Reactions of Aromatic Molecules
Electrophilic Aromatic Substitution: The Mechanism
Last updated: May 29th, 2026 |
Electrophilic Aromatic Substitution: The Mechanism
- Electrophilic aromatic substitution (EAS) reactions proceed through a two-step mechanism.
- In the first step, the aromatic ring, acting as a nucleophile, attacks an electrophile (E+).
- This is the slow (rate-determining) step since it disrupts aromaticity and results in a carbocation intermediate.
- In the second (fast) step a C-H bond is deprotonated to re-form a C-C pi bond, restoring aromaticity.
- The first step resembles attack of an alkene on H+, and the second step resembles the second step of the E1 reaction. The end result is substitution. (Break C-H, form C-E).
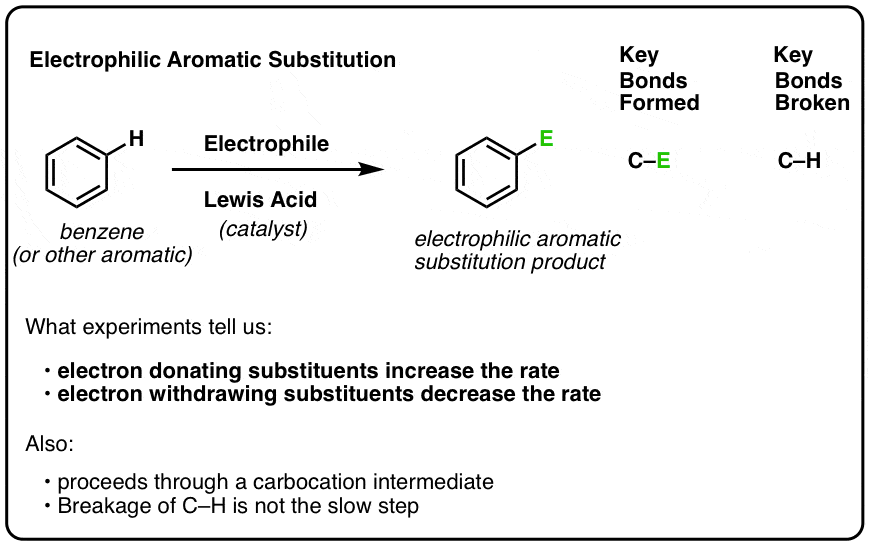
Table of Contents
- Electrophilic Aromatic Substitution Mechanism, Step 1: Attack of The Electrophile (E) By a Pi-bond Of The Aromatic Ring
- Electrophilic Aromatic Substitution Mechanism, Step 2: Deprotonation Of The Tetrahedral Carbon Regenerates The Pi Bond
- Putting Two Steps Together: The General Mechanism
- The Reaction Energy Diagram of Electrophilic Aromatic Substitution
- Beyond Benzene: Formation Of Ortho, Meta, and Para Disubstituted Benzenes
- EAS On Monosubstituted Benzenes: The Distribution Of Ortho, Meta and Para Isomers Is NOT Random
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Electrophilic Aromatic Substitution Mechanism, Step 1: Attack of The Electrophile (E) By a Pi-bond Of The Aromatic Ring
Last post in this series on reactions of aromatic groups we introduced activating and deactivating groups in Electrophilic Aromatic Substitution (EAS). We learned that electron-donating substituents on the aromatic ring increase the reaction rate and electron-withdrawing substituents decrease the rate. [In the fine print, we also mentioned that evidence strongly suggests that the reaction proceeds through a carbocation intermediate, and that breakage of C-H is not the slow step.]
Having established these facts, we’re now ready to go into the general mechanism of this reaction.
It’s a two-step process.
The good news is that you’ve actually seen both of the steps before (in Org 1) but as part of different reactions!
The first step of electrophilic aromatic substitution is attack of the electrophile (E+) by a pi bond of the aromatic ring. [Note: the identity of the electrophile E is specific to each reaction, and generation of the active electrophile is a mechanistic step in itself. We’ll cover the specific reactions next. This post just covers the general framework for electrophilic aromatic substitution].
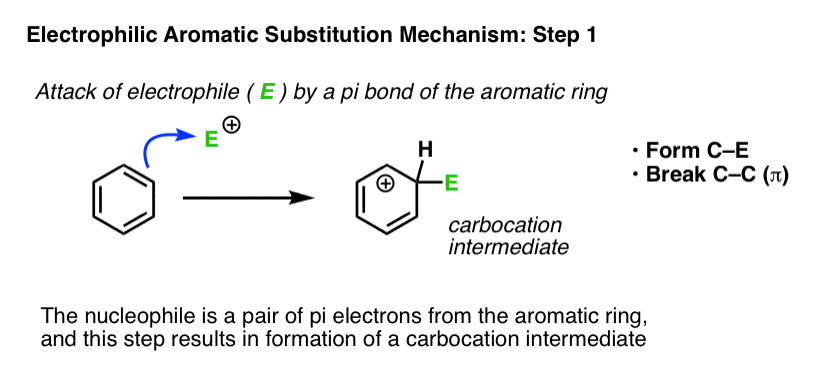
Where have we seen this type of step before? In the chapter on alkenes, we saw a whole series of reactions of pi bonds with electrophiles that generate a carbocation. A common example is the reaction of alkenes with a strong acid such as H-Cl, leading to formation of a carbocation. The reaction above is the same step, only applied to an aromatic ring.
