Substitution Reactions
Comparing the SN1 and SN2 Reactions
Last updated: June 2nd, 2026 |
Comparing the SN1 and SN2 Reactions
In nucleophilic substitution reactions, a bond between carbon and a leaving group (C–LG) is broken, and a new bond between carbon and a nucleophile (C–Nu) is formed.
Nucleophilic substitution reactions of alkyl halides occur through two main pathways. The key difference lies in the timing of the bond-forming and bond-breaking steps.
- The SN1 mechanism (Substitution, Nucleophilic, UNImolecular rate determining step) generally passes through two steps; first, a (slow, rate-determining) breaking of the C–LG bond on the substrate to form an intermediate carbocation, followed by (fast) addition of a nucleophile to the carbocation (form C–Nu) to give the substitution product (there is often a third acid-base step which follows the substitution reaction when neutral nucleophiles like H2O or ROH are used)
- The SN2 mechanism (Substitution, Nucleophilic, Bimolecular rate determining step) occurs in a single, concerted step: attack of the nucleophile on the backside of the C–LG bond, passing through a transient five-membered transition state en route to a tetrahedral product where configuration at the carbon has been inverted.
Since the rate-determining step in the SN1 is formation of a carbocation, it can be helpful to think of the “big barrier” to the SN1 reaction as being carbocation stability, That is, any factor which leads to the increased stability of a carbocation intermediate will increase the rate of the SN1. That’s why this pathway tends to be favored by tertiary alkyl halides, since the order of carbocation stability generally proceeds tertiary > secondary > primary (See article: Carbocation Stability)
Similarly, since the rate-determining step of the SN2 is the backside attack of a nucleophile on carbon, it can be helpful to think of the “big barrier” to the SN2 reaction as being steric hindrance (See article: Steric Hindrance). Any factor which increases the difficulty with which the nucleophile can access the sigma* orbital of the C–LG bond will result in a slower reaction, which helps us to rationalize why the SN2 is faster with methyl and primary alkyl halides than for secondary and tertiary alkyl halides.
Owing to these two different mechanisms, the SN1 and SN2 reactions exhibit key differences in
- observed rate laws (unimolecular for SN1, bimolecular for SN2)
- patterns of stereochemistry (retention + inversion for SN1, inversion for SN2)
- relative rates of reaction for primary, secondary, and tertiary alkyl halides (tertiary > secondary > primary for SN1, primary > secondary > tertiary for SN2
Additionally, the SN1 and SN2 reactions are sensitive to the identity of the solvent and of the strength of the participating nucleophile. More detail below!

Table of Contents
- But First: The Story Of The Cats And The Comfy Chair
- The SN1 Proceeds Through A Stepwise Mechanism. The SN2 Proceeds Through a Concerted Mechanism
- Reaction Coordinate Diagrams of the SN1 and SN2 Reactions
- The Rate Laws of the SN1 and SN2 Reactions
- Primary, Secondary, and Tertiary Alkyl Halides in SN1 and SN2 Reactions
- Comparing the Stereochemistry of SN1 and SN2 Reactions
- Solvents and Nucleophiles – SN1 Reaction
- Solvents and Nucleophiles – SN2 Reaction
- Back To The Cats
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. But First: The Cats And The Comfy Chair
But first – have you ever heard the story of the cats and the comfy chair?

Cat #1 finds Cat #2 on his comfy chair and wants to sit. He has two options.
- He can wait for Cat #2 to leave, and then sit in the comfy chair.
- He can kick the Cat #2 out of his comfy chair.
2. The SN1 Proceeds Through A Stepwise Mechanism. The SN2 Proceeds Through a Concerted Mechanism
Let’s compare the mechanisms of the SN1 and SN2 reactions first, since every other difference we will observe is a consequence of their different mechanisms. (Generally, proposing a mechanism only comes after collecting a lot of experimental evidence (e.g. rate laws, stereochemistry, relative rates, etc.) , but since this isn’t an Agatha Christie novel, we’re giving away the ending first).
The SN1 generally passes through a two-step “stepwise” mechanism where
- The leaving group leaves (break C–LG) to give a carbocation intermediate (slow, rate-determining step)
- The resulting carbocation intermediate is attacked by a nucleophile to give a new product (form C–Nu) (fast step)
(In many cases, there is often a third step involving deprotonation of the nucleophile to give a neutral product, especially if the nucleophile is neutral)
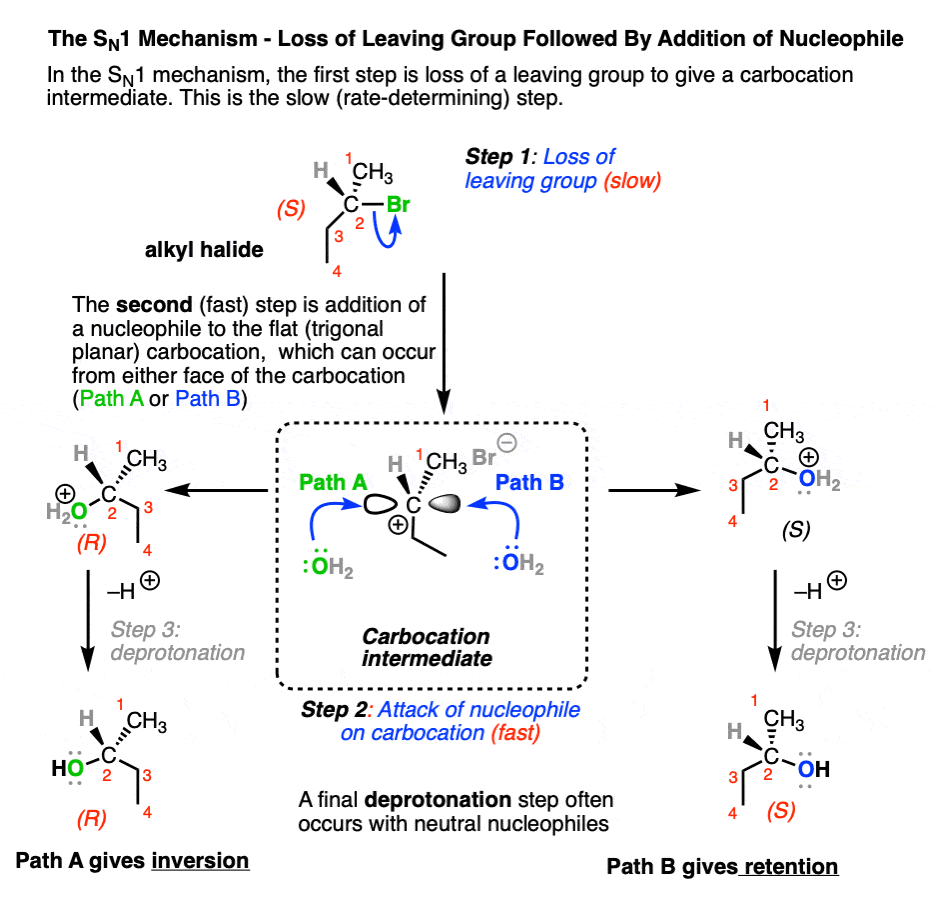
Note that the carbocation intermediate has a trigonal planar geometry at carbon, and its empty p-orbital can undergo addition by a nucleophile equally well at either face. In cases where substitution occurs on a stereogenic carbon (aka “chiral carbon”) this can result in a mixture of retention and inversion of stereochemistry, relative to the configuration of the original alkyl halide. [Note 1]
In contrast, the SN2 reaction passes through a one-step concerted mechanism where one equivalent of nucleophile adds to one equivalent of substrate, resulting in formation of a new bond to the nucleophile and the loss of a leaving group from carbon.
