).
In the first step, the alkene reacts with Hg2+ to give a three membered ring with a positive formal charge on mercury:
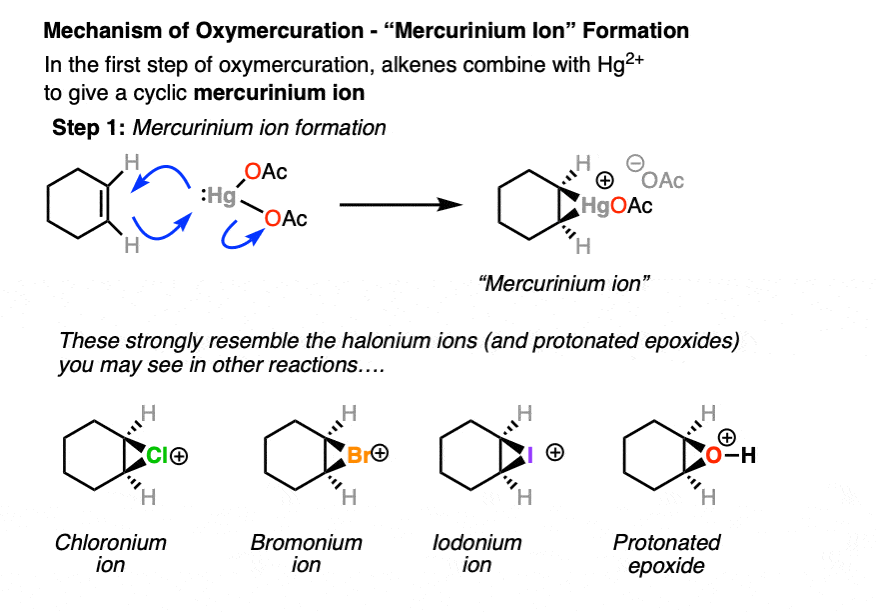
If you have previously covered halogenation of alkenes, this pattern might strike you as familiar, since the mercurinium ion resembles the chloronium, bromonium, and iodonium ion intermediates in the halogenation of alkenes! [See article – Halogenation of Alkenes] It is also similar to the structure of protonated epoxides. (This article might also be helpful: Alkene Addition Pattern #2 – The “3-Membered Ring” Pathway] )
In acyclic alkenes, this 3-membered bridging ring prevents bond rotation and is responsible for the high stereoselectivity of the reaction.
In the next step, the nucleophile (H2O) attacks the C-Hg bond from the back, resulting in formation of C-O and breakage of C-Hg.
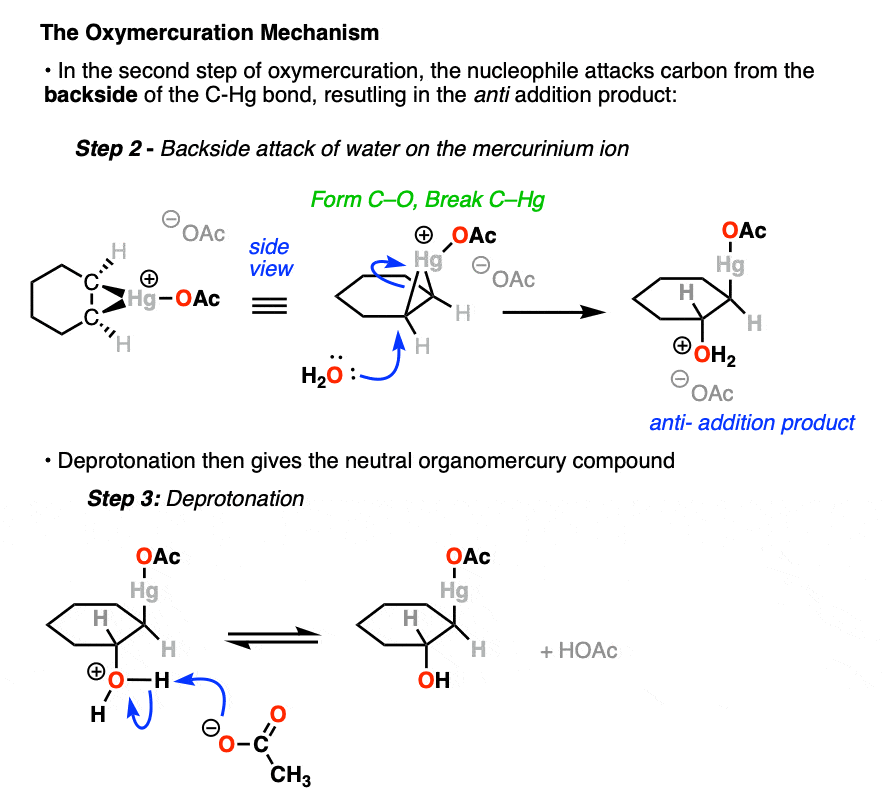
Why from the back and not the front? Because formation of a bond requires donation of a pair of electrons into an empty orbital, which happens to be the C-Hg sigma* (antibonding) orbital. The C-Hg sigma (bonding) orbital is already full.
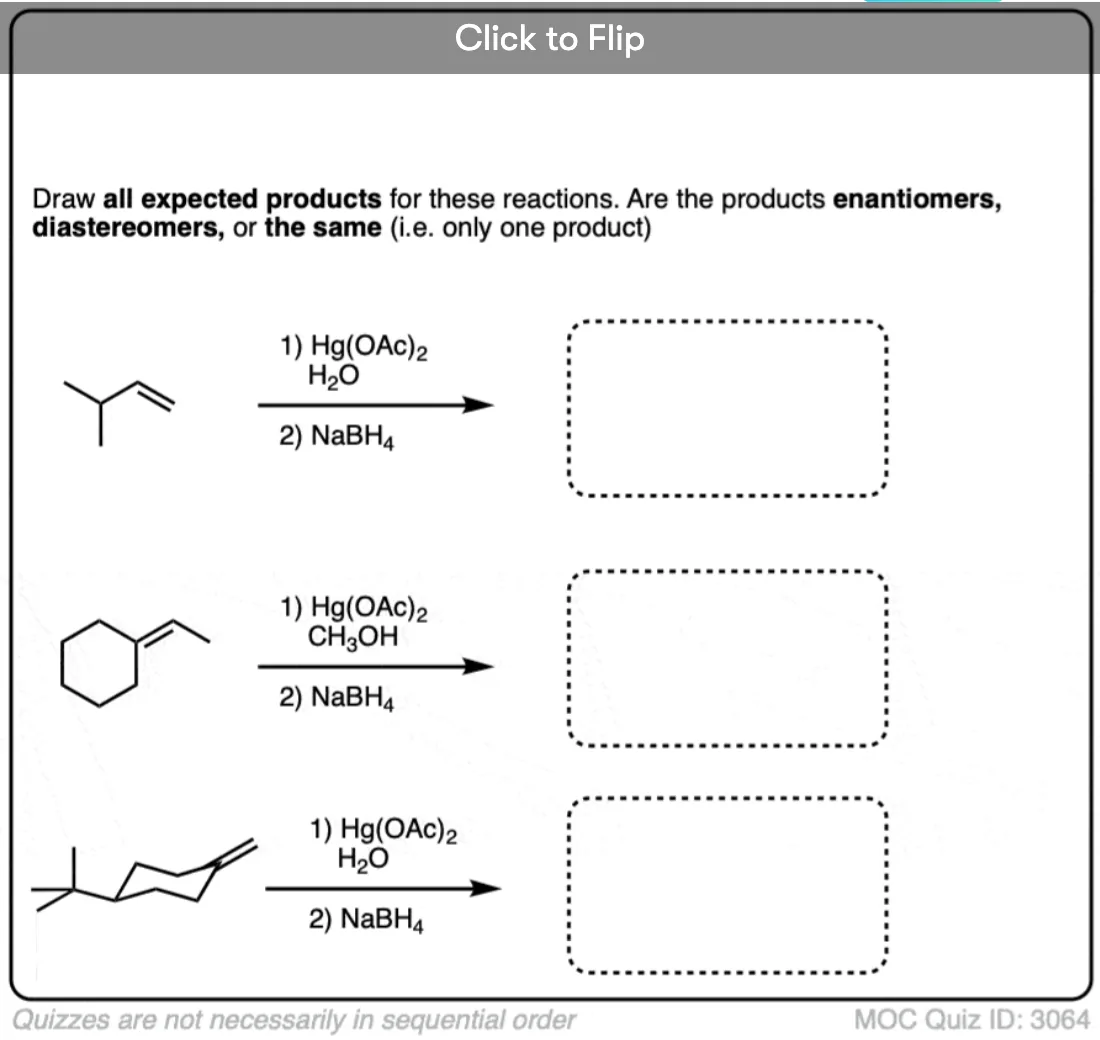
In the case of cyclohexene, addition of H2O could happen at either carbon with equal likelihood. This will therefore give a racemic mixture of enantiomers.