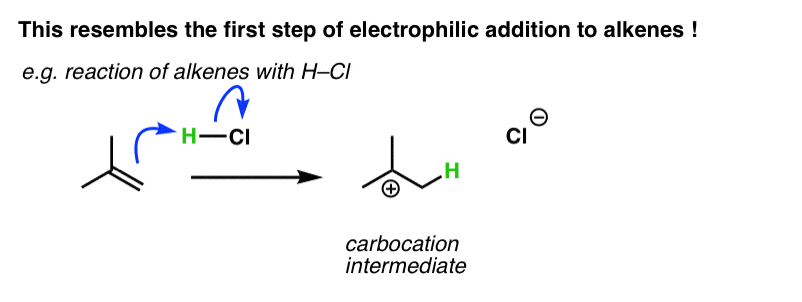
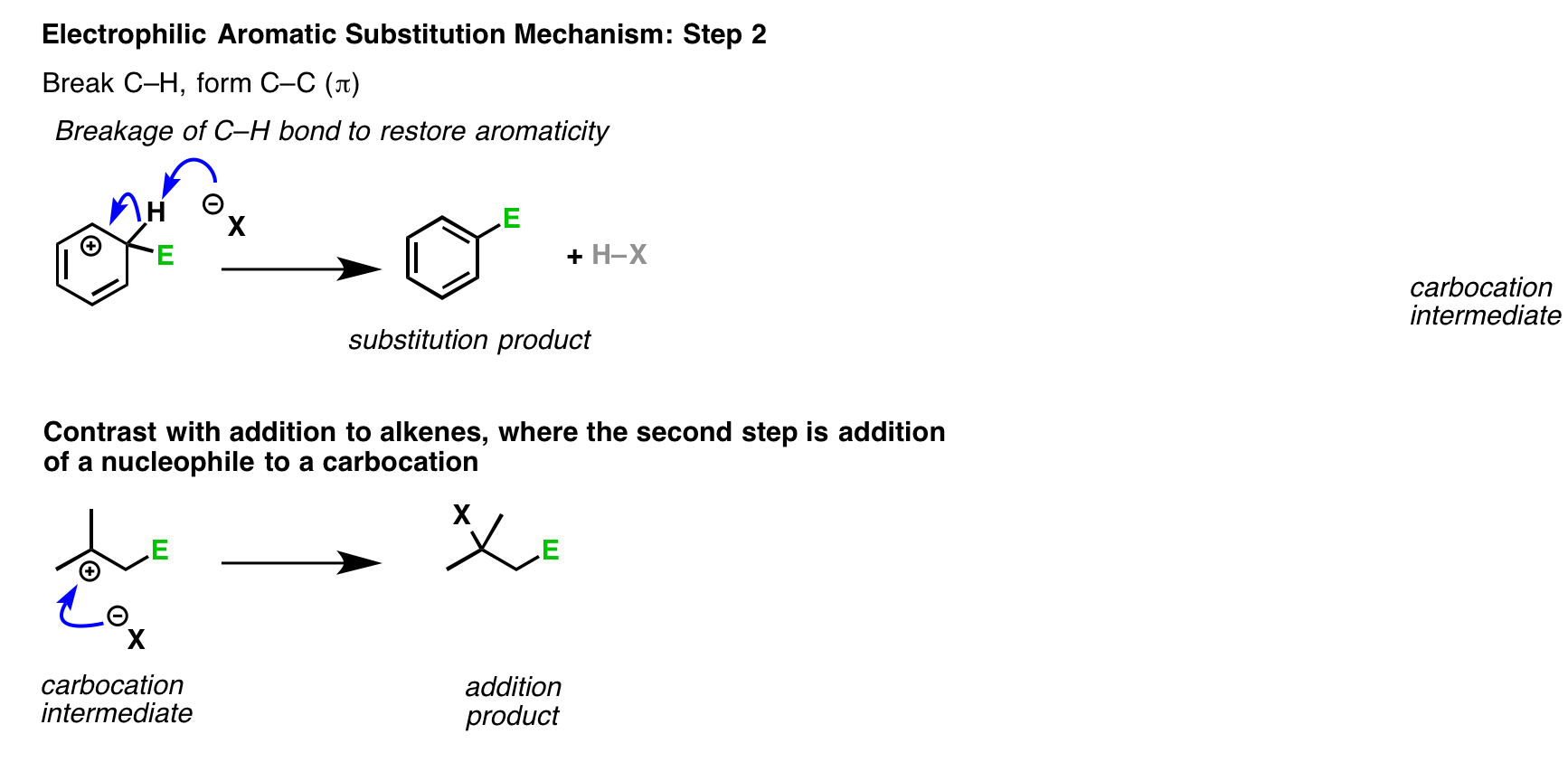
You might recall that the second step of addition of HCl to alkenes is the attack of Cl on the carbocation, generating a new C-Cl bond. That’s not what happens in electrophilic aromatic substitution. [Note 1]
2. Electrophilic Aromatic Substitution Mechanism, Step 2: Deprotonation Of The Tetrahedral Carbon Regenerates The Pi Bond
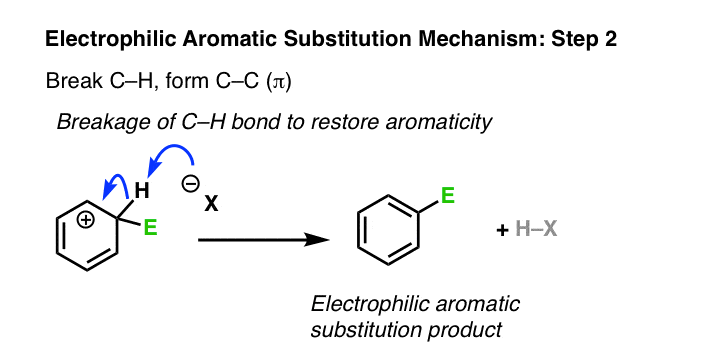
The second step of electrophilic aromatic substitution is deprotonation. This breaks C–H and forms C–C (π), restoring aromaticity. You may recall that this is strongly favored – the resonance energy of benzene is about 36 kcal/mol. [This is the type of phenomenon chemists like to call a “thermodynamic sink” – over time, the reaction will eventually flow to this final product, and stay there. ]
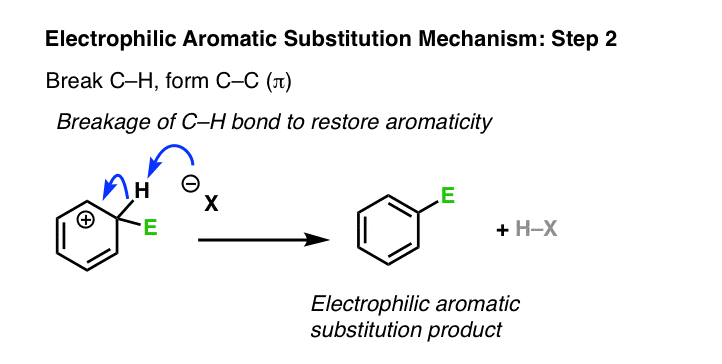