This is achieved through donation of a pair of electrons from the nucleophile into the empty, antibonding sigma-star (σ*) orbital on the backside of the C–LG bond. The carbon-nucleophile bond (C-Nu) forms at the same time that the carbon-leaving group bond (C-LG) breaks.
Since carbon cannot comfortably accommodate more than four bonding partners at one time these two bonds are considered to have partial bonding character in the transition state (note the dashed lines, below) where carbon adopts a “trigonal bipyramidal” geometry.
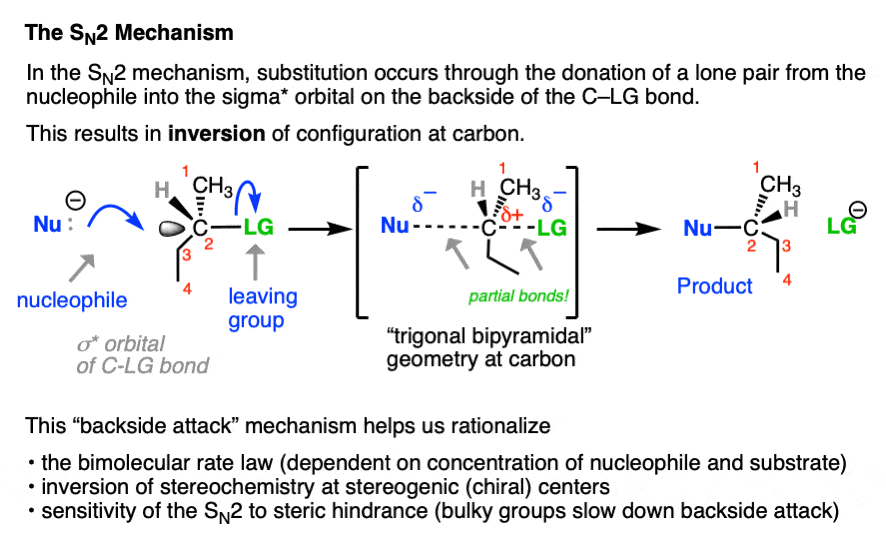
As the C-Nu bond forms and the C-LG bond breaks, the three “trigonal” substituents on the “equator” of the carbon relax to a tetrahedral geometry, with the difference that they are completely inverted from their original position (like the often-invoked metaphor of the “umbrella turning inside-out in a strong wind”) .
3. Reaction Coordinate Diagrams Of The SN1 and SN2 Reactions
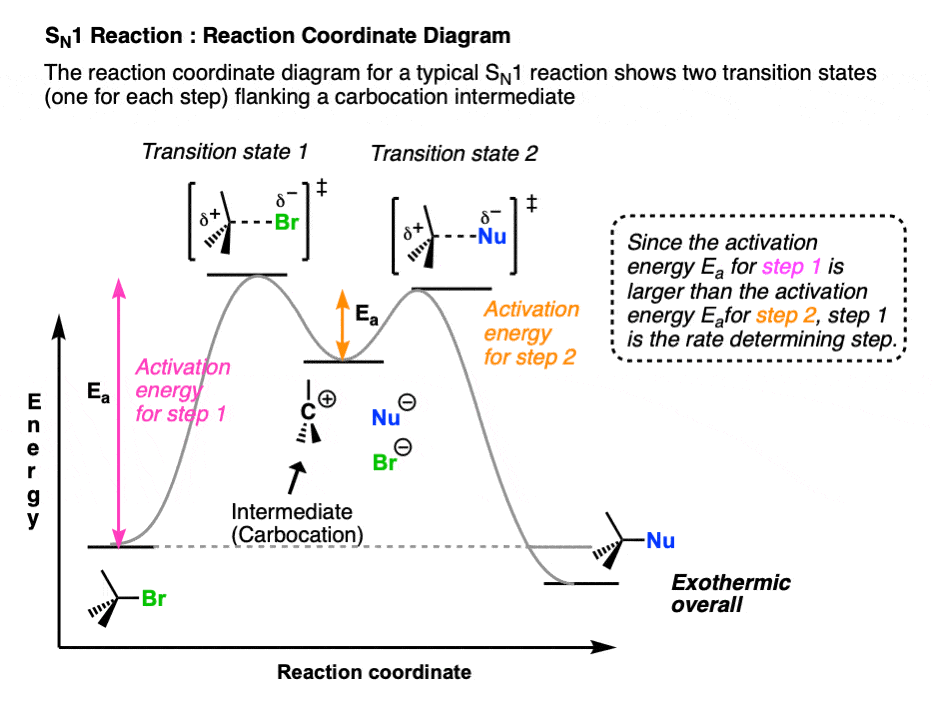
One way to visualize the differences between these two mechanisms is to sketch out their reaction coordinate diagrams, where we graph changes in potential energy (vertical axis) the starting materials pass along the “reaction coordinate” toward their conversion into products (horizontal axis) (These diagrams resemble a graph of changes in altitude (also potential energy!) experienced by a hiker navigating a mountain pass between two destinations.)
In these diagrams the “peaks” (local maxima) represent transition states whereas “valleys” (local minima) represent intermediates. (A transition state is a transient species with partial bonds. An intermediate is a potentially isolable species. )
The reaction coordinate diagram of the SN1 reaction has a two peaks, representing the two transition states (Step 1 and Step 2, respectively) flanking a single “valley” representing the carbocation intermediate.
Each step of the process has an activation energy represented by the difference in energy between the reactant and the transition state.
The rate-determining step of a reaction is the step requiring the highest activation energy, that is, the largest change in potential energy from reactant to transition state. In the SN1 reaction, the rate determining step is (illustrated in pink) loss of the leaving group from the alkyl halide to give the carbocation.
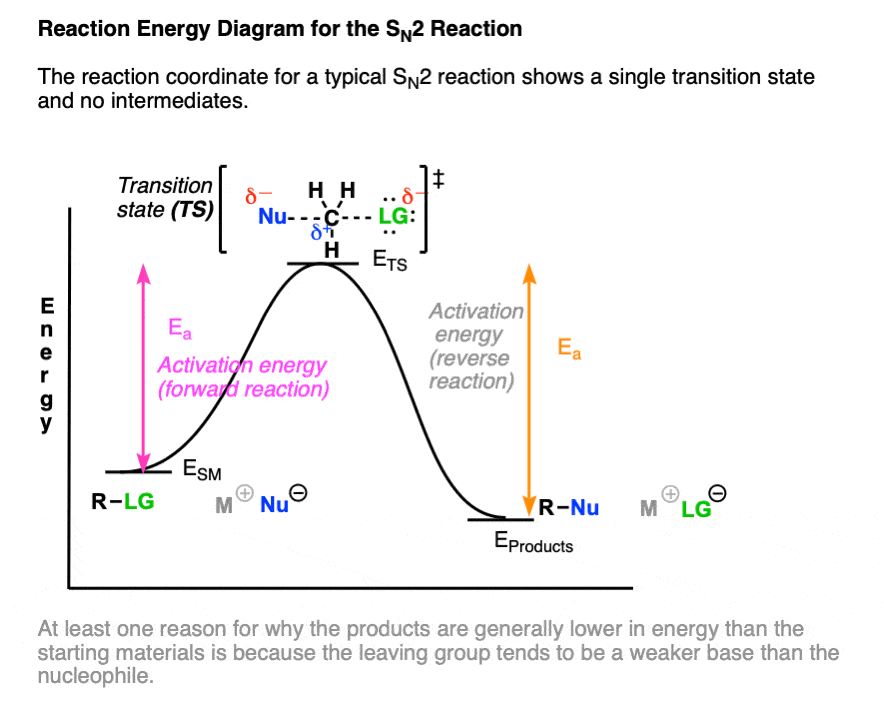
The reaction coordinate diagram of the SN2 reaction shows only a single transition state (one “peak”) corresponding to the concerted formation of C-Nu and breakage of C-LG without any intermediate. (Note 2)
One note – the difference in energy between the starting material and the product reflects the fact that the leaving group is generally a weaker base than the nucleophile, as well as the difference in bond strengths to carbon. (See article: What Makes A Good Leaving Group)
4. The Rate Laws Of The SN1 and SN2 Reactions
One way of probing the mechanism of a given substitution reaction is to measure the changes in reaction rates when the concentration of both nucleophile and substrate are varied.