The final step here is deprotonation of -OH2(+) to give -OH and one equivalent of acetic acid.
4. Understanding Markovnikov Selectivity
In the case of non-symmetrical mercurinium ions, we are left having to explain why the nucleophile (H2O) attacks the more substituted carbon and not the less substituted carbon. Furthermore, we have to explain why addition is occurring preferentially via a backside attack on a tertiary carbon, when those who have already learned the SN2 reaction have been taught that tertiary carbons are more sterically hindered.
I think it’s helpful to think of the mercurinium ion as being more like a carbocation than like a tertiary alkyl halide. [Note 3] We don’t really worry about steric hindrance when thinking about the nucleophile approaching a tertiary carbocation. In the case of the mercurinium ion, the neighboring mercury is capable of donating a pair of electrons to one face of the carbocation, resulting in a long and weak C-Hg bond.
Think of it as a “carbocation with benefits”. :-)
In this vein, imagine some “resonance forms” of the mercurinium ion where there is a carbocation on the more substituted carbon and another one with the carbocation on the less substituted carbon. I put “resonance forms” in quotes here because we teach that you aren’t really supposed to break single bonds when interconverting resonance forms, but we’re making an exception here. [Ref]
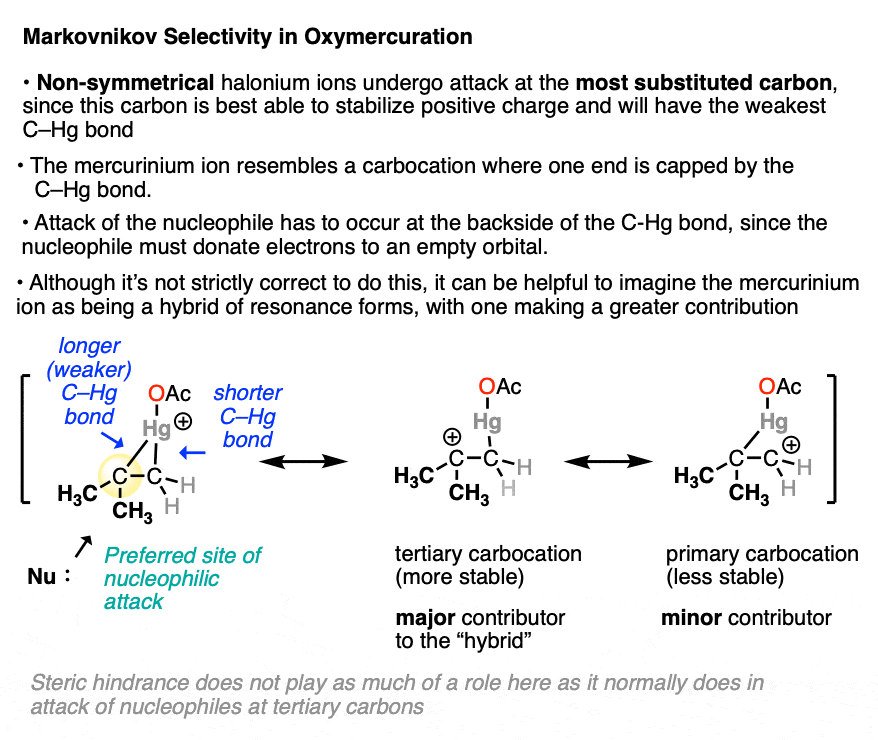
The resonance form where the positive charge is on the most substituted carbon will make a greater contribution to the hybrid. It will also have the weakest C-Hg bond and the greatest positive charge density on carbon – i.e. the greatest electrophilicity.
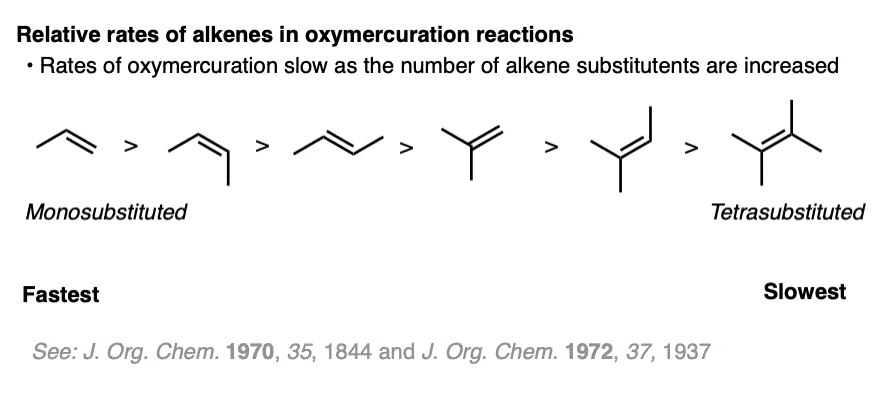
But… why does it still perform a backside attack at the more substituted carbon? Shouldn’t this be more sterically hindered? Perhaps this is why the rate of oxymercuration reactions go down as substitution on the alkene is increased. More discussion here.
It’s worth noting that in certain cases where backside attack would be extremely hindered, syn addition is observed. See Ref.