Have we seen this type of step before? Yes – it’s essentially the second step of the E1 reaction, (after loss of a leaving group) where a carbon adjacent to a carbocation is deprotonated, forming a new C-C pi bond.
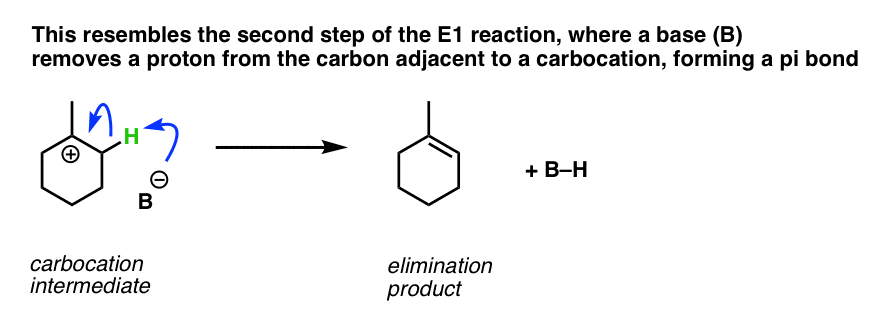
{kind=link}
{kind=link}
Just as in the E1, a strong base is not required here. A halogen atom (such as Cl– ) will usually suffice, as will any number of other weak bases, such as H2O. The exact identity of the base depends on the reagents and solvent used in the reaction.
3. Putting Two Steps Together: The General Mechanism
Let’s combine both steps to show the full mechanism. Again, we won’t go into the details of generating the electrophile E, as that’s specific to each reaction.
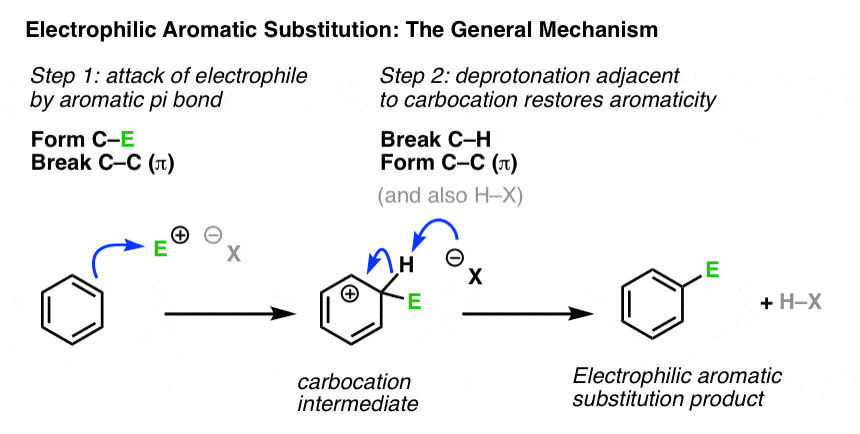
Note that attack could have occurred at any one of the six carbons of benzene and resulted in the same product.