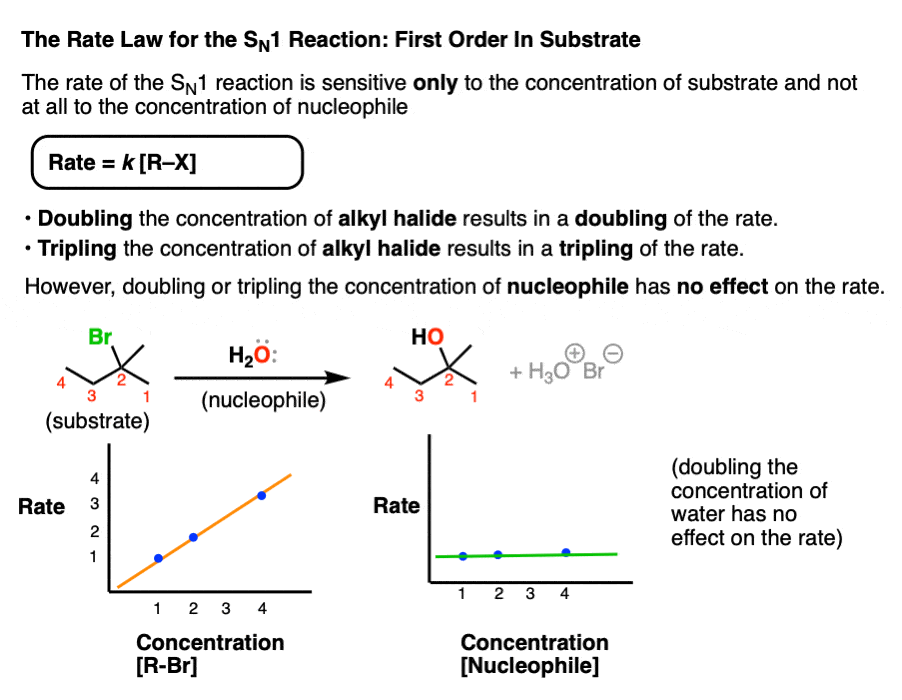
Since the rate-determining step of the SN1 reaction is loss of a leaving group from the substrate – a unimolecular reaction – the rate of product formation in the SN1 should depend only on the concentration of substrate.
Rate = k [Concentration of alkyl halide]
Doubling, tripling, or quadrupling the concentration of substrate should thus result in a doubling, tripling, or quadrupling of the rate of product formation, respectively.
Doubling, tripling, or quadrupling the concentration of nucleophile on the other hand should have no effect on the reaction rate since the nucleophile is not involved in the rate-determining step.
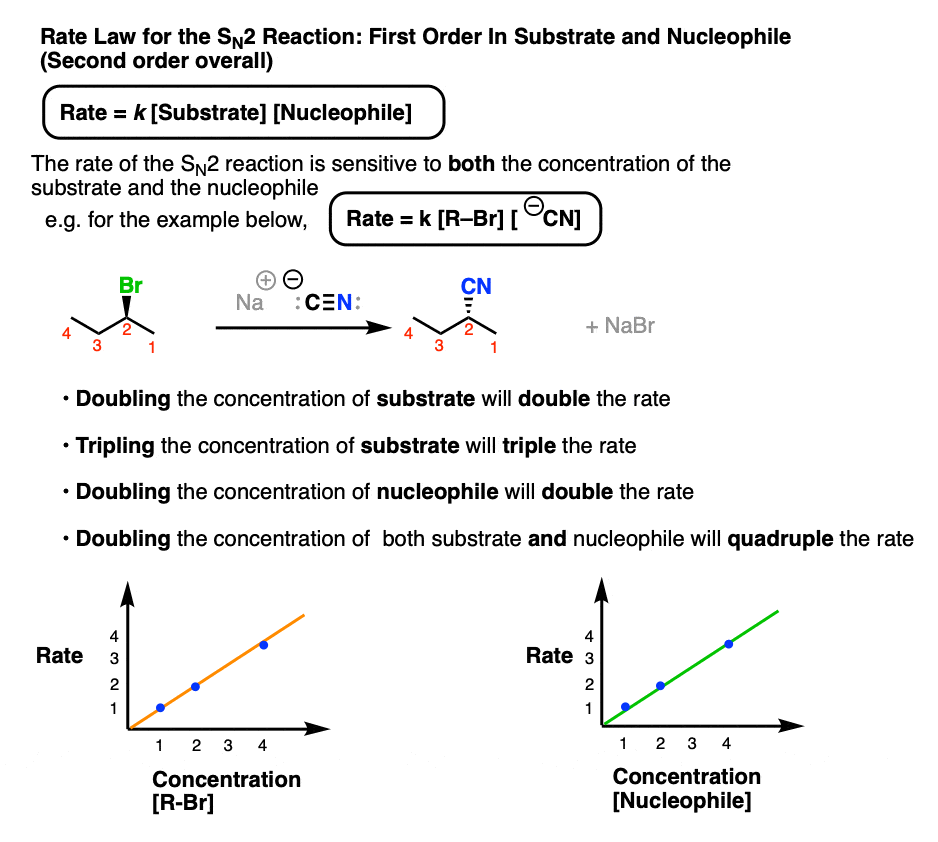
The SN2 reaction, by contrast, has a bimolecular rate-determining step where one equivalent of nucleophile combines with one equivalent of substrate.
The overall rate law of the SN2 is thus dependent on both the concentration of substrate and the concentration of nucleophile, taking the form:
Rate = k [Concentration of alkyl halide] [Concentration of nucleophile]
We say this is “first-order” in nucleophile and “first-order” in substrate, or “second order” overall.
- Doubling the concentration of substrate will double the rate of formation of product.
- Likewise, doubling the concentration of nucleophile will also double the rate of product formation.
- Doubling the concentration of substrate and nucleophile will result in a quadrupling of the rate of product formation.
5. The Effect of Substrate On The Rate of SN1 and SN2 Reactions
The dependence of the rate of the SN1 on carbocation stability and the rate of the SN2 on steric hindrance means that the trends of their reaction rates with primary, secondary, and tertiary alkyl halides proceeds in opposite directions.
The rate-determining step of the SN1 reaction is formation of a carbocation. Since tertiary carbocations are more stable than secondary carbocations which are in turn far more stable than primary (and methyl) carbocations, we should observe that the rate of SN1 reactions is fastest with tertiary alkyl halides.