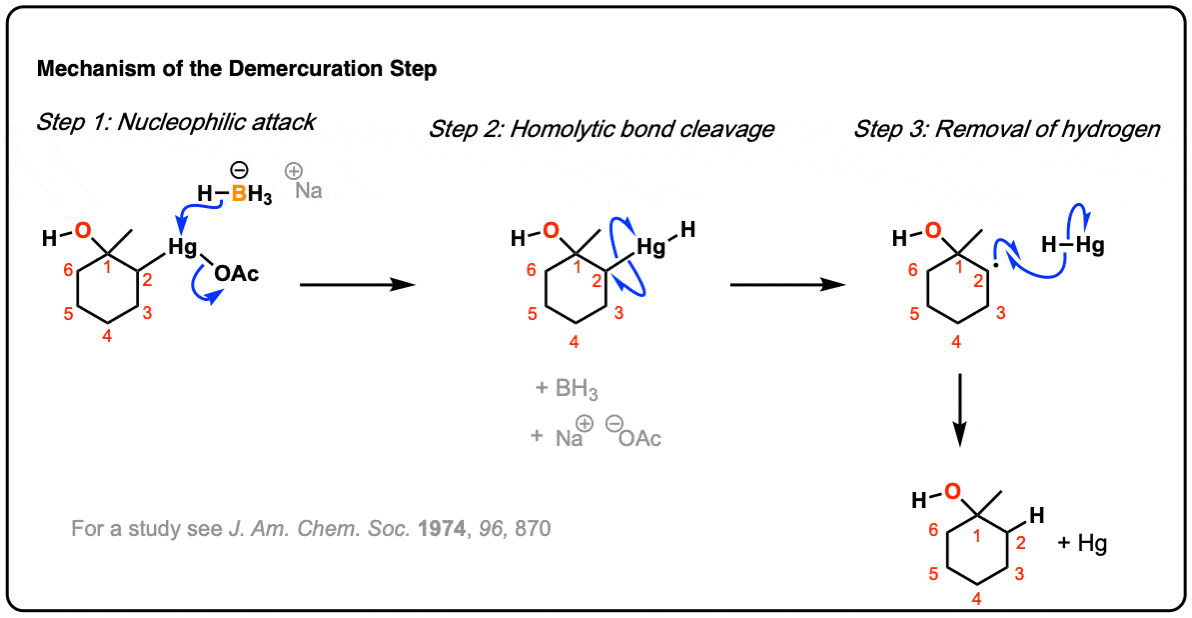
5. Demercuration
Once oxymercuration is complete, the intermediate organomercury compound is almost never isolated. Instead, demercuration is performed with sodium borohydride (NaBH4), which rapidly breaks the C-Hg bond and forms a new C-H bond.
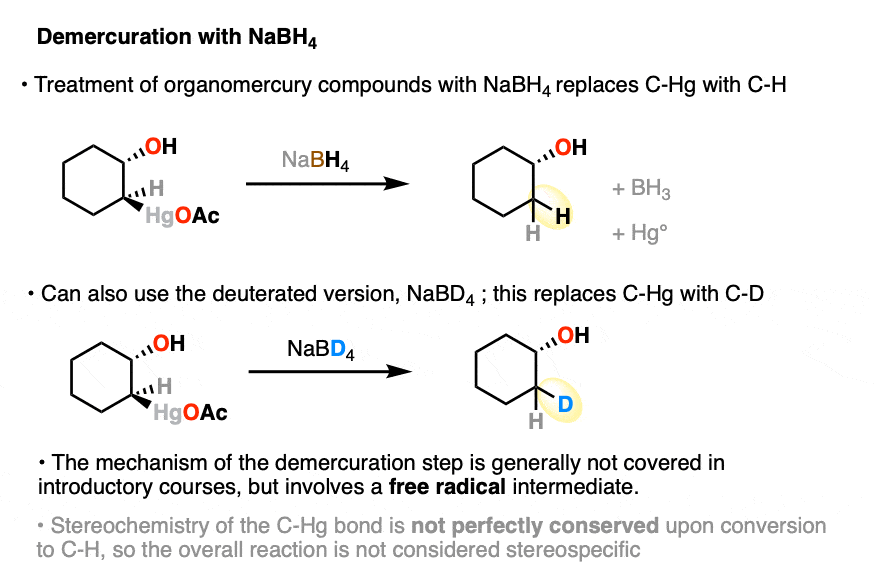
Alternatively, demercuration can be carried out with sodium borodeuterohydride (NaBD4) which allows formation of the deuterated product. (Remember that D is just the heavy isotope of H).
The mechanism of the demercuration step is often not covered in introductory textbooks, but it goes through a free-radical intermediate. To see a plausible mechanism, hover here or click this link.
{kind=link}
Free radicals have a shallow trigonal pyramidal structure and can undergo rapid inversion of their stereochemistry. Therefore, in most cases, generation of the free radical at carbon results in scrambling of its stereochemistry (much like the first step of the SN1). Thus, while the oxymercuration step is stereoselective, the demercuration step is not. [Note 5]
6. Oxymercuration Provides Markovnikov Alcohols Without Rearrangement
We’ve already learned that one way to get the Markovnikov addition of water across alkenes is just to add aqueous acid.
So why bother having another method?
Well, acid-catalyzed hydration has a serious drawback in that the carbocation intermediate can sometimes undergo rearrangements such as hydride and alkyl shifts. [See article – Rearrangements in Alkene Addition Reactions]
See if you can draw the mechanism of this rearrangement, for example.