4. The Reaction Energy Diagram of Electrophilic Aromatic Substitution
What might the reaction energy diagram of electrophilic aromatic substitution look like?
First, the overall appearance is determined by the number of transition states in the process.
Recall that transition states always have partial bonds and are at the “peaks” of a reaction energy diagram, and intermediates such as carbocations are in the “valleys” between peaks. Intermediates can be observed and isolated (at least in theory); in contrast, transition states have a lifetime of femtoseconds, and although they may fleetingly be observed in certain cases, they can never be isolated.
Electrophilic aromatic substitution has two steps (attack of electrophile, and deprotonation) which each have their own transition state. There is also a carbocation intermediate. This means that we should have a “double-humped” reaction energy diagram.
Second, the relative heights of the “peaks” should reflect the rate-limiting step.
What’s the slow step? In other words, which of the two steps has the highest activation energy?
One clue is to measure the effect that small modifications to the starting material have on the reaction rate.
We showed in the last post that electron-donating substitutents increase the rate of reaction (“activating”) and electron-withdrawing substituents decrease the rate of reaction (“deactivating”). [Conversely, substitution of hydrogen for deuterium has very little effect on the reaction rate, which leads us to conclude that the second step is not rate-determining. ]
Since electron-donating and electron-withdrawing substitutents affect the nucleophilicity of the pi bond (through pi-donation and pi-acceptance) as well as the stability of the intermediate carbocation, the logical conclusion is that attack on the electrophile (step 1) is the rate-determining step. We therefore should depict it with the higher “hump” in our reaction energy diagram, representing its higher activation energy.
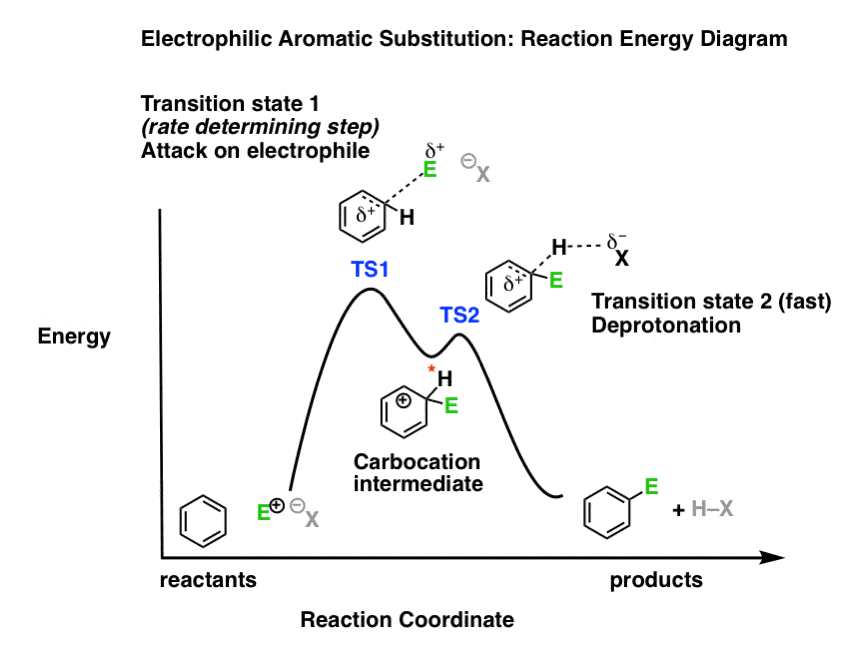
Note that this reaction energy diagram is not to scale and is more of a sketch than anything else. A truly accurate reaction energy diagram can be modelled if one had accurate energies of the transition states and intermediates, which is sometimes available through calculation.
5. Beyond Benzene: Formation Of Ortho, Meta, and Para Disubstituted Benzenes
So that’s all there is to electrophilic aromatic substitution? Yes and no.
Yes, this addresses electrophilic aromatic substitution for benzene.
But, as you’ve no doubt experienced, small changes in structure can up the complexity a notch.
Imagine we start not with benzene, but with a mono-substituted derivative, such as methylbenzene (toluene).
What are the possible products of electrophilic aromatic substitution on a mono-substituted benzene derivative?
Unlike with benzene, where only one EAS product is possible due to the fact that all six hydrogens are equivalent, electrophilic aromatic substitution on a mono-substituted derivative can yield three possible products: the 1,2- isomer (also called “ortho“), the 1,3-isomer (“meta“) and the 1,4-isomer (“para“).
If we assume that the reaction obeys the laws of statistics, we might therefore expect that the product distribution should be 40% ortho. 40% meta, and 20% para.