This is indeed the case, as illustrated when various alkyl halides are subjected to typical SN1 conditions (a poorly nucleophilic, polar protic solvent such as H2O) [Ref]
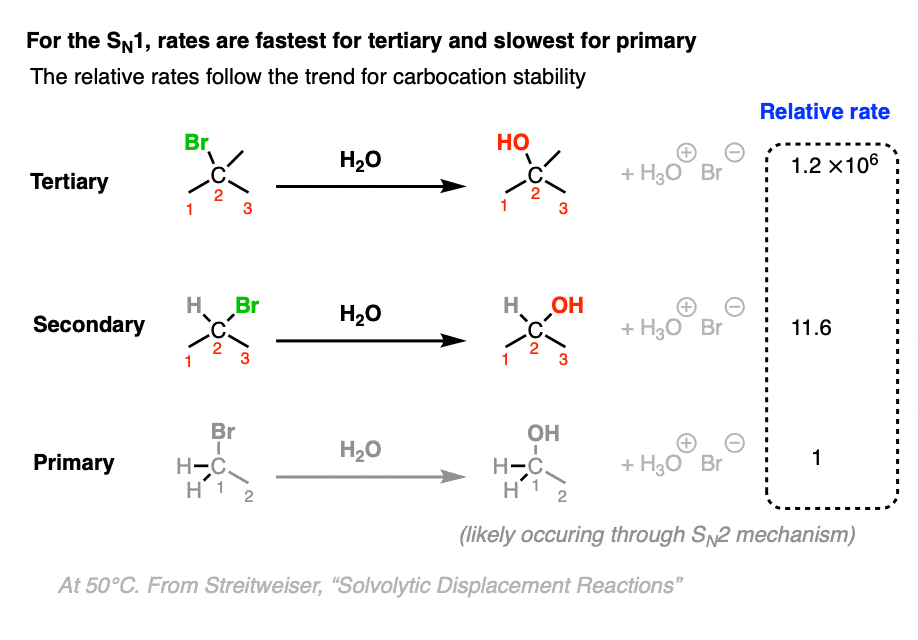
(Note that these are relative rates, where the rate of substitution at t-butyl bromide 1.2 × 106 is measured relative to the rate of substitution at ethyl bromide (1))
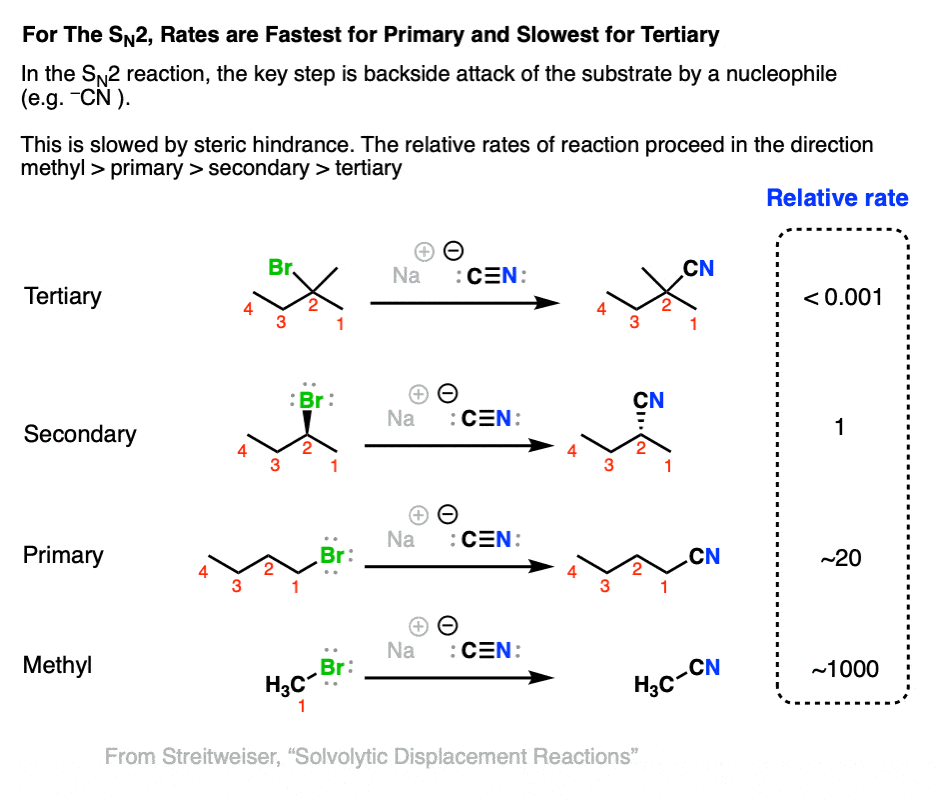
The rate determining step of the SN2 reaction is backside attack of a nucleophile on an alkyl halide. Since hydrogen atoms are smaller than carbon atoms, we should expect that the rate of SN2 reactions is fastest with methyl and primary alkyl halides and slowest with tertiary alkyl halides.
This also agrees with experiment, as shown below when a variety of alkyl halides are treated with the strong nucleophile NaCN.
6. Comparing The Stereochemistry of SN1 and SN2 Reactions
The stereochemistry of the products relative to those of the starting material are also a useful probe of SN1 versus SN2 pathway.
When a stereogenic center loses a leaving group to become a trigonal planar carbocation, it loses chirality.
Since the resulting carbocation can be attacked on either face by a nucleophile, the resulting product will be a mixture of retention and inversion of stereochemistry.
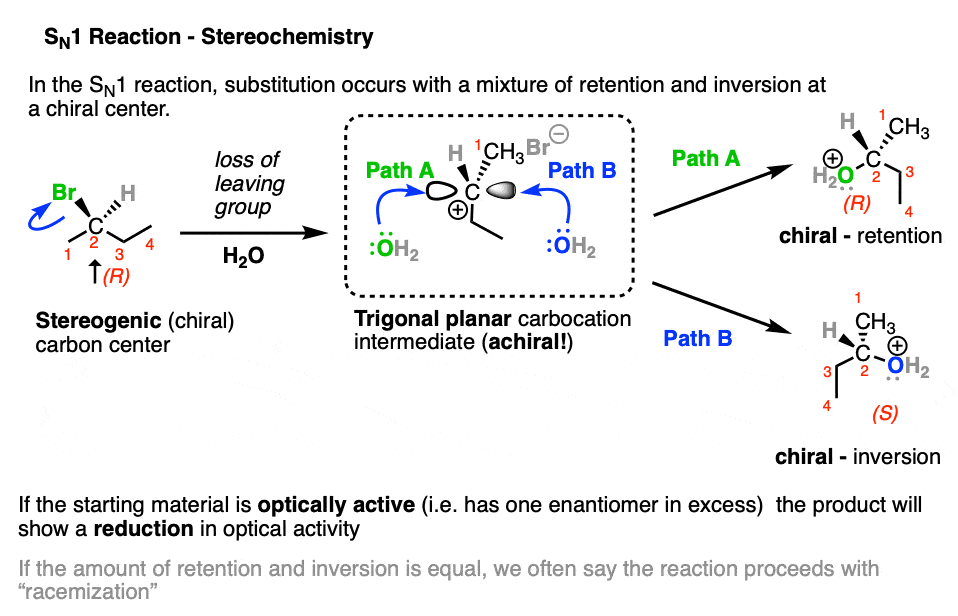
An SN2 reaction that occurs on a stereogenic carbon will result in inversion of configuration, but will retain optical purity.