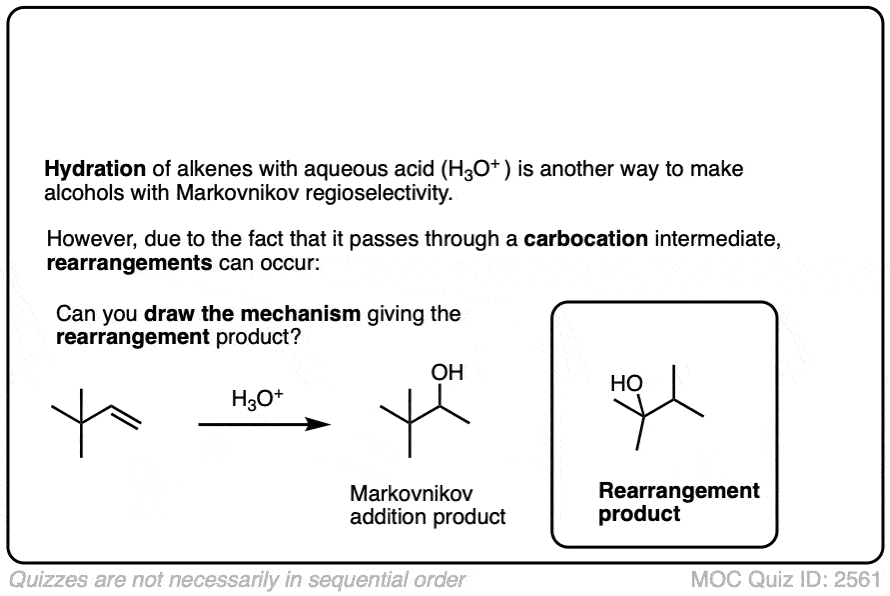 Click to Flip
Click to Flip
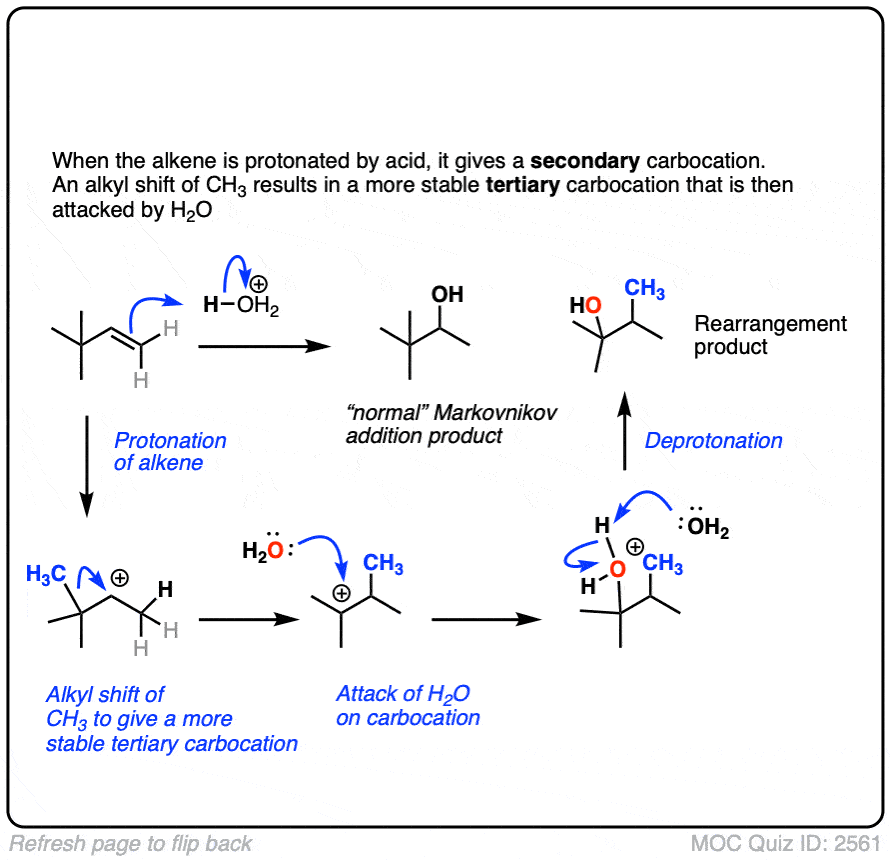
One advantage of oxymercuration-demercuration is that rearrangement products are only rarely observed.
7. Addition of Alcohols to Give Ethers
Mercurinium ions will react with many different nucleophilic solvents (which is why the reaction is often called, “solvomercuration”)
A useful method for making ethers involves using alcohols as the solvent (“alkoxymercuration”)
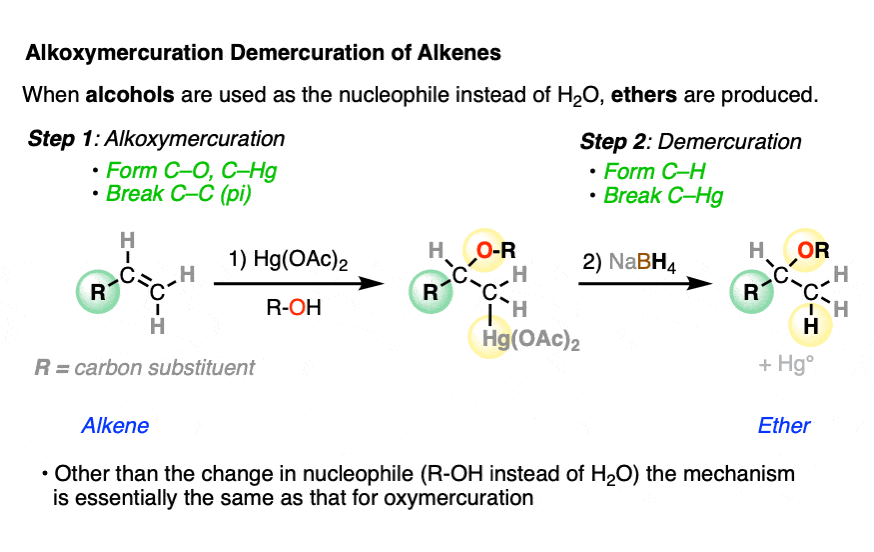
Other than the identity of the nucleophile, the mechanism of the reaction is otherwise the same as that for the reaction with water.
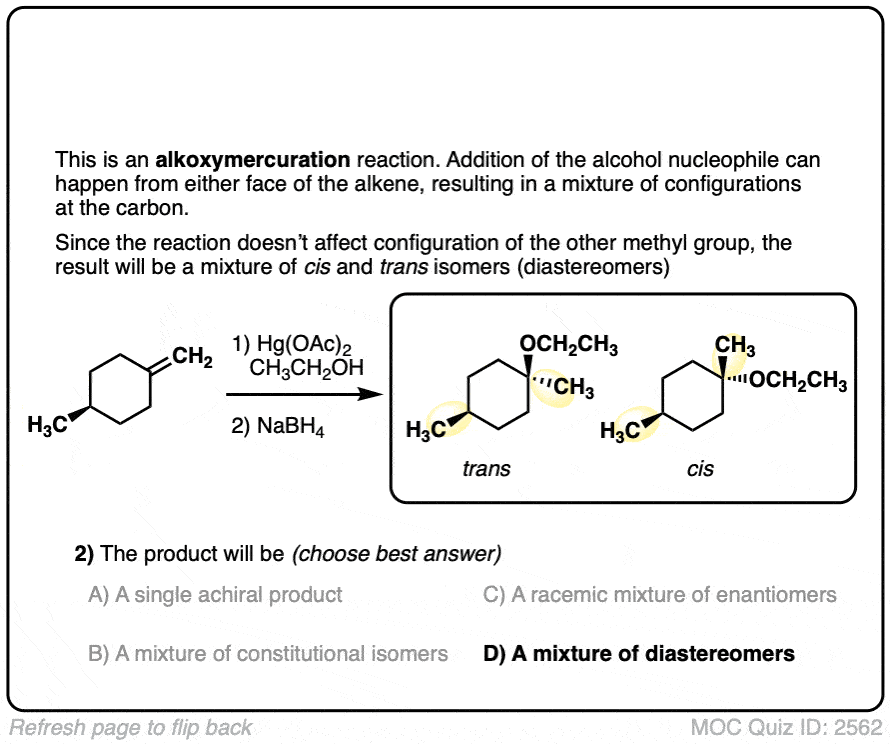