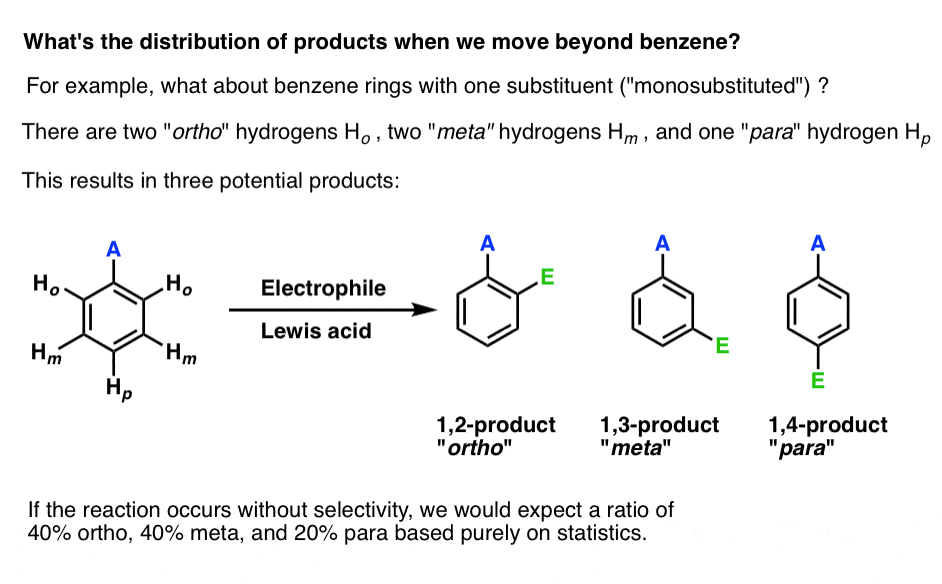
So is that what happens?
No! Two important examples are illustrative.
6. EAS On Monosubstituted Benzenes: The Distribution Of Ortho, Meta and Para Isomers Is NOT Random!
In the nitration of toluene, the product distribution is far from statistical. We get much less meta (5%) than expected, and more ortho (57%) and para (37%) than expected.
In this sense we can say that the methyl group tends to act as an ortho- para- director: it “directs” the electrophile to these positions at the expense of the meta position.
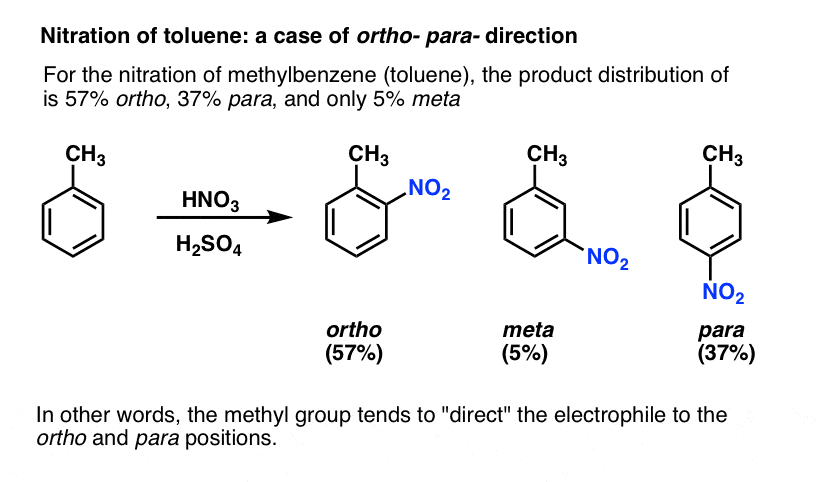
Is this the case for all substituents? No.
In the nitration of nitrobenzene, the opposite result is obtained. Much less ortho and para is produced than expected, and the meta product is major (93%).
In this case the nitro group is said to be acting as a meta- director.
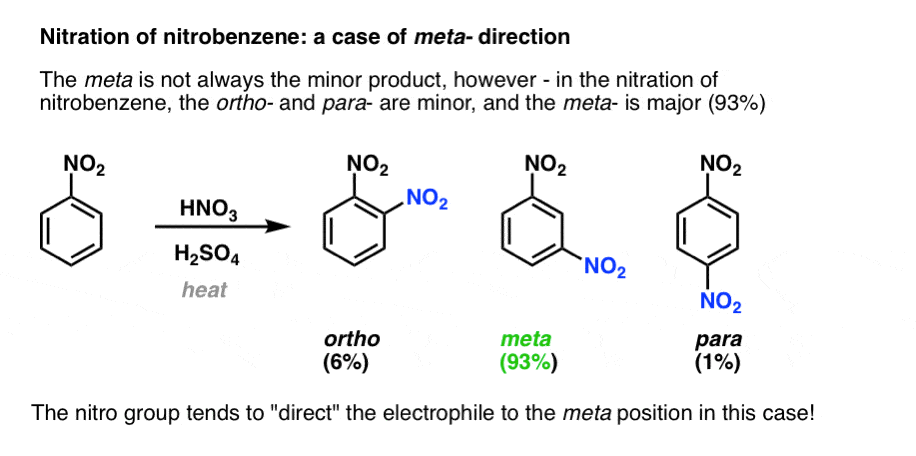
Substituents on benzene tend to fall into one of two categories: ortho–para directors, or meta directors.