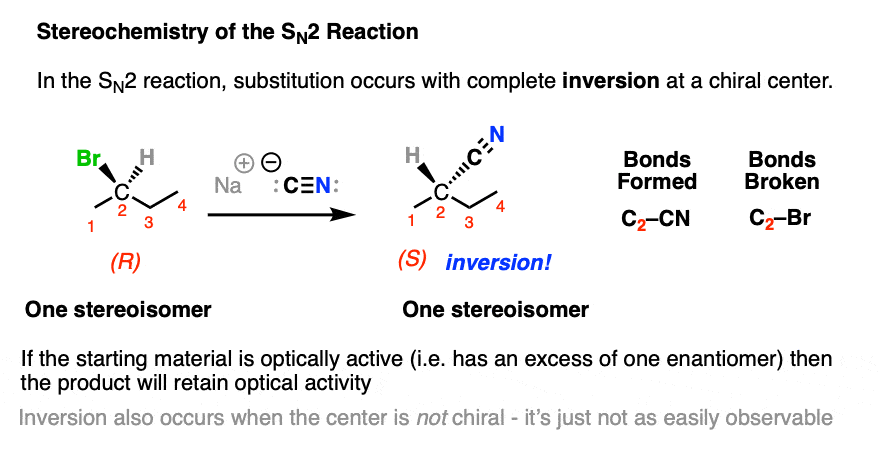
Note that inversion still happens whether or not the starting material is chiral or not; it just won’t be observable.
7. Solvents And Nucleophiles – SN1
The SN1 reaction tends to occur when alkyl halides capable of forming reasonably stable carbocations are dissolved in polar protic solvents that are capable of acting as nucleophiles.
Loss of a leaving group to give a carbocation results in the formation of a transient ion pair (i.e. the carbocation and the leaving group) from a neutral species (i.e. the alkyl halide)
Just as salts like NaCl are unlikely to dissolve in non-polar solvents like hexane (dielectric constant, ε = 2), carbocation formation is much more favorable in polar solvents like water (ε = 78), alcohols (ε ~ 20-40), or carboxylic acids (e.g. formic acid, ε = 51) , which are able to stabilize charges through hydrogen bonding or other dipolar interactions.
Carbocations resemble neutral compounds of boron (e.g. BF3), in that they are excellent Lewis acids containing 6 valence electrons and an empty p-orbital. Once formed, carbocations readily undergo addition even with poor Lewis bases to give products with a full octet around carbon.
So in practice, this generally means the solvent is the nucleophile, since it is present in much higher concentrations relative to anything else. [Note 3]

8. Solvents and Nucleophiles – SN2
SN2 reactions can certainly be carried out in polar protic solvents like alcohols, especially with primary alkyl halides.
But since the rate of the SN2 isn’t dependent on carbocation formation, a much wider variety of (less polar) solvents may be used.
Furthermore, alkyl halides are poorer electrophiles than carbocations. This is actually a good thing, since they are much less likely to undergo side reactions like rearrangement or elimination, or react with the first Lewis base they see.
For practical purposes, SN2 reactions tend to be carried out with stronger (i.e. charged) nucleophiles. A wide variety of nucleophilic partners can be used, which makes the SN2 extremely versatile, especially with primary alkyl halides. (See article – Why The SN2 Reaction Is Powerful)
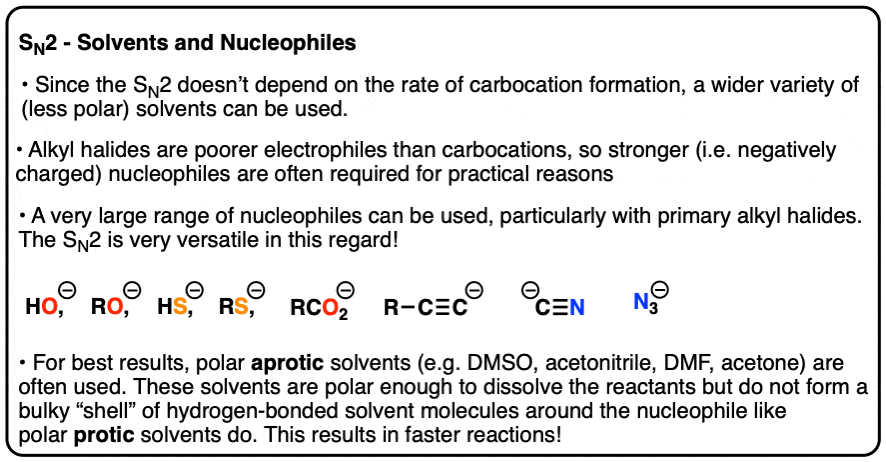
Polar aprotic solvents such as DMSO, acetone, DMF and acetonitrile are often chosen for SN2 reactions since they are polar enough to dissolve the reaction partners, but cannot form hydrogen bonds to the nucleophile. This has the practical effect of making the nucleophile less bulky, since the nucleophile isn’t surrounded by a shell of solvent molecules everywhere it goes.
9. Back To The Cats
- In the SN2, the nucleophile (Cat #1) forms a bond to the substrate (comfy chair) at the same time the leaving group (Cat #2) leaves.
- In the SN1, the leaving group (Cat #2) leaves the substrate (comfy chair), and then the nucleophile (Cat #1) forms a bond.
Don’t forget – you can download a free 1-page Summary Sheet of SN1 vs SN2 reactions containing all the material on this blog post here: Download SN1 vs SN2 Summary Sheet PDF
Notes
Cat Illustration by my talented cousin, political cartoonist Graeme MacKay
Note 1. [Intimate ion pairs can affect this].
Note 2. [If the nucleophile is neutral, e.g. H2O, then there will be a second, lower-energy transition state corresponding to the deprotonation step]
Note 3. [It is possible to trap carbocations with external nucleophiles.]
UPDATE . The most perfect cat video ever. Thanks to Alex Roche (Rutgers U.) for sending.
Quiz Yourself
Become a MOC member to see the clickable quiz with answers on the back.
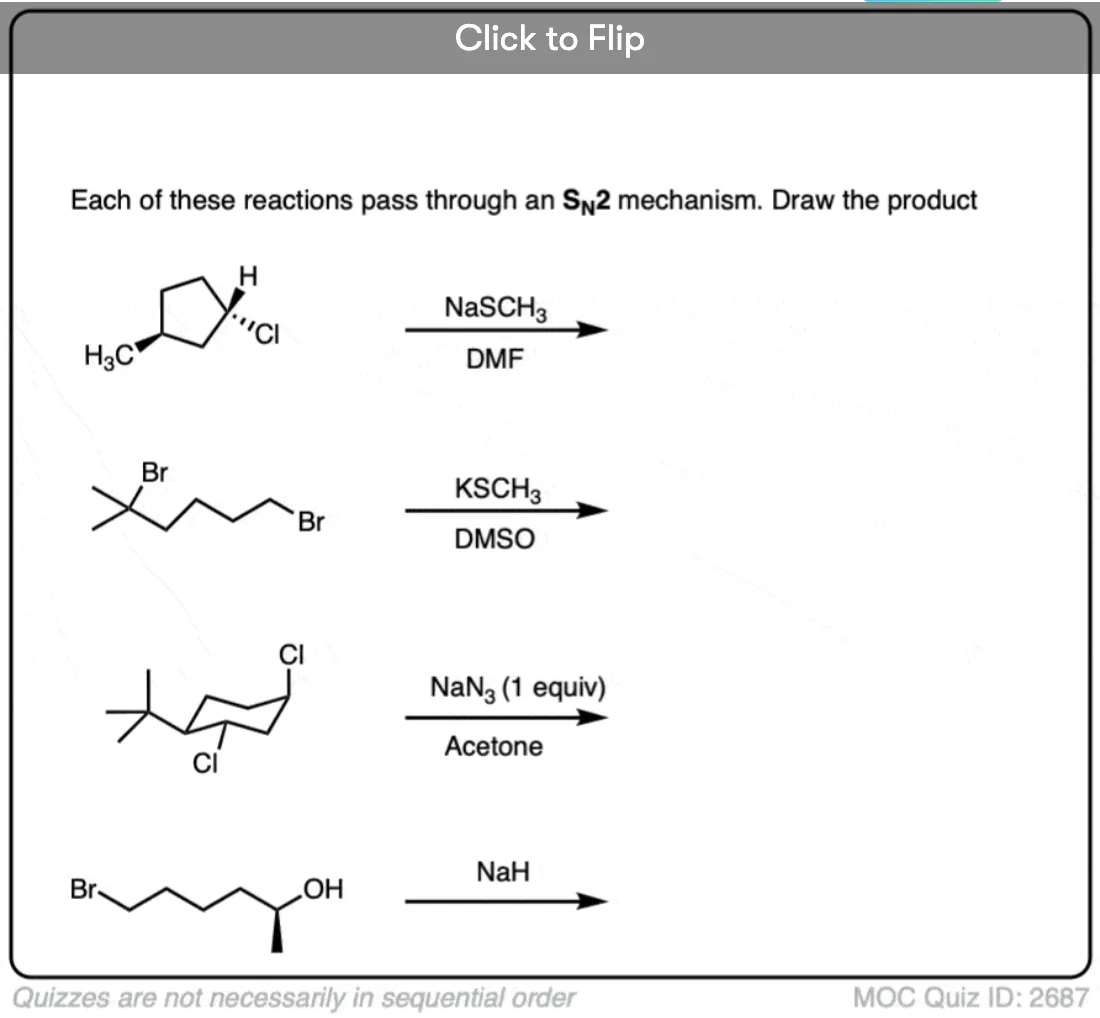
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
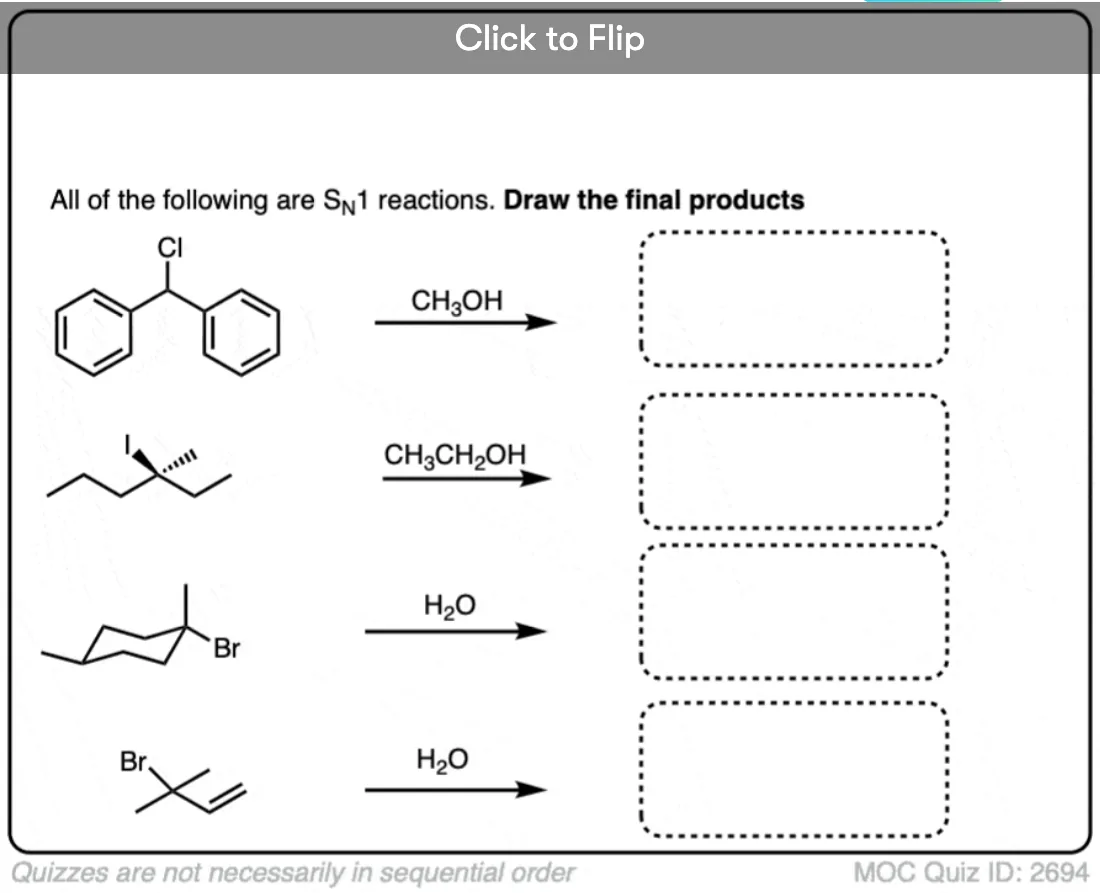
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