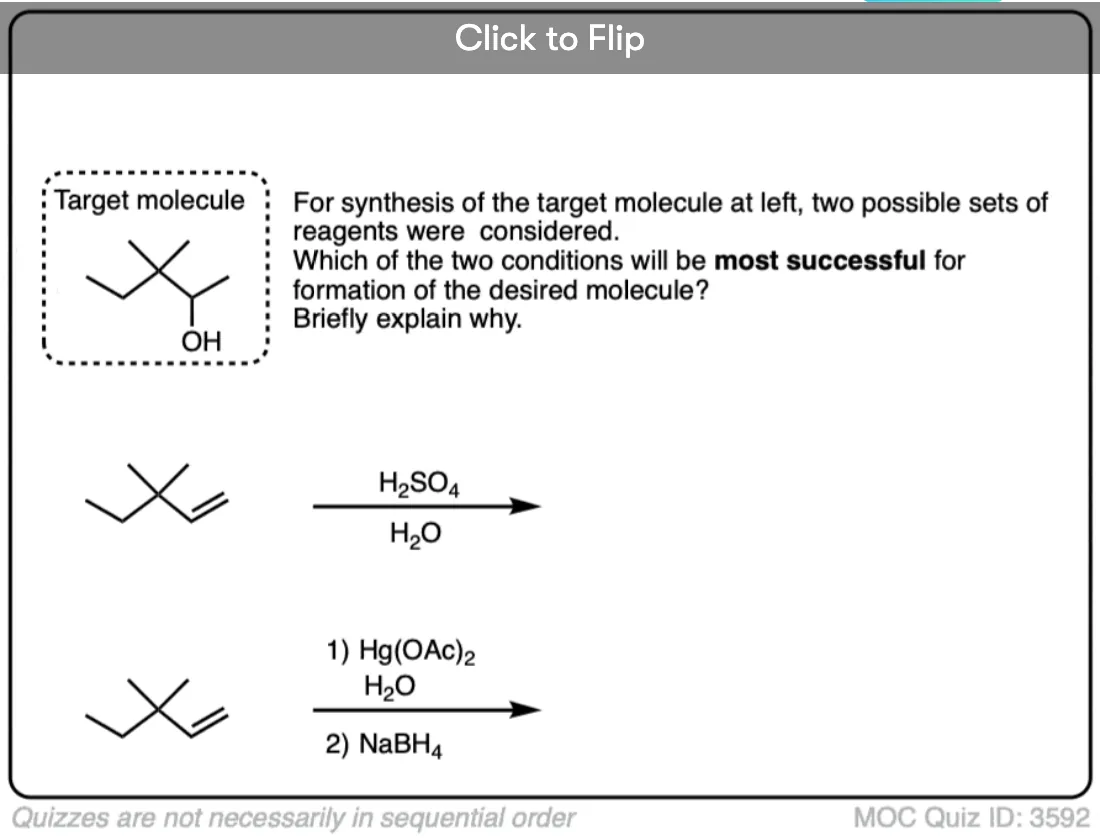
See if you can draw the product of the following reaction:
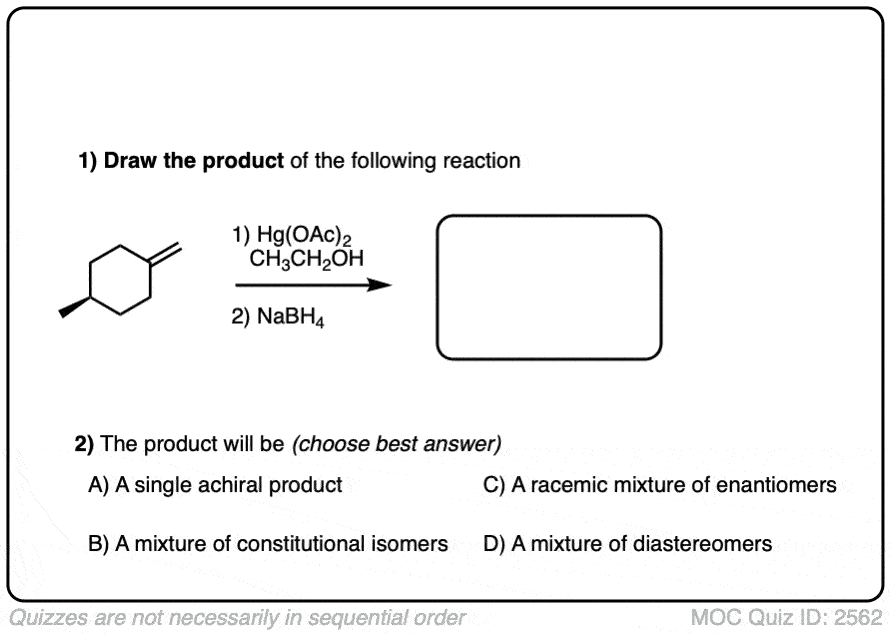 Click to Flip
Click to Flip
An advantage of oxymercuration over some other methods of ether formation is that it can work well for making ethers of secondary and tertiary alcohols.
SN2 reactions between alkoxide nucleophiles (RO(-)) and secondary alkyl halides can run into problems from competing elimination reactions (E2). See this article on The Williamson Ether Synthesis.
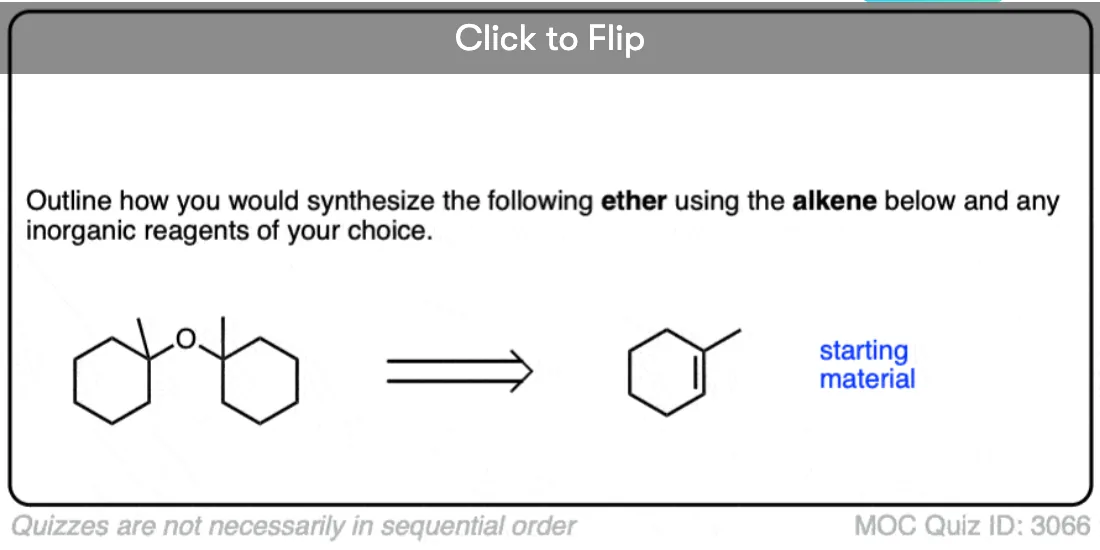
See if you can identify some starting materials for the formation of this ether from an alkene and an alcohol.