If you’re sharp, you might have already made an intuitive leap: the ortho- para- directing methyl group is an activating group, and the meta- directing nitro group is deactivating. So, therefore, are all activating groups ortho- para- directors and all deactivating groups meta- directors?
It’s a good guess – and almost accurate! The fly in the bourbon is the halogens (F, Cl, Br, I) which are deactivating ortho-para directors.
Why? What leads some substituents to be ortho-para directors, and others to be meta-directors?
That’s going to have to wait until the next post for a full discussion. But here’s a hint: it has to do with our old friend, “pi-donation”.
Thanks to Mattbew Knowe for valuable assistance with this post.
Notes
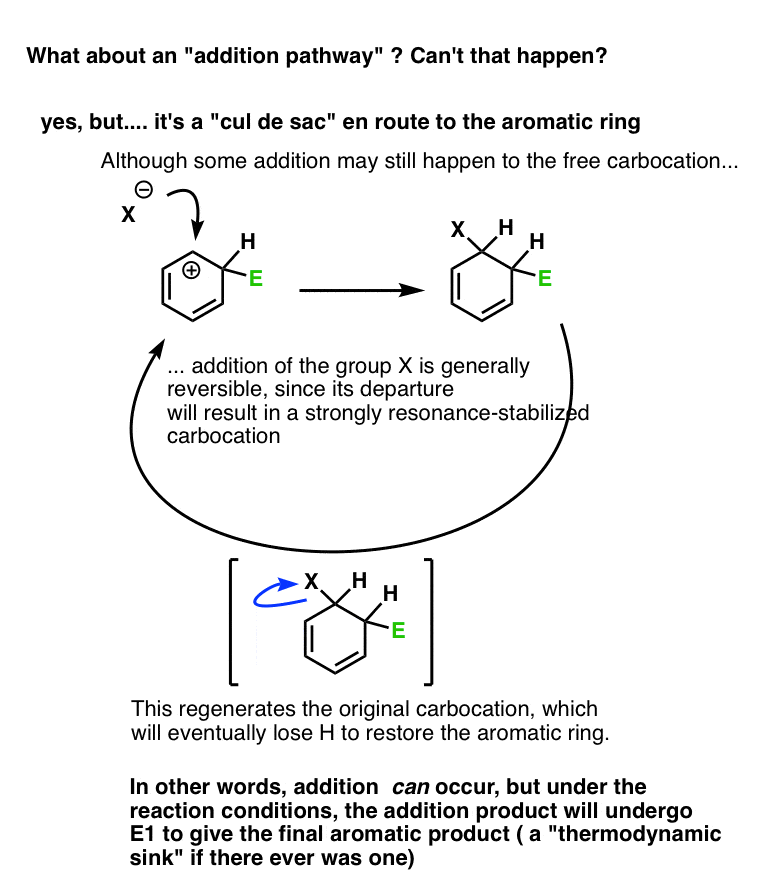
Note 1 – Why can’t the counterion attack the aromatic ring carbocation? Does that happen?
Yes, but it’s a dead end.
Let’s say we form the carbocation, and it’s attacked by a weak nucleophile (which we’ll call X).
This gives us the addition product.
However, it’s rarely a very stable product. X is typically a weak nucleophile, and therefore a good leaving group. Furthermore, loss of the leaving group will result in a highly resonance-stabilized carbocation. (Think of the first step in the SN1 or E1 reaction).
This would re-generate the carbocation, which could then undergo deprotonation to restore aromaticity. Once that aromatic ring is formed, it’s not going anywhere. : – )
To make a long story short, yes, addition could occur, but the addition product will eventually undergo E1 to form the aromatic product.
(figure below)
Quiz Yourself!

Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
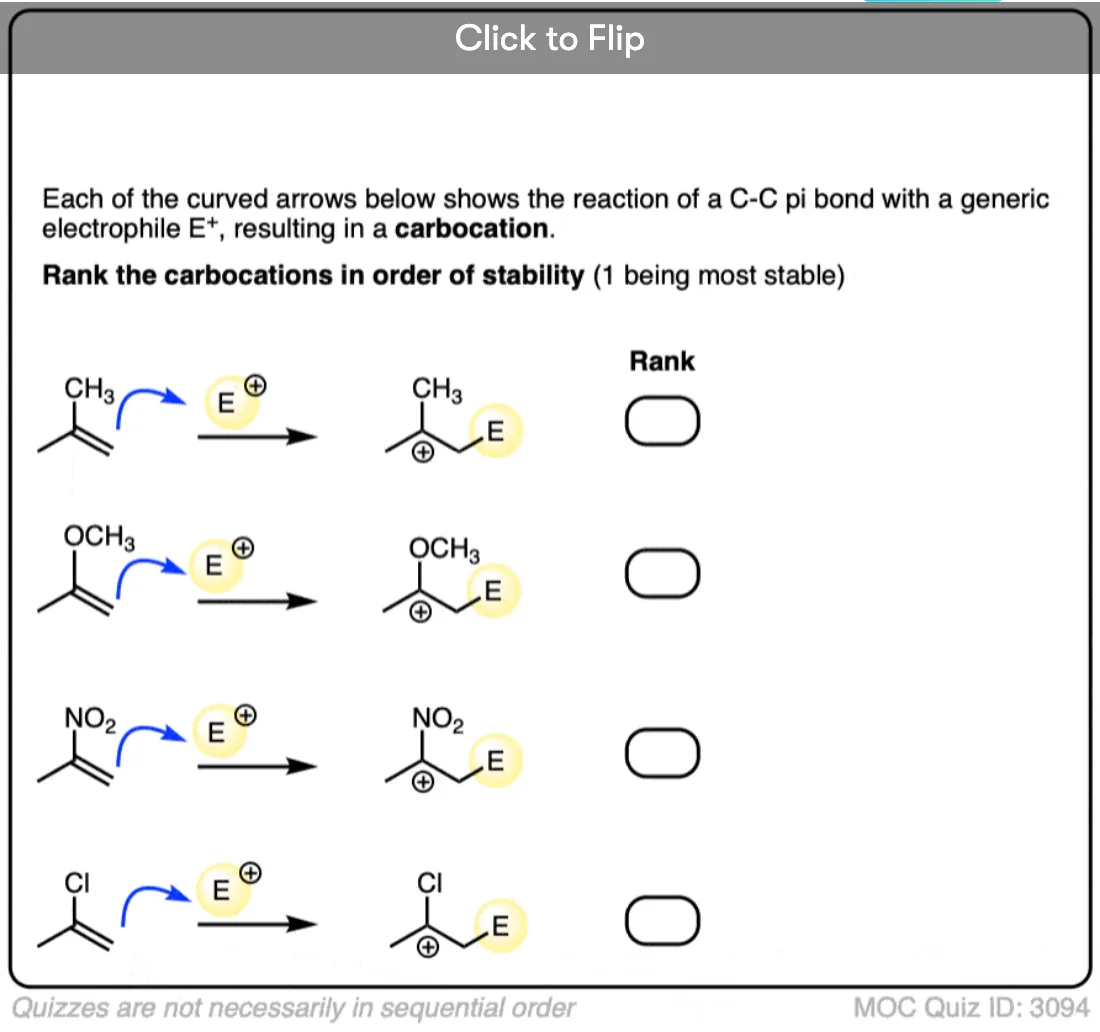
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
The EAS mechanism covers a variety of reactions – Friedel-Crafts substitutions, halogenation, nitration, and many others.
- A Quantum Mechanical Investigation of the Orientation of Substituents in Aromatic Molecules
G. W. Wheland
Journal of the American Chemical Society 1942, 64 (4), 900-908
DOI: 10.1021/ja01256a047
This discusses the structure of the arenium ion that gets formed in EAS reactions, also known as the s-complex or Wheland intermediate, after the author here who first proposed it. - A Quantitative Treatment of Directive Effects in Aromatic Substitution
Leon M. Stock, Herbert C. Brown
Phys. Org. Chem. 1963, 1, 35-154
DOI: 10.1016/S0065-3160(08)60277-4
This is a very comprehensive review for its time, summarizing work on directing effects in EAS (e.g. determining which groups are o/p-directing vs. meta-directing, and to what extent they direct/deactivate). - Aromatic substitution. XXVIII. Mechanism of electrophilic aromatic substitutions
George A. Olah
Acc. Chem. Res., 1971, 4 (7), 240-248
DOI: 10.1021/ar50043a002
An account by Prof. Olah on the work he had carried out studying the mechanism of various types of electrophilic aromatic substitution reactions – nitration, halogenation, as well as Friedel-Crafts acylation and alkylation. - Aromatic substitution. XXXVI. Aluminum trichloride and antimony pentafluoride catalyzed Friedel-Crafts alkylation of benzene and toluene with esters and haloesters
George A. Olah and Jun Nishimura
Journal of the American Chemical Society 1974, 96 (7), 2214-2220
DOI: 10.1021/ja00814a035
In this case, carboxylic esters are not studied (as those would lead to acylation rather than alkylation). This covers other types of esters in Friedel-Crafts alkylation: alkyl chlorosulfites, arenesulfinates, tosylates, chloro- and fluorosulfates, trifluoromethanesulfonates (triflates), pentafluorobenzenesulfonates, and trifluoroacetates. - Aromatic substitution. XXXVII. Stannic and aluminum chloride catalyzed Friedel-Crafts alkylation of naphthalene with alkyl halides. Differentiation of kinetically and thermodynamically controlled product compositions, and the isomerization of alkylnaphthalenes
George A. Olah and Judith A. Olah
Journal of the American Chemical Society 1976, 98 (7), 1839-1842
DOI: 10.1021/ja00423a032
This is a similar paper by Prof. Olah and his wife, Judith Olah, on the mechanism of Friedel-Crafts alkylation, except using naphthalene instead of benzene. Naphthalene is different in that there are two sites for monosubstitution – the a and b positions. - Stable carbocations. CLXX. Ethylbenzenium ions and the heptaethylbenzenium ion
George A. Olah, Robert J. Spear, Guisseppe Messina, and Phillip W. Westerman
Journal of the American Chemical Society 1975, 97 (14), 4051-4055
DOI: 1021/ja00847a031
This paper discusses the characterization of benzenium ions, which are intermediates in EAS, and the characterization of the heptaethylbenzenium ion, which is a stable species because it lacks a proton and therefore eliminates with difficulty. - The Anomalous Reactivity of Fluorobenzene in Electrophilic Aromatic Substitution and Related Phenomena
Joel Rosenthal and David I. Schuster
Journal of Chemical Education 2003, 80 (6), 679
DOI: 10.1021/ed080p679
A very interesting paper, suitable for curious undergrads, and discusses something that most practicing organic chemists will know empirically – fluorobenzene is almost as reactive as benzene in EAS or Friedel-Crafts reactions, which is counterintuitive when one considers electronic effects. - Electrophilic Aromatic Substitution: New Insights into an Old Class of Reactions
Boris Galabov, Didi Nalbantova, Paul von R. Schleyer, and Henry F. Schaefer, III
Accounts of Chemical Research 2016, 49 (6), 1191-1199
DOI: 10.1021/acs.accounts.6b00120
The late Prof. P. v. R. Schleyer was a giant in Physical Organic chemistry, and this paper, published posthumously, covers work done towards the end of his life in re-determining the mechanism of EAS. - Unified Mechanistic Concept of Electrophilic Aromatic Nitration: Convergence of Computational Results and Experimental Data