Solvolytic Displacement Reactions by Andrew Streitweiser is an oldie but a goodie in terms of compiling a lot of information (particularly reaction rates) of SN1 and SN2 reactions. Available on the Internet Archive for 1-hour loans here.
- Reaction kinetics and the Walden inversion. Part VI. Relation of steric orientation to mechanism in substitutions involving halogen atoms and simple or substituted hydroxyl groups
W. A. Cowdrey, E. D. Hughes, C. K. Ingold, S. Masterman, and A. D. Scott
J. Chem. Soc. 1937, 1252-1271
DOI: 10.1039/JR9370001252
The points listed in the summary are worth reading for understanding what influences the SN1 and SN2 pathways. - Mechanism of substitution at a saturated carbon atom. Part XXVI. The rôle of steric hindrance. (Section A) introductory remarks, and a kinetic study of the reactions of methyl, ethyl, n-propyl, isobutyl, and neopentyl bromides with sodium ethoxide in dry ethyl alcohol
I. Dostrovsky and E. D. Hughes
J. Chem. Soc. 1946, 157-161
DOI: 10.1039/JR9460000157
Table I in this paper shows the reduction in reaction rate for the SN2 reaction of R-Br with OEt- when R goes from methyl -> ethyl -> n-propyl -> isobutyl -> t-amyl. This can be attributed to steric hindrance, as backside attack of the substituted carbon becomes increasingly challenging. - Mechanism of substitution at a saturated carbon atom. Part III. Kinetics of the degradations of sulphonium compounds
John L. Gleave, Edward D. Hughes and Christopher K. Ingold
J. Chem. Soc. 1935, 234-244
DOI: 10.1039/JR9350000236
This is a useful paper – in the beginning the terms “SN1” and “SN2” are introduced and defined, and Figs. 1 and 2 depict how the two mechanisms can compete depending on the structure of the substrate. - Influence of poles and polar linkings on the course pursued by elimination reactions. Part XVI. Mechanism of the thermal decomposition of quaternary ammonium compounds
E. D. Hughes, C. K. Ingold, and C. S. Patel
J. Chem. Soc. 1933, 526-530
DOI: 10.1039/JR9330000526
At the end of this paper, the authors make an important point: “When the various series can be more fully filled in, what has been described as a “ point ” of mechanistic change will probably appear as a region, and thus, just as with reaction (A), we now generalise the original conception of reaction (B) by the contemplation of a range of mechanisms, (Bl)-(B2), both extremes of which have been experimentally exemplified”. Basically, the SN1 and SN2 mechanisms as taught are two extremes of a continuum, and in practice most reactions lie somewhere in between. - Mechanism of substitution at a saturated carbon atom. Part IX. The rôle of the solvent in the first-order hydrolysis of alkyl halides
Leslie C. Bateman and Edward D. Hughes
J. Chem. Soc. 1937, 1187-1192
DOI: 10.1039/JR9370001187 - The Common Basis of Intramolecular Rearrangements. VI.1 Reactions of Neopentyl Iodide
Frank C. Whitmore, E. L. Wittle, and A. H. Popkin
Journal of the American Chemical Society 1939, 61 (6), 1586-1590
DOI: 1021/ja01875a073
An early paper demonstrating that SN1 reactions can be induced by reaction of an alkyl halide with silver salts. In this case, the neopentyl cation quickly rearranges to the significantly more stable t-amyl cation, and those products are obtained. - Reaction kinetics and the Walden inversion. Part I. Homogeneous hydrolysis and alcoholysis of β-n-octyl halides
Edward D. Hughes, Christopher K. Ingold and Standish Masterman
J. Chem. Soc. 1937, 1196-1201
DOI: 10.1039/JR9370001196 - Reaction kinetics and the Walden inversion. Part IV. Action of silver salts in hydroxylic solvents on β-n-octyl bromide and α-phenylethyl chloride
Edward D. Hughes, Christopher K. Ingold and Standish Masterman
J. Chem. Soc., 1937, 1236-1243
DOI: 10.1039/JR9370001236
These two papers examine reactions of 2-octyl halides in an attempt to see if pure SN1 or SN2 pathways on the same substrate can be favored simply by varying the reaction conditions.
Thanks for the sn1 and sn2 comparisons
Ah, I’m glad it isn’t the “hobo on the bench” analogy anymore, thanks for being considerate enough to change it to cats
I love how they use their tails to balance in the video!
Hi,thanks for the post. But here is question.why protic solvent are favored by SN1 and vice versa?
In SN1 reactions the rate limiting step is loss of leaving group to give a carbocation. Polar protic solvents have a higher dielectric constant and can stabilize the resulting carbocation species that results from ionization. Secondly, polar protic solvents are often the nucleophiles in these reactions and can be neutralized through deprotonation.
In SN2 reactions the rate limiting step is attack of the nucleophile at the alkyl halide. Using a polar aprotic solvent results in a more free nucleophile (free from hydrogen bonding) resulting in a higher reaction rate than if a polar protic solvent were used.
What should i do when both strong and weak nucleophile are present in the reaction??? Should i proceed through SN1 OR SN2???
If the substrate is primary, it will be SN2, since the reaction rate will be faster with a stronger nucleophile.
Nicely Explained really impressed…
**Now, gaining confidence in organic chemistry**
THANK YOU ,
plzz make more blogs on organic chemistry thank you once again…
Cool, thanks for letting me know Sanket. So what could be better about the site?
Why reactivity in SN1 reaction is directly proportional to the on stability of carbonation ? And how ?
Please respond me.
Wonderfully describe. I am a chemistry teacher; I teach that topic to my students as per your article. I like your way of understanding. Thank you so much for sharing your this information about two reactions.
Your blog posts are so helpful, please keep doing what you’re doing! Also love the summary sheet for this subject, super helpful as well.
Glad you found it useful Ky!
Wow.this is very useful site for everyone but please add reactions with mechanisms..
Glad you find the site useful Shailenda!
Is it possible a trans compound react in a SN2 reaction? (eg.trans -1-iodo-4-ethylcyclohexane and methoxide ion)
Or just tehe cis ones react because its possible ” the carbon bonded to the leaving group is attacked by the nucleophile on its back side”? How do we know if its possible to have the mixture of the 2 configurations?
It’s completely possible. With cyclohexanes, the important thing to note is that the leaving group must be axial in order for the SN2 to occur.
Your website is a Godsend. Thank you!
Thanks Blake!
This is very impressive. I’m having a Msc. In applied organic chemistry. And it helped me to explain this to my students. The cat example is nice. Thank you.
Ha! Thanks Shiral!
Just wanna say that I already bought all your cheat sheets and it is a big help!! Thank you for clear explanations :)
Thank you so much for supporting the site Cacia!
Good explanation. Specially the story of the cats and the relation with the reactions is very good. Thank you
Thank you very much for your views regarding the Sn1 and Sn2 reaction……..This will help me alot…..
Thank you so much for great revision of Sn1 and Sn2 reactions .!
Really helpful, especially with the cartoon illustrations. Thank you
Thank my cousin Graeme for that, ha!
I know primary substrates favor SN2 and E2 reactions – but my book talks about how a primary carbocation can form in an SN1 reaction if it is accompanied by a simultaneous rearrangment. The book specifically says it can happen as a result of a methyl shift. My question is: can it also happen via a simultaneous hydride shift? Or only through a methyl shift? My teacher has given us some problems to work through that would require a primary carbocation to form through a hydride shift, and I just want to make sure that is feasible.
Yes, it can certainly happen through a simultaneous hydride shift.
SN1 mechanism — the rate of reaction depends on substract. It is independent of nucleophile. So it can show ist order mechanism (unimolecular). It is more than 1 step mech.
Most stable carbo cation will favour sn1 mech.
Polar protic solvent will favour
Best solvolysis can favour
More steric hindered can show sn1 mech.
What about the effect of the polar aprotic solvent on the rate of rxn? That’s a valid question, and it’s unfortunately not addressed.
I’d love to see direct data on that. At the moment I haven’t been able to find anything that would provide a direct comparison. If I do, I’ll put it in.
The link for the exercises is broken :(
I’ve taken it down. ASU has changed their website.
Studying for my DAT and this gave so much of everything I needed and nothing I didn’t! Thank you!
Glad to hear it, Tye!
thank you so much this was the best help i could find on the subject – turned me around on understanding substitution rxns!!
why tertiary alkyl halides prefer nucleophilic reactions more easily via sn1 mechanism ?
Carbocation stability is the brief answer.
Personally, I think steric effects should also be considered.
Even with the help given, I am having trouble with how kinetics can be used to tell the difference between SN1 and SN2.
Double the concentration of substrate and double the concentration of nucleophile. If the reaction is first order overall, the reaction rate will only double. If the reaction rate is second order overall, the reaction rate will quadruple.
Great explanation.. simple, precise and easy to understand. thanks a bunch :)
what is the reason for the recemized product in SN1 reaction? to be precise with the question.. the no. of front side and back side attack is same. why it doesn’t differ?
Great analogy and summary! Thank you so much! I shall definitely be reading more of your work!
But I am little bit confused in trend of nucleophilisity of halogens in polar aprotic solvents.
Thank you
Thanks Peaches!
Thanks for this great explanation! If you didnt know whether the reaction was Sn1 or Sn2, what physical property could you use to distinguish them?
If it’s a chiral secondary alkyl halide, then you could use optical rotation to distinguish them.
you are great!! Thank u very much
Which one allows for a better control over the configurations of products SN1 or SN2?
SN2, of course, because it is stereospecific.
Here what do you mean by inversion of configuration.Is it relative or absolute
Relative.
It is very un-PC to be using homeless people in this analogy, imagine if you changed the analogy to a black person on the bench and a white person wanting to sit down (quite offensive). You have a cartoon of two cats wanting to sit in a basket, why not just use that analogy; how about just two people, one sitting on the bench and the other wanting to sit on the bench, no socio economic issues implied. Apart form that very useful information.
This analogy is fab…Its so clever and easy to remember! Thank you so much…I made use of it in my oxford interview!
I like the article very much, but I would draw your attention to the paragraph
”The dependence of rate on the substrate”
Given that for the Sn1 reaction the big barrier is carbocation stability,that is to say that
the carbocation stability increases with increasing substituents on the carbon
( tertiary>secondary>> primary……instead of saying that resonance is a factor,
I would like to make this clearer by saying that tertiary groups are EWG groups
and because they are electron withdrawing they make the carbocation a better
electrophile.This in turn renders the cation strong enough to react with a weak
nucleophile.
Note….the above is not a correction ..just for clarity
Hey in SN1 why tertiary is more reactive though it is relatively stable
The stability of the tertiary carbon allows it to be a stronger carbocation compared to primary carbon which is very unstable as it the carbocation formed would be unstable. If the carbocation is unstable then it can just react in reverse with the leaving group and the reaction wouldn’t proceeed. So reactivity of SN1 reaction increases as the groups around carbon increase
Hello, I have a question about steric hindrance for an Sn2 reaction. Specifically with cycloalkanes. I thought that the higher the number of carbons a cycloalkane has (the more corners it has) the more likely it was for a nucleofile to attack it from the inside. In my chemistry book on the other hand there’s a table with relative reactivity. And it puts Cyclopentane (more reactive) before Cyclohexane. Why doesn’t this one follow the rule? Many thanks in advance! Love your website!
Hi – not sure exactly what you mean by the “inside” . In the case of cyclopentane and cyclohexane, the ring isn’t big enough for the nucleophile to fit in the “inside” of the ring.
Hard to say re: cyclopentane vs. cyclohexane. I can’t imagine there’s a huge difference. cyclohexane has the barrier of requiring the leaving group to be axial.
Could it have something to do with angle strain? increased angle strain in the cyclopentane could lead to a higher energy of the starting materials, and thus, a lower barrier to cross to undergo an SN2 reaction?
Thank you so much. I love organic chemistry but it’s very hard at times. My professor talks waaay too fast so I’m missing out on important details. Love this piece of text…
This is absolutely wonderful resource! I understand it now, and it’s nowhere as complex that my lecturer made it look!
Thanks so much!
Glad you found it useful Michal!
Awesome explaination with some simple but effective intellectual ideas!!!!!!……
What’s with the hobo story huh?
Am a student taking organic chemistry at Kenyatta University-Kenya, this is so helpful to me
Thank You so much.
i need the example and difference btween SN1 and SN2
i need example for both SN1 and SN2 to differentiate between them
Thanks for the great explanation. I have a question about the rate of Sn1 reaction, how would a primary carbocation that can undergo an alkyl shift to become tertiary fit in, I know that a primary carbocation is slower than secondary, but the shift would stabilize it. Or does the shift take enough time that it wouldn’t end up being faster than a secondary?
If talking about the rate of formation of a free carbocation, formation of primary carbocations is slower than that of secondary. However, it is very rare that primary carbocations form – when alkyl shifts occur to a primary carbon, it is usually a concerted rearrangement mechanism that doesn’t strictly go through a free carbocation. That makes it difficult to strictly compare the rates since they occur through different mechanisms.
Thank you so much for the nice explanations. Your explanations helped me get several difficult points cleared.
Thanks for the write up. Truly helpful.
all of these come out in our quiz. this is very accurate and well explained (;
Glad you were well prepared!
I’m studying Organic Chemistry from Clayden, but I really like this website to have some extra background, mnemonics and nice summaries.
I wanted to check the exercises pointed out at the end of this lecture, but the link gives an error (404 – File or directory not found.).
Oh, thank you!
This was so helpful!! I love how simple you break it down. THANK YOU SO MUCH!!!
in General chemistry, there is chapter about kinetics. If you guys are confused about rate determining steps, I would encourage reading that chapter or review it thoroughly. I will try to explain it here a little.
1) SN1 ,
Since the rate determining steps depend on the carbocations, so we look at 1st order kinectic , which can be found by.
k= [Electrophile] , where k is rate of reaction , as the the concetration of electrophile goes down, the reactions is reaching towards end, or stopping or decreasing, whatever you think is appropriate at given electrophile concentration.
2) SN2, you will need good Nuceophile and electrophile, thus intermediate stage is 5 ligands, and conculsion is four , sp3 to sp3, but remember it does have 5 ligands, intermediate, which is VSPER Theory, 5 ligands, is Trigonal bipyramidal.
K = [electrophile] [nucelphile]
k is rate of the reaction, depends on both electrophile and nucelophile, so it is second order, 1 step, fast reaction.
So as the both increases the reaction rate will go up, if one goes down, it is kind of like limited reagents , which one exhaust first etc, if one is exhausted, does not matter, how much you have the other, the reaction WILL NOT Proceed.
So I would say, conclusion to this summary, Relate Both, general chemistry and organic chemistry, it will make MUCH MORE SENSE, and you will never forget :)
Sir Thank you, I was looking for this article. You’d explained it very nice. Even my teacher couldn’t.
Sir will you please explain me why alpha-halocarbonyl compounds are not much reactive with Sn1 mechanism?
The carbocation that forms is destabilized by the adjacent electron withdrawing C=O group, making this a very unstable carbocation.
Thank you sir, it was very helpful.
Wow – such a good website and so well explained thank you sooooo much. Way better then my lectures :)
This was so outrageously helpful. I will definitely be using this site for much of my orgo work this year.
Thank you, very glad to hear it.
in problem 10 you mention that allylic halide is more reactive and rxn is SN2. Allylic system are more reactive becoz of resonance for which there must be formal charge intermediate as in SN1 not SN2. So why is it that without any charged intermediate the left bromine is favored.
Thanks very much, this was a great review of the topic!
Hey, just a question here. I know that the branching of the base/nucleophile will direct the reaction towards E2 or Sn2, where steric hinderance of the base/nu: will most likely lead to an E2 rxn, b/c the H+ protons are more accessible.
Does branching of the base/nucleophile have any affect on E1 or Sn1?
I do know that branching of the substrate helps stabilize the carbocation…
Since the rate-determining step of SN1 and E1 reactions is formation of the carbocation, an event independent of the nucleophile, branching of the base/nucleophile does not have a significant effect on these reactions.
Well actually the branching of a base/nucleophile can have an effect. Lets think about the carbocation during its transition state. It positively charged and thus in an ideal world it would want to be stabilised, thus reducing its energy. If you have large bases this can almost protect the transition state, providing mixed effects with the stabilisation of the cation being good, the steric hindrance for the nucelophile being bad.
For the SN2, since steric hindrance decreases as we go from primary to secondary to tertiary, the rate of reaction proceeds from primary (fastest) > secondary >> tertiary (slowest).
*Shouldn’t this be “steric hindrance increases as we go from *
Fixed. thanks for pointing that out!
actually steric hinderence increases as we move from primary to secondary. hope this will help….
Some of my students challenged me to do the upcoming class in haiku. Since a fractonal distillation lab is pretty dull to supervise after everyone is up and running, I put the introduction to SN1 in haiku format. (Each done on a powerpoint slide with a pretty background…)
Leaving group breaks off
Forming carbocation
SN1, first step
very reactive
intermediate species
they need electrons
tertiary good
hyperconjugation helps
resonance does too
add more Nu? No help.
the rate is independent
that’s kinetic proof
climbing two mountains
reaction coordinate
C+ is high pass
how do you decide?
SN1 or SN2
there are many factors.
Nicely done!
Crystal clear. Beautiful analogy. Here comes James, frighten the poor hobo away so he can seat himself on the bench. James must be big and intimidating.
I can’t claim credit for the analogy – I heard it secondhand. But it’s effective isn’t it?
It is and thanks for making it comprehensible. I v already covered this with my students but I ll use this to refresh them again.
im a little confused with the analogy though. according to the purpose of the lucas test (which determines if the substrate is primary, secondary or tertiary) tertiary rxns undergoing sn1 mechanisms are faster than secondary rxns undergoing sn1 rxns and yet faster than primary substrates undergoing sn2 mechanisms. it seems to me like kicking the hobo off the bench (sn2) would be a lot faster than waiting for him to leave (sn1). flip flopped from the actual speeds of the mechanisms. can u elaborate on the analogy. b/c i would rlly love to use something like that to go by on the MCAT. thanks