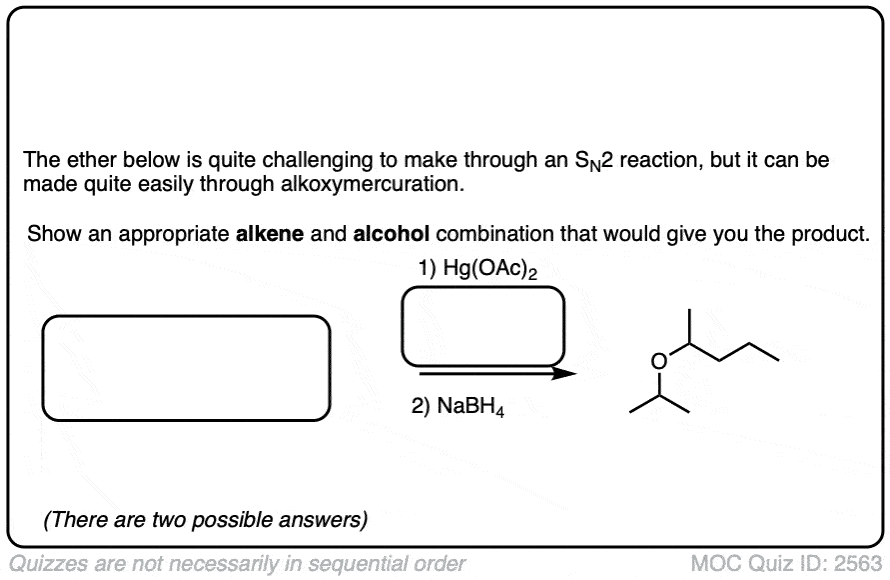 Click to Flip
Click to Flip
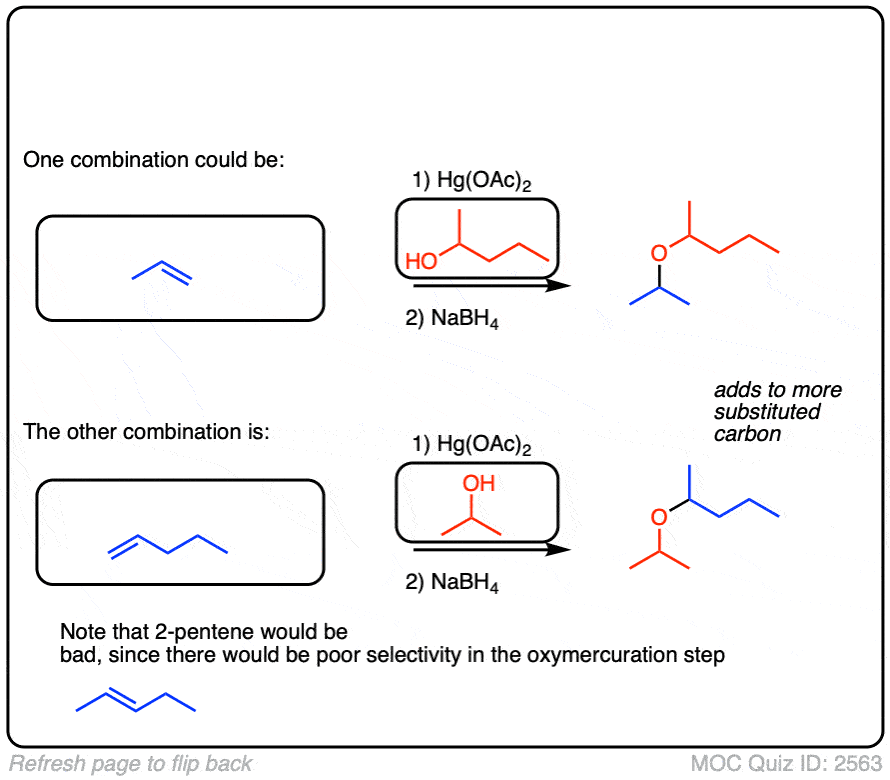
8. Intramolecular Examples
If the molecule has an alkene and and alcohol, then it can react with itself.
This results in the formation of a cyclic product. Five- and six-membered rings work best.
Instructors love testing their students’ understanding of reactions by surprising them with intramolecular versions of reactions, since they involve no new concepts but are a bit tricky if you haven’t practiced them.
See if you can draw the product of the reaction below.
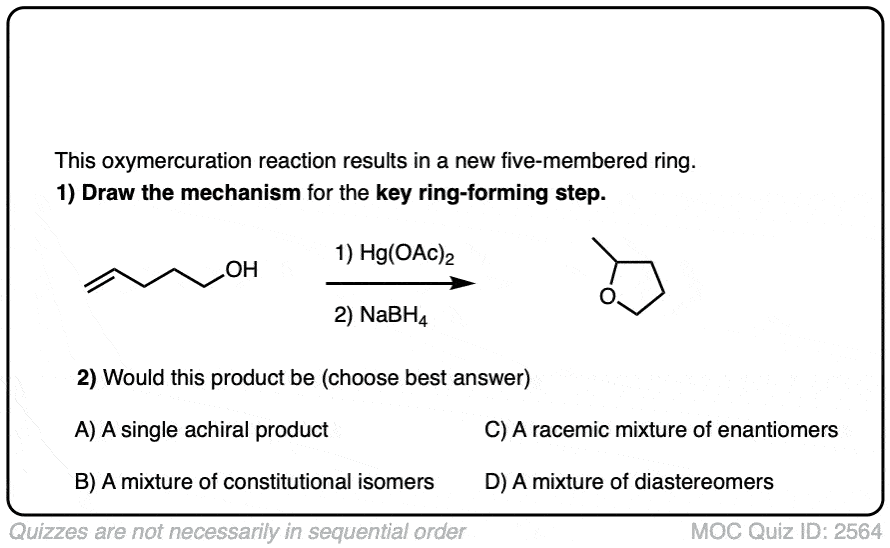 Click to Flip
Click to Flip
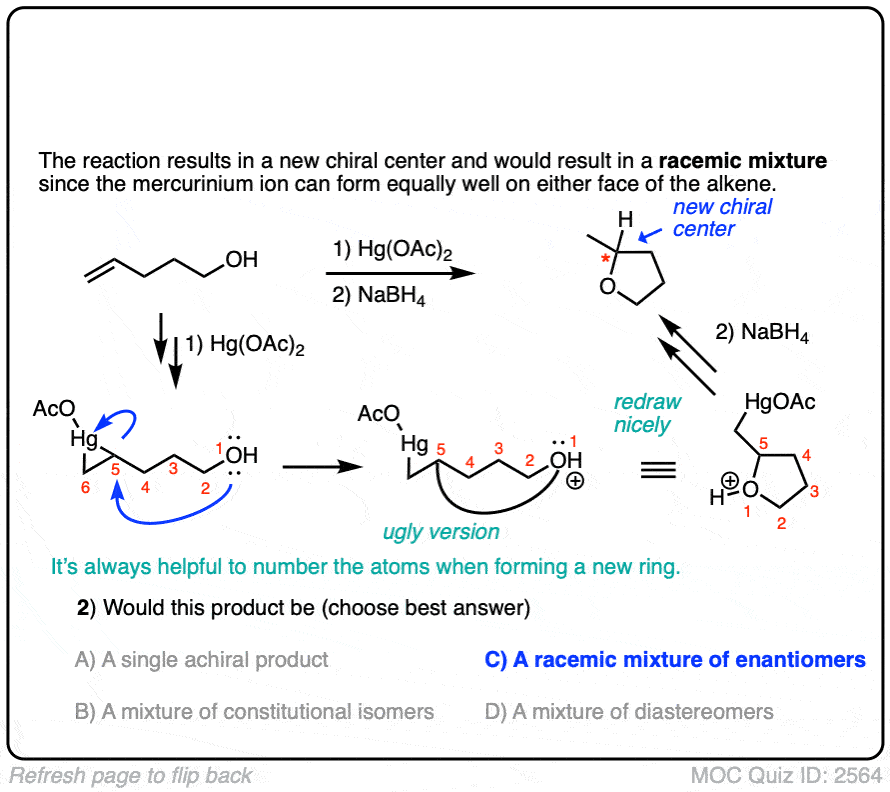
When drawing the products of intermolecular reactions, it can be very helpful to number the atoms to make sure you don’t accidentally add (or subtract) any atoms. It can also be helpful to draw an ‘ugly version’ of the product first just to get the connectivity right, and then re-draw it nicely.
9. Summary
- One of the key takeaways from this article is to understand the contrast between oxymercuration and acid-catalyzed hydration. Acid-catalyzed hydration will potentially give carbocation rearrangements, but oxymercuration will not.
- Another is the contrast between the Markovnikov selectivity of oxymercuration and the anti-Markovnikov selectivity of hydroboration.
- A third key point is to see the similarity in mechanism between oxymercuration of alkenes, halogenation of alkenes, and the opening of epoxides under acidic conditions.
Notes
Note 1. Alkylmercury compounds are highly toxic and should not be worked with without proper training and safety equipment. Dimethylmercury (CH3)2Hg is particularly dangerous and has been responsible for numerous fatalities. [See safety bulletin]
Note 2. Chatt says:
“Both cis- and trans-1-methoxy-2-chloromercuri)cyclohexanes prepared from cyclohexene can be distilled under reduced pressure without change. Except for their greater density they are typical organic compounds. “
Note 3. The structure of the carbon in the mercurinium ion is flatter and more sp2 like than the tetrahedral geometry of the sp3-hybridized carbon in an alkyl halide, which alleviates steric crowding somewhat.
Note 4. Interesting to note the relative reactivity of various alkenes towards oxymercuration. Monosubstituted alkenes are best. Tetrasubstituted alkenes are much slower. With tri- and tetrasubstituted alkenes it is sometimes necessary to use the more electrophilic Hg(OTFA)2.
Note 5. A useful scheme from Whitesides’ studies on the mechanism of demercuration that demonstrates a free-radical intermediate was present: demercuration with NaBH4, followed by trapping with oxygen (O2) led to a mixture of cis and trans stereoisomers. (J. Am. Chem. Soc. 1974, 96, 870)
Note 6. Here is an interactive model of the oxymercurinium ion formed from 2-methylpropene, courtesy of Rowan.
Note that this is the oxymercurinium ion of an unsymmetrical alkene. The C-Hg bond to the least substituted carbon is considerably shorter (2.25 Å) than the C-Hg bond to the more substituted carbon (2.76 Å – such a long bond that it does not even register as a bond in the modeling software).
This goes a long way towards explaining why attack occurs at the more substituted carbon; the C-Hg bond is much longer (and weaker!).
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
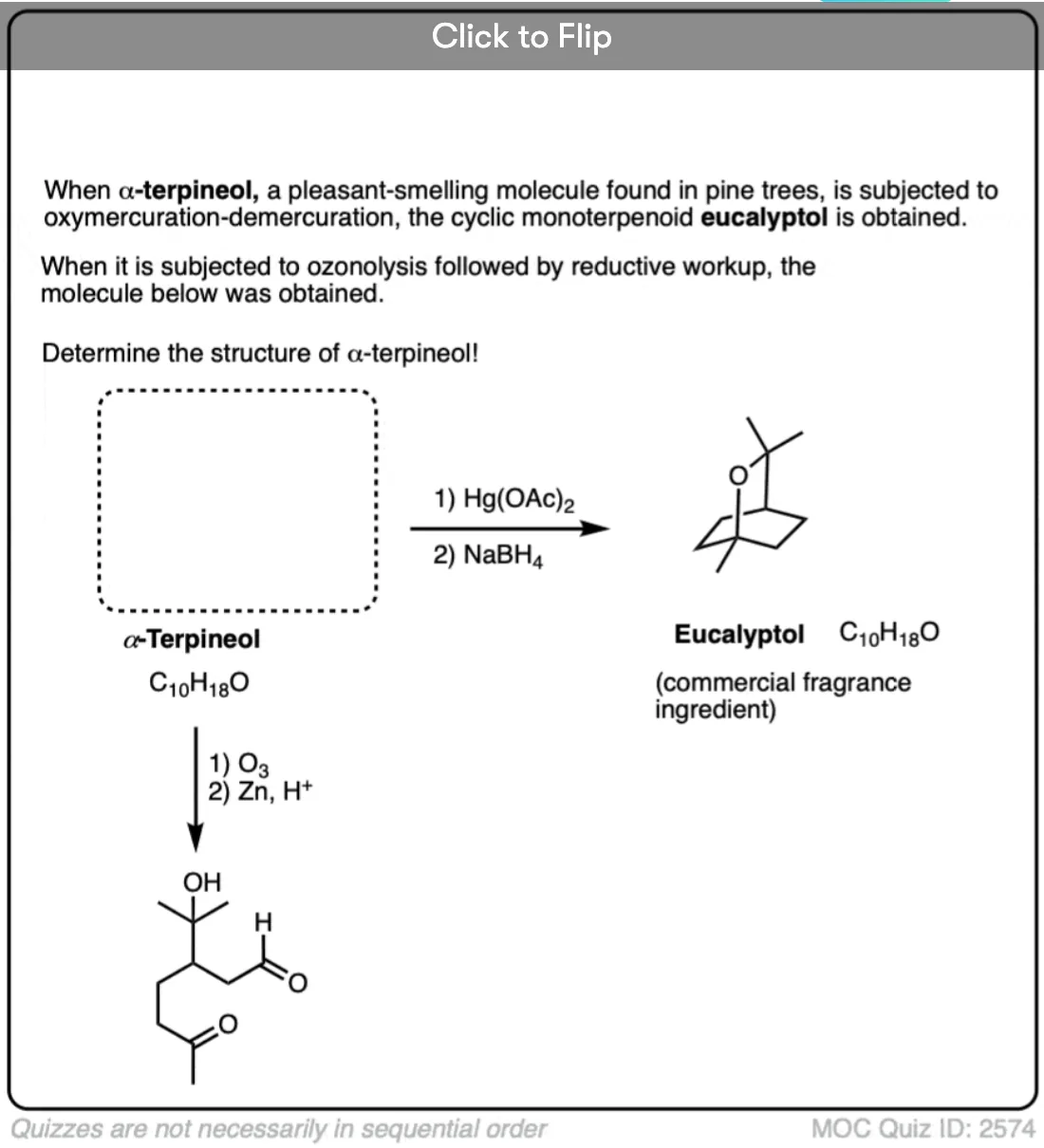
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
-
- Ueber das Verhalten von Mercurisalzen gegen Olefine
K. A. Hofmann, Julius Sand
Chem. Ber. 1900, 33, 1340-1353.