Pierre M. Esteves, José Walkimar de M. Carneiro, Sheila P. Cardoso, André H. Barbosa, Kenneth K. Laali, Golam Rasul, G. K. Surya Prakash, and George A. Olah
Journal of the American Chemical Society 2003, 125 (16), 4836-4849
DOI: 10.1021/ja021307w
This is the grand-daddy paper on nitration, summarizing a lifetime’s worth of work on the subject.
Hello,
In some books they describe electrophilic aromatic substitution as the electrophile attack the benzene ring. However, they still draw the arrow from the pie electrons to the electrophile. Is it correct to state that the electrophile is attacking the nucleophile in this case? Thank you.
Strictly speaking, no, since the electrophile is not providing the source of the electrons, the aromatic ring is.
It’s common to say things like “metals are attacked by strong acid” for example, when in reality it’s the metal providing the electrons and the acid is accepting them. It doesn’t really matter so long as you understand where the electrons are coming from.
Hello thanks for the information…using curly curves to show the mechanism of electrophyllic substitution of benzene,show how the products of the following processes are formed: 1: chlorination
2: nitration
3: friedel craft alkylation
hello, thank you first! i have a question, in the example of methyl benzene, why did the reaction give the product where nitro group is in ortho position in a greater yield than the one where it is in para position?
There are two ortho positions and only one para position. So if it were completely random (and there were no meta) the yield would be 66%. The fact that the yield is 57% shows that the methyl group has some steric influence. The effect becomes much greater as the alkyl group is increased in size.
Have you seen the section on EAS from “Modern Physical Organic Chemistry” (p. 608) by Anslyn? The description is quite clear. In most cases the reaction will be second-order overall, but when there is a bulky group (e.g. t-butyl) the rate of the reverse reaction can be significant. In those cases the reverse reaction starts to have about the same barrier as the rate of deprotonation, which leads to secondary isotope effects.
Hey! Great post however a few questions come to my mind from the texts I have. They have been great and clear but how primary isotopic effects and secondary isotopic effects are affected by the bulkiness of the electrophile have not been described very clearly, and other texts don’t seem have any information of them.
It mentions that primary isotopic effect is seen when the loss of an alpha-hydrogen happens in the slower than the formation of the Wheland-complex. It says this is only the case when the electrophile is a significantly bulkier group.
I’d also like to know how charge-transfer complexes are formed from pi-complexes, but that is a little off-topic and at the risk of digressing I wont talk too much about them (I dont understand them well enough anyway)
Assalam o alaikum.very good explanation sir.i enjoyed a lot reading it because it is giving me strong concepts.much helpful!!
I appreciate sir.
The energy of second intermiediate is more than first in the given energy diagram? ….
There is only one intermediate – the carbocation. Are you referring to the two transition states?
Very nice information about of nitration reaction……👌👌👌👌
OK, glad you found it useful Ajit.
Very good post. Very accurate and extremely detailed. I would suggest that you maybe include an example of electrophilic aromatic substitution using acetic anhydride. It’s a very good test question, and (if the school is legit probably tested it many times) because the mechanism is literally the same, you treat it the same as you would for another compound but people have trouble with it.
Thank you Eric! I agree that it’s a common exam question (and a good one). On a related post on the intramolecular Friedel-Crafts reaction I do include an example of that. Good tip!
Can you give me the mechanism of