Alcohols, Epoxides and Ethers
Elimination Reactions of Alcohols
Last updated: May 28th, 2026 |
All About Elimination Reactions of Alcohols (With Acid)
- The hydroxyl group of alcohols is normally a poor leaving group.
- However, when treated with strong acid, R-OH is converted into R-OH2(+) and H2O is a much better leaving group.
- With tertiary alcohols, H2O can then leave, resulting in a carbocation.
- If a strong acid such as H2SO4 or p-TsOH is used, the most likely result is elimination, since the conjugate bases of these acids (HSO4 – and TsO– ) are poor nucleophiles and unlikely to add to the carbocation.
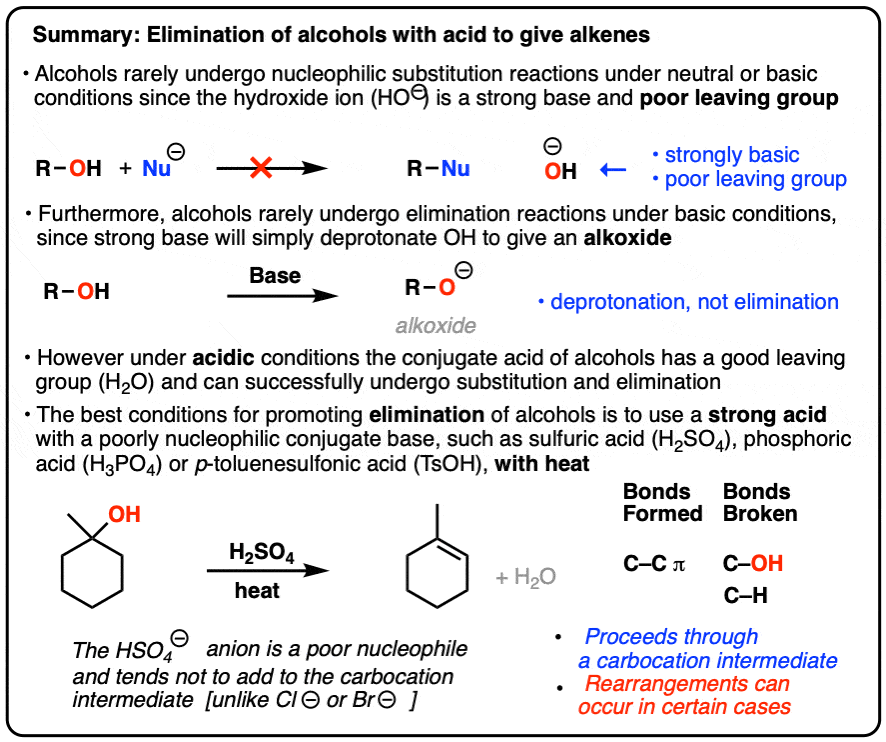
Table of Contents
- Hydrohalic Acids (HX) Plus Alcohols Give Substitution Products…
- …But H2SO4, H3PO4, and TsOH Give Elimination Products!
- Elimination of Tertiary Alcohols Proceeds Through an E1 Mechanism
- Why Do H2SO4, H3PO4 and TsOH Give Elimination Products?
- What About Secondary Alcohols?
- Elimination Reactions With Carbocation Rearrangements
- Ring Expansion Followed By Elimination
- Primary Alcohols and H2SO4 Can Form Alkenes (via E2)
- Summary: Elimination Reactions of Alcohols
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Hydrohalic acids (HX) plus alcohols give substitution products…
Previously (See post: Making Alkyl Halides from Alcohols) we saw that treating an alcohol with a strong hydrohalic acid – think HCl, HBr, or HI – resulted in the formation of alkyl halides. With a tertiary alcohol like the one drawn below, this proceeds through an SN1 mechanism. [Protonation of alcohol, then loss of H2O to form a carbocation, then attack of nucleophile on carbocation].
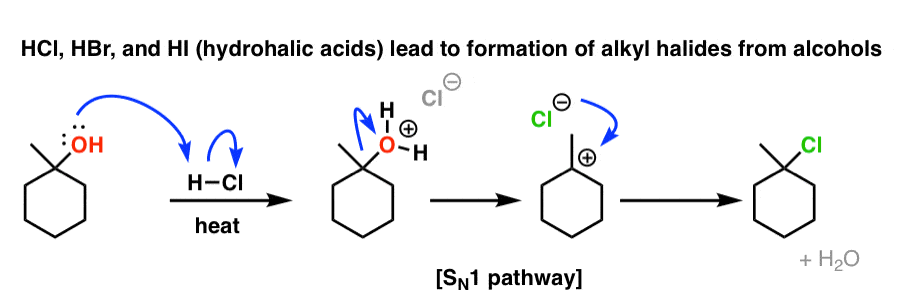
2. …But H2SO4, H3PO4, and TsOH Give Elimination Products!
You’d be forgiven for thinking that if we treated an alcohol with H2SO4 (sulfuric acid) the same type of thing would occur, and the carbocation would be attacked by the (-)OSO3H anion to make the product below. Very reasonable to propose. In practice, however, it doesn’t work that way!
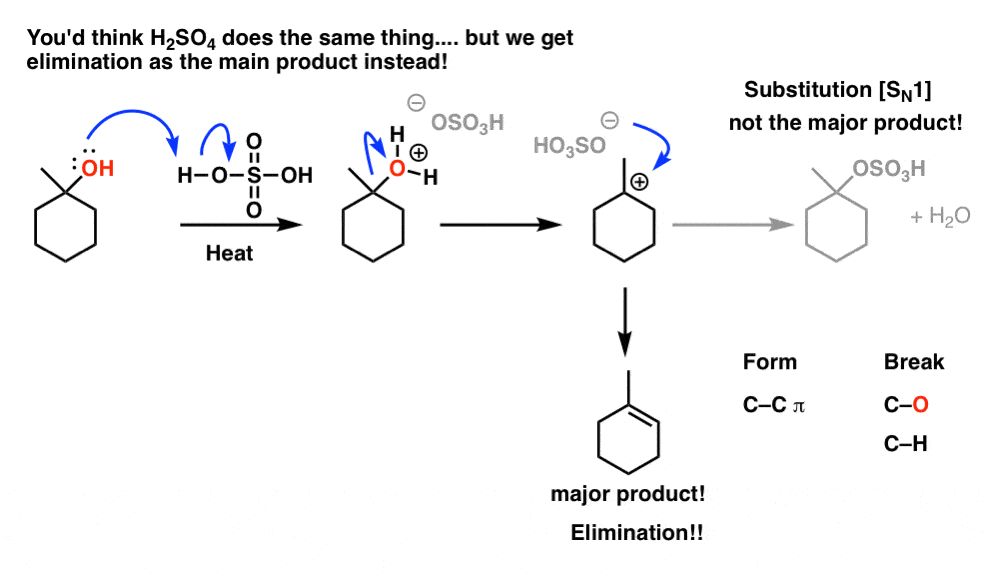
So what happened here?
First, look at what bonds formed and broke. We formed C-C (π) and broke C-OH and C-H. (We also formed H-O , in that molecule of water that forms as a byproduct). This is the pattern of an elimination reaction.
Now let’s ask: How could this have formed? If you look closely, note that we’ve broken a C-H bond on the carbon adjacent to the carbocation and formed a new C-C π bond at that spot. It’s reasonable to propose that instead of attacking the carbocation to form a new substitution product, a base removed a proton adjacent to the carbocation and formed the alkene. [That carbon adjacent to the carbocation is often referred to as the “β (beta) carbon”. The carbocation itself is the “α (alpha) carbon”].
3. Elimination of Tertiary Alcohols Proceeds Through an E1 Mechanism
We’ve seen this type of process before actually! This is an E1 process [elimination (E) , unimolecular (1) rate determining step]. You might also remember that elimination reactions tend to follow “Zaitsev’s rule” – we always form the most substituted alkene [or to put it another way, we remove a proton from the carbon with the fewest attached hydrogens] because alkene stability increases as we increase the number of attached carbons.
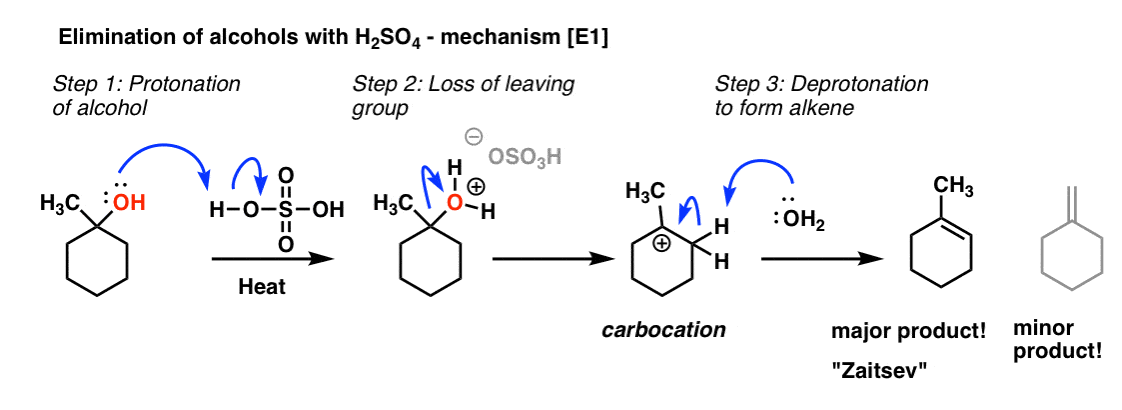
[By the way, you might ask – why “heat” ? Heat generally tends to favour elimination reactions.]
By no means is H2SO4 the only acid that does this. Phosphoric acid (H3PO4) as well as “tosic acid” (p-toluenesulfonic acid) also tend to form elimination products.
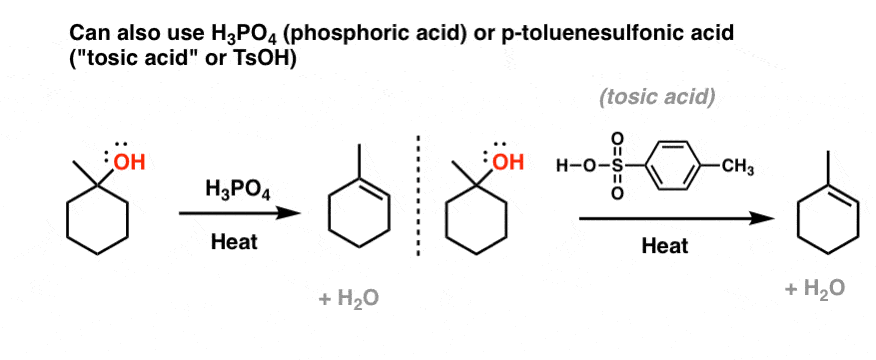
4. Why Does H2SO4 (Or H3PO4 or TsOH) Give Elimination Products But HCl, HBr, HI give Substitution Products?
So why do we get elimination reactions with H2SO4 as acid (or H3PO4, or TsOH) whereas we get substitution reactions with HCl, HBr, and HI?
The answer is that the HSO4– anion is a very poor nucleophile , being quite stabilized by resonance. In the diagram below, note how that negative charge is delocalized over three different oxygens [the same is true for the TsO– and H2PO4– anions].
Compare that to halide anions, where the negative charge cannot be spread over more than one atom. The upshot is that delocalization of charge results in a slower reaction of HSO4– as a nucleophile compared to deprotonation of C-H by a base, and the alkene product dominates.
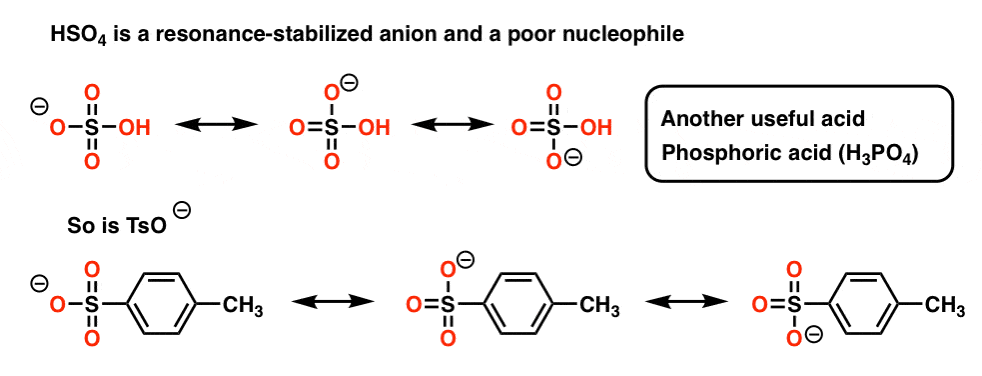
So the bottom line here is that heating tertiary alcohols with these acids will result in loss of water [“dehydration”] and formation of an alkene [elimination].
5. What About Elimination Reactions Of Secondary Alcohols?
Heating a secondary alcohol with sulfuric acid or phosphoric acid? Same deal as with tertiary alcohols: expect an alkene to form. As with all elimination reactions, there are two things to watch out for: first, the “most substituted” alkene (Zaitsev) will be the dominant product, and also, don’t forget that trans alkenes will be favoured (more stable) than cis alkenes due to less steric strain.
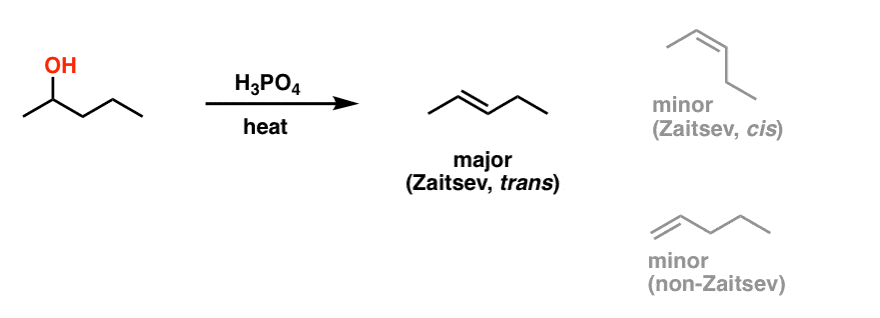
There is one last thing to watch out for with secondary alcohols, though… like a bad nightmare, they keep coming back.
6. Elimination Reactions With Carbocation Rearrangements
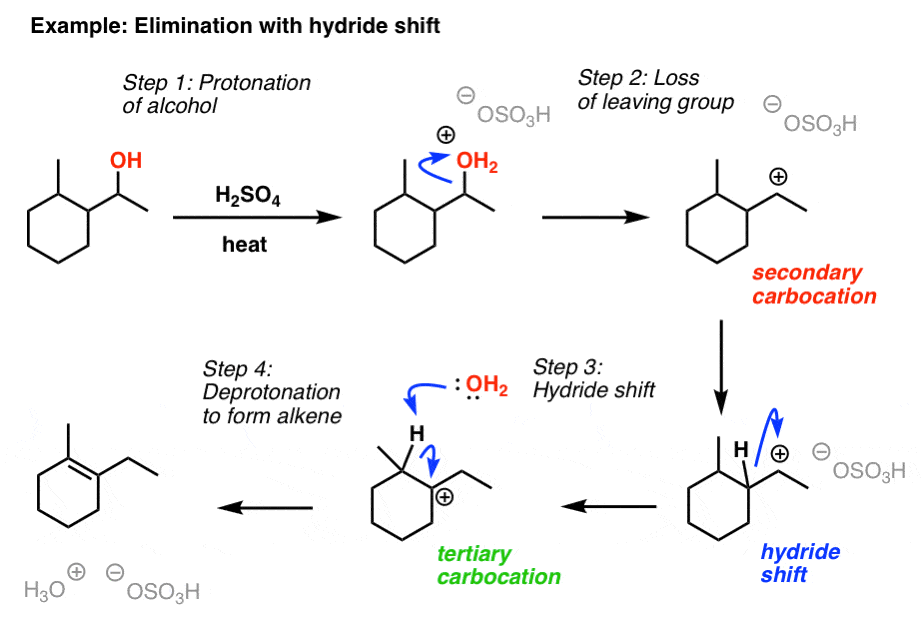
As we saw with the reactions of HCl, HBr, and HI with secondary alcohols, we have to watch out for carbocation rearrangement reactions. If a more stable carbocation can be formed through migration of an adjacent hydride (H- ) or an alkyl group, then that migration will occur.
For example, treatment of the alcohol below with H2SO4 leads to formation of a secondary carbocation, followed by a hydride shift to give a tertiary carbocation, followed by deprotonation at whichever β carbon leads to the most substituted alkene.
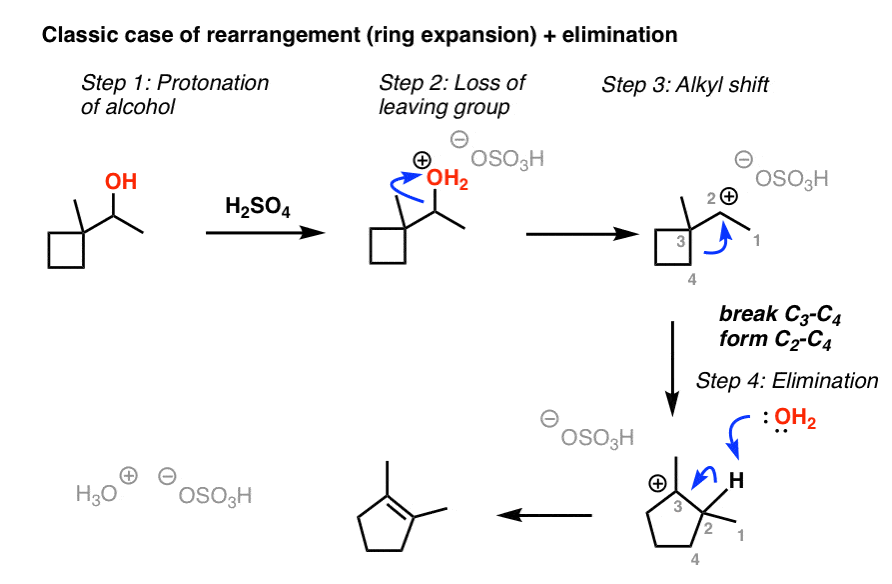
7. Ring Expansion Followed By Elimination
It’s also possible for alkyl shifts to occur to give a more stable carbocation. A classic example of this are expansions of strained rings (like cyclobutanes) to give less strained rings (like cyclopentanes).
For example in the case below the key step is where the C3-C4 bond breaks to form the C2-C4 bond, resulting in a new (tertiary) carbocation on C-3 as well as a less strained ring. Since there isn’t a good nucleophile around, elimination occurs in such a way that the most substituted alkene is formed.
8. Primary Alcohols And H2SO4 Can Form Alkenes (E2)
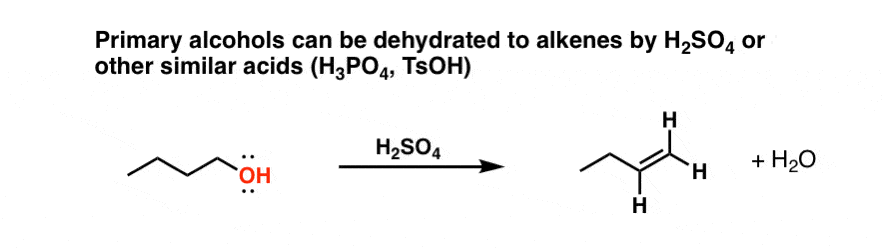
The final class of alcohols to be concerned about is primary alcohols. You might ask: if we treat a primary alcohol (say, 1-butanol) with a strong acid like H2SO4, will also get elimination to an alkene?
Yes, alkenes can be formed this way (along with some formation of symmetrical ethers [see this previous post]). Here’s an example.
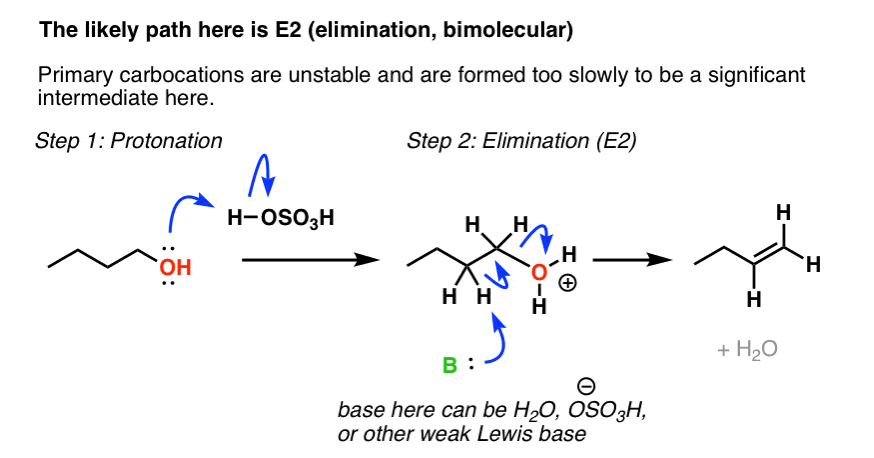
There is a catch however: the E1 pathway (formation of a primary carbocation) is not the most likely pathway here. Primary carbocations tend to be extremely unstable, and it’s more likely that the reaction passes through an E2 mechanism where the transition state will be lower in energy. Notice what happens here: first we protonate the alcohol to give the good leaving group OH2+ , and then a weak base (which I’m leaving vague, but could be H2O, (-)OSO3H, or another molecule of the alcohol) could then break C-H, leading to formation of the alkene.
9. Summary: Elimination Reactions of Alcohols (E1)
If you see a tertiary or secondary alcohol with H2SO4, TsOH, or H3PO4 (and especially if you see “heat”) think: carbocation formation followed by elimination reaction (E1).
And if you see that a more stable carbocation could be formed through migration of an adjacent H or alkyl group, expect that to happen.
If you see a primary alcohol with H2SO4, TsOH, or H3PO4, expect symmetrical ether formation accompanied by elimination to form the alkene.
Next Time…
So far we’ve learned two ways to convert alcohols to alkenes:
- convert them to a good leaving group, and then add base (2 steps)
- add strong acid with heat (one step)
Ideally, we’d like to just use one step. But strong acid can lead to complications (carbocation rearrangements, cough cough) and we might ask: “isn’t there an easier way”?
There is! That’s what we’ll cover in the next post.
Next Post: Elimination Of Alcohols To Alkenes With POCl3
Notes
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
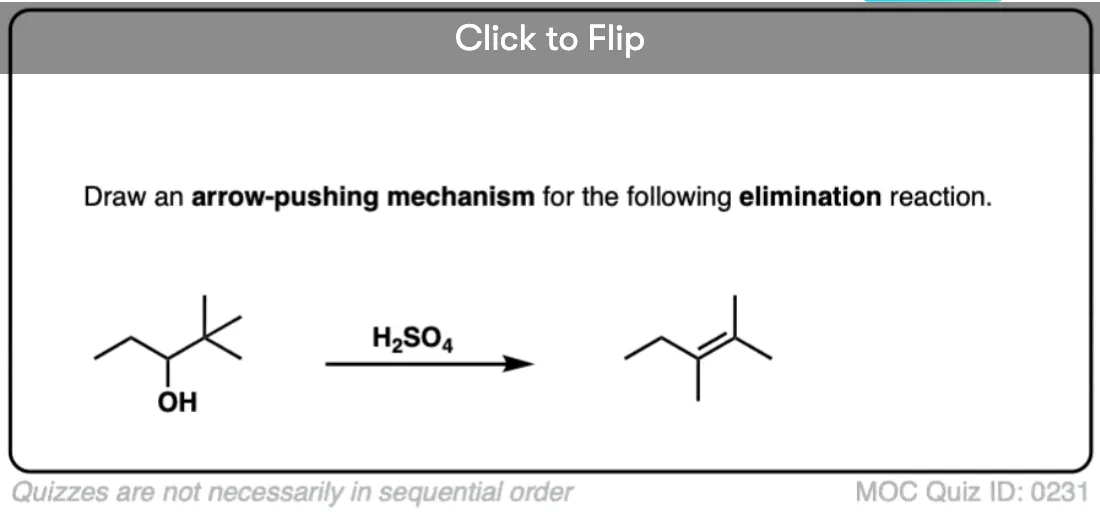
Become a MOC member to see the clickable quiz with answers on the back.
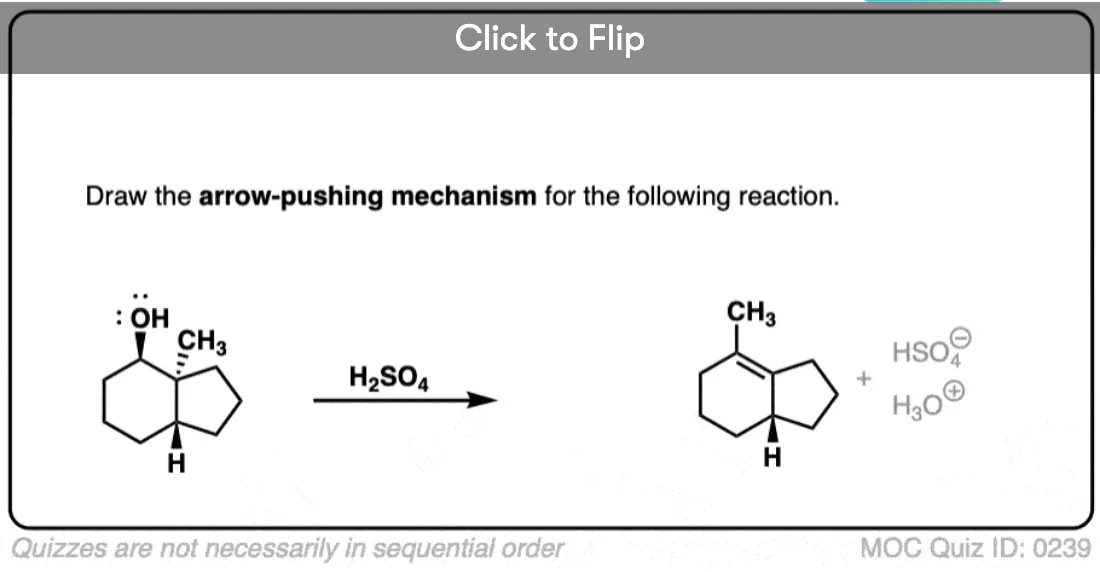
Become a MOC member to see the clickable quiz with answers on the back.
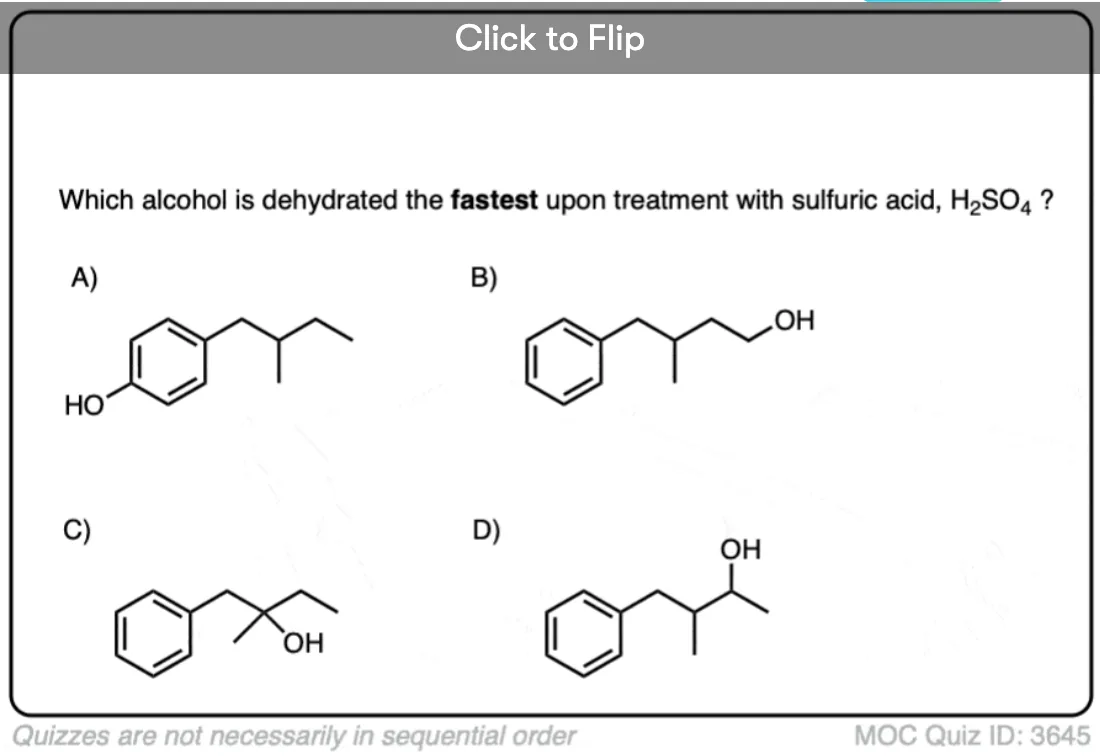
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
A variety of conditions are possible for this transformation (alcohol -> alkene), all of which involve converting the -OH into a better leaving group. The use of acid is the simplest method to achieve this, as protonation of -OH gives -OH2+, an excellent leaving group (water).
Elimination with POCl3:
- The Effect of Structure on the Course of Phosphoryl Chloride-Pyridine Dehydration of Tertiary Alcohols
Ronald R. Sauers
Journal of the American Chemical Society 1959 81 (18), 4873-4876
DOI: 1021/ja01527a028 - Stereospecificity and regiospecificity of the phosphorus oxychloride dehydration of sterol side chain alcohols
Jose Luis Giner, Christian Margot, and Carl Djerassi
The Journal of Organic Chemistry 1989 54 (2), 369-373
DOI: 1021/jo00263a020
This article by the legendary chemist Carl Djerassi (who developed norethindrone, the first female contraceptive) describes the selectivity of POCl3-pyridine dehydration conditions in steroid synthesis. It also has a general procedure for POCl3-pyridine dehydration in the experimental section. - A general approach to linearly fused triquinane natural products. Total syntheses of (.+-.)-hirsutene, (.+-.)-coriolin, and (.+-.)-capnellene
Goverdhan Mehta, A. Narayana. Murthy, D. Sivakumar. Reddy, and A. Veera. Reddy
Journal of the American Chemical Society 1986 108 (12), 3443-3452
DOI: 1021/ja00272a046
This paper by Prof. Goverdhan Mehta (considered the ‘Indian E. J. Corey’) demonstrates the applicability of the POCl3-pyridine dehydration in natural product total synthesis. - The 3-methylcholestanols and their derivatives
D. H. R. Barton, A. da S. Campos-Neves and R. C. Cookson
J. Chem. Soc., 1956, 3500-3506
DOI: 10.1039/JR9560003500
This paper by Nobel Laureate Prof. Derek H. R. Barton has a POCl3-pyridine dehydration (see p. 3504-3505 in the experimental section).Using a Brønsted acid: - THE COMPOSITION OF BUTENE MIXTURES RESULTING FROM THE CATALYTIC DECOMPOSITION OF THE NORMAL BUTYL ALCOHOLS
William G. Young and Howard J. Lucas
Journal of the American Chemical Society 1930, 52 (5), 1964-1970
DOI: 1021/ja01368a030
While this would seem to be an easy thing to study today, with NMR and GC-MS, in the early days this was not so easy. The butenes were converted to dibromides, distilled, and then the three-component dibromide mixture analyzed by density, refractive index, and determination of the second-order rate constants with potassium iodide in acetone. Both Bill Young and H. J. Lucas contributed greatly to the development of chemistry in Southern California – H. J. Lucas was a professor of chemistry at Caltech, and Bill Young later became a professor of chemistry at UCLA, and was the advisor for Prof. Saul Winstein’s M.S. in chemistry (Prof. Winstein ended up joining Bill Young at UCLA after his PhD!). - The Dehydration of Secondary and Tertiary Alcohols
Albert L. Henne and Alfred H. Matuszak
Journal of the American Chemical Society 1944, 66 (10), 1649-1652
DOI: 1021/ja01238a012
An early paper that demonstrates the E1 nature of this reaction, by demonstrating that dehydration of various secondary and tertiary alcohols give products obtained through rearrangement. - Tracer studies on alcohols. Part II. The exchange of oxygen-18 between sec.-butyl alcohol and water
C. A. Bunton and D. R. Llewellyn
J. Chem. Soc., 1957, 3402-3407
DOI: 10.1039/JR9570003402
This paper provides experimental evidence that stronger acids favor elimination over substitution. As the Hammett acidity (-H0) of the medium increases, carbocation formation is increasingly favorable, which promotes elimination over substitution. Sulfuric acid and perchloric acid are much stronger acids than the hydrogen acids (HCl, HBr, HI), which explains why sulfuric acid is commonly used to make olefins from alcohols. - Reactions of n-butene and butan-2-ol in dilute acid. The elucidation of the mechanism and the intermediate in elimination from secondary alcohols and in the hydration of olefins
Joost Manassen and Fritz S. Klein
J. Chem. Soc., 1960, 4203-4213
DOI: 10.1039/JR9600004203
The authors use radiolabeling to study both the forward and reverse reactions (hydration of alkene and elimination of alcohol), to prove that they both go through a common carbocation intermediate. - The mechanism of the acid-catalyzed dehydration of 1,2-diphenylethanol
Donald S. Noyce, Donald R. Hartter, and Ralph M. Pollack
Journal of the American Chemical Society 1968, 90 (14), 3791-3794
DOI: 1021/ja01016a034
Interestingly, in the reaction of 1,2-diphenylethanol in acid, formation of the intermediate carbocation is fast and reversible, and proton loss to the olefin is rate-determining. - The effect of substituents on the rate of the acid-catalyzed dehydration of 1,2-diarylethanols
Donald S. Noyce, Donald R. Hartter, and Frank B. Miles
Journal of the American Chemical Society 1968, 90 (14), 3794-3796
DOI: 1021/ja01016a035
In this reaction, the effect of substitution on the phenyl ring a to the -OH is greater than that of substitution on the other ring. - Catalytic, Tunable, One-Step Bismuth(III) Triflate Reaction with Alcohols: Dehydration Versus Dimerization
Laura E. Kolsi, Jari Yli-Kauhaluoma, and Vânia M. Moreira
ACS Omega 2018, 3 (8), 8836-8842
DOI: 1021/acsomega.8b01401
A modern variant of this reaction using Lewis Acids. The authors also show that these conditions can be used for forming dimers vs. simple elimination products. The dimer is formed by the carbocation reacting with the alkene.
Hi James, If I got any doubt in organic chemistry, I look upto your work. I need to know, Does primary alcohols on acid catalysed elimination produces any rearranged products. In this webpage (http://www.columbia.edu/itc/chemistry/c3045/client_edit/ppt/PDF/05_08_13.pdf), Butan-1-ol gave 2-butene as a major product. In wade Jr text book 1-pentanol produced 2-pentene as major product.
Between substitution and elimination reactions in alcohols which one is catalyzed with acid or a base?
Acid makes the OH a better leaving group, since the new leaving group will be the weaker base H2O, not HO(-).
Base makes the OH a better nucleophile, since RO(-) is a better nucleophile than the neutral alcohol ROH.
Both substitution and elimination reactions of alcohols can be catalyzed by acid.
HELLO. WOULD YOU MIND TELLING ME THE MECHANISM OF ALCOHOL and Me2C(OMe)2 and p-TsOH(CATALYST)?Thanks in advance
It’s a way of forming a cyclic acetonide from a diol. https://en.wikipedia.org/wiki/Acetonide
Is there a way to convert a diol to alkene from ways mentioned above?
Not in one step. It’s necessary to do a reduction of some kind.
However, there is a reaction called the Corey-Winter reaction that will reduce diols to alkenes. https://en.wikipedia.org/wiki/Corey%E2%80%93Winter_olefin_synthesis
Hi James. Was just wondering if HNO3 would cause the same reaction to occur as H2SO4 or H3PO4 (an E1 rxn)?
Quick answer: yes .
Longer answer: yes, but it depends on the concentration of HNO3 and the type of alcohol. Dilute HNO3 by itself is probably fine. Concentrated HNO3 contains some NO2+ which is an excellent electrophile, which the alcohol can add to, leading to R-ONO2 . If the alcohol is a primary or secondary alcohol, this can then be oxidized to an aldehyde or ketone, or onwards. Tertiary alcohols don’t oxidize.
Another problem with alcohols: you’ve heard of nitroglycerin? That’s made by adding HNO3 (as well as a bit of H2SO4) to the tri-ol glycerin, which leads to potentially explosive results.
For that reason we usually just stick to H2SO4 or H3PO4!
James
Thermal stability vs migratory aptitude:
I have this doubt. Cant find a solution anywhere. Please help
What would be the elimination product of 2-methyl-2-phenylpropan-1-ol?
Migration of Ph- is faster than R- but will lead to a less stable intermediate and vice versa.
Is it an example of kinetic vs thermodynamic control?
What would be the more suitable answer?
This would be an example of anchimeric assistance (neighboring group participation). After protonation of OH, the phenyl group acts as an internal nucleophile, leading to a bridged intermediate. Attack of water on the bridged intermediate gives 2-methyl-1-phenyl-2-ol, which then undergoes a normal dehydration to give 2-methyl-1-phenyl-1-propene
In what cases does rearrangement take place ? When a more stable carbocation is formed or are there any other criteria as well ?
Can alcohols undergo an E2 reaction? Since it requires deprotonation to create a better leaving group, I would think not but I’m not sure.
Not conventional E2 reactions. Deprotonation of the hydroxyl group would make the resulting species (O-) an even worse leaving group!
Concerning the 4th picture (Elimination of alcohols with H2SO4 – mechanism [E1]), why does water deprotonate the carbocation in step 3? If Kw = 1.0 x 10^-14 then shouldn’t the formation of H3O+ be very unfavorable?
In the last example, E2 reaction with a primary alcohol, why does 2-butene (the more stable alkene) not formed from 1-butanol?
Because in order for elimination to occur, the C-H bond has to break on the carbon next to the carbon bearing the leaving group. The leaving group is on C1, the CH bond must therefore break on C2, and the bond forms between C1 and C2, giving 1-butene.
just want to thankyou for this clear explanation. i was really confused why H2SO4 was only explained as forming E1 E2 products but not SN1 SN2. couldnt find the answer anywhere until i stumbled on this page.
Why we use H2SO4 in case of alcohols reacting with HBr and that of we use H3PO4 in case of alcohols reacting with HI . why
They should be equivalent.
What happens if you use two cis or trans OH in the educt?
It’s somewhat possible that you might get some epoxide formation, or even formation of a ketone/aldehyde. Depends on the structure of the substrate.
When ethanol is heated at 140*C in the presence of conc. Sulphuric acid. Is this a beta elimination reaction??
Likely formation of diethyl ether.
I would assume that secondary alcohols can undergo both E1 and E2 reactions. In your post, you are suggesting that secondary alcohols favor an E1 mechanism. Is that true only if a secondary carbocation can rearrange to give a tertiary? Is it safe to say that otherwise, secondary alcohols can undergo both E1 and E2?
The identity of the acid is important. In the case of H2SO4 or H3PO4, there simply is no sufficiently strong base present to cause an E2 reaction to occur. Loss of H2O to form a carbocation followed by elimination will be the favoured pathway.
Note that secondary alkyl halides can undergo E2 reactions just fine. The issue with alcohols here is that we are using strong acid to turn the OH into a good leaving group. If we add a strong base here (to perform an E2) it will just end up neutralizing this species.
As far as rearrangement is concerned, it will generally only be favoured in a situation where a more stable carbocation will form. That is true for the conversion of secondary carbocations to tertiary carbocations. It *can* be true that rearrangements of tertiary carbocations occur, but generally only in situations where they would be more stabilized (e.g. tertiary carbocation to a resonance-stabilized tertiary carbocation )
sorry I put my e mail wrong, posting my question again.
why not a SN2 reaction after protonation of primary alcohols??? H2O is a good leaving group and primary carbon is not hindered, a perfect recipe for SN2. HSO4- can attack through SN2, why not? why elimination? given that HSO4- is a week base too.
HSO4- is an extremely poor nucleophile for the SN2.
Plus there is heat involved in the reaction..which is favourable for elimination reactions…thank u n feel free to correct if wrong
thank you so much for these information but i have a small question …is there a difference between Elimination and dehydration ??
Dehydration specifically refers to loss of water. Elimination in the sense of this post refers to formation of a double bond. There is overlap between the two when dehydration leads to formation of a double bond.
I posted a message a few days ago, but somehow it was erased. There it goes again:
– “… we remove a proton from the carbon with the most attached hydrogens”; it’s the carbon with the FEWEST attached hydrogens!
– please check the formulas of acids and their corresponding anions in the text; some appear like this: “H2SO4 as acid (or H3PO4” (they are written correctly in the images).
Don’t know why that comment didn’t post. Thank you for your keen eye, as always!