DOI: 10.1002/cber.190003301231
One of the earliest reports on the oxymercuration of alkenes, from 1902. - The Coördination Complexes of Mercuric Ion with Cyclohexene
H. J. Lucas, F. R. Hepner, and S. Winstein
Journal of the American Chemical Society 1939 61 (11), 3102-3106
DOI: 10.1021/ja01266a036
Possibly the first proposal for the mercurinium ion. From the doctoral research of Saul Winstein, who went on to have an exceptionally successful career in physical organic chemistry. Many familiar terms in common usage – “neighboring group participation”, “nonclassical ions”, “anchimeric assistance”, “intimate ion pair”, “internal return”, and “homoaromaticity” are his coinings. See here for a biography. - Solvomercuration-demercuration. I. Oxymercuration-demercuration of representative olefins in an aqueous system. Mild procedure for the Markovnikov hydration of the carbon-carbon double bond
Herbert Charles Brown and Philip J. Geoghegan Jr.
The Journal of Organic Chemistry 1970 35 (6), 1844-1850
DOI: 10.1021/jo00831a028
The use of NaBH4 for removing mercury was a relatively late development. This paper from H.C. Brown’s group is an excellent introduction to oxymercuration with lots of examples on different classes of olefins, relative rate studies, and Markovnikov vs. anti-Markovnikov selectivities. If you were to read just one article on this list, this would be the one to read.
(See also JACS 1967 1522 for the initial communication) - Reactions of alkylmercuric halides with sodium borohydride in the presence of molecular oxygen
Craig L. Hill and George M. Whitesides
Journal of the American Chemical Society 1974 96 (3), 870-876
DOI: 10.1021/ja00810a036
Study of the mechanism of the demercuration step clearly shows that it goes through a free-radical mechanism. The intermediate radical can be trapped with molecular oxygen and gives a mixture of stereoisomers. - Organomercury Compounds in Organic Synthesis
Richard C. Larock
Angewandte Chemie International Edition in English, 1978, 27, 17
DOI: 10.1002/anie.197800271
Fairly short and useful review on organomercury compounds in organic chemistry; section 4 deals with oxymercuration. - Addition Compounds of Olefins With Mercuric Salts
J. Chatt
Chemical Reviews, 1951, 48, 7
DOI: 10.1021/cr60149a002
Review of the older literature on oxymercuration up until about 1950, covering different mechanistic proposals. - Studies on the Mechanism of the Oxymercuration of Substituted Cyclohexenes
Daniel J. Pasto and John A. Gontarz
J. Am. Chem. Soc. 1971, 93, 6902.
DOI: 10.1021/ja00754a035
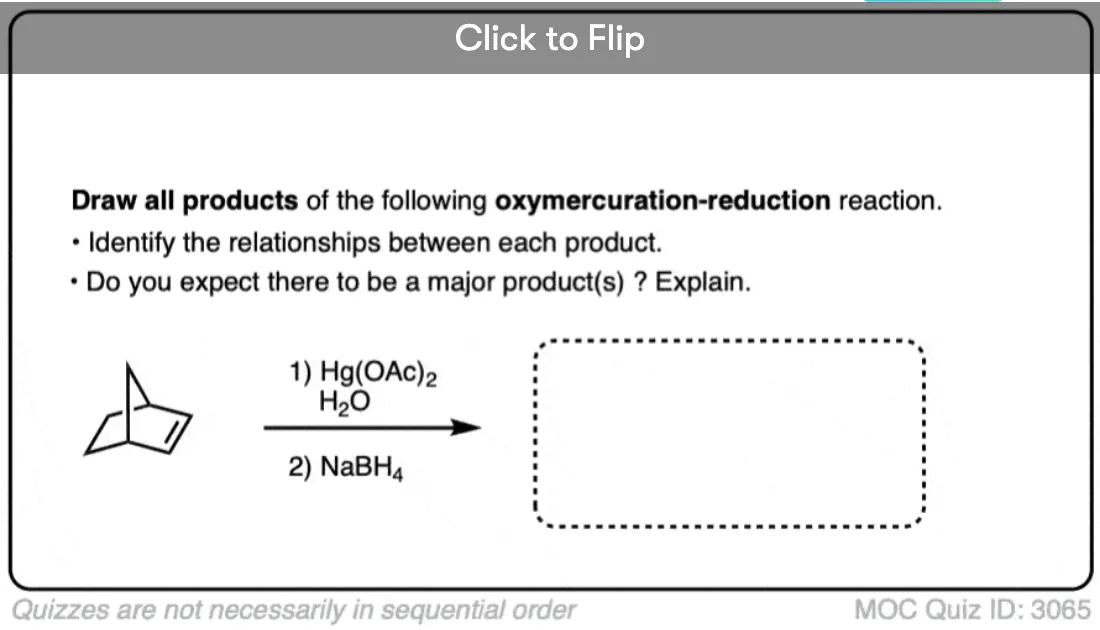
Good discussion here on the regioselectivity of oxymercuration to mercurinium ions with 4-t-butyl cyclohexenes. Includes a few examples of “anti-Markovnikov” addition products that form for stereoelectronic reasons. - Solvomercuration-demercuration. V. Additions to bicyclic olefins. VI. Stereochemistry of the oxymercuration-demercuration of norbornene, 7,7-dimethylnorbornene, and related olefins
Herbert C. Brown and James H. Kawakami
Journal of the American Chemical Society 1973 95 (26), 8665-8669
DOI: 10.1021/ja00807a027
Oxymercuration on very hindered norbornene system gives syn products. The authors propose that the mercurinium ion is in equilibrium with a transient cation. - Mechanism of the reduction of organomercurials with sodium borohydride
Daniel J. Pasto and John A. Gontarz
Journal of the American Chemical Society 1969 91 (3), 719-721
DOI: 10.1021/ja01031a036
The reduction of organomercurials with NaBH4 goes through a free-radical intermediate. In some cases (e.g. cyclopentene) , retention of configuration is high (>95%). In others (e.g. 2-butene) bond rotation is fast relative to recapture.
- Ueber das Verhalten von Mercurisalzen gegen Olefine
Which step in oxymercuration.. can be considered as RDS
Mercurinium ion formation, which is very dependent on the substitution pattern of the alkene. Significant rate differences between mono, di, tri, and tetrasubstituted alkenes.
The “hover here” button doesn’t work.
Fixed. Thanks for the heads up!
It seems you have a duplicate quiz in section 8; it is testing ether formation instead of intramolecular reactions. Lovely site, thanks!
Good catch – fixed! – thank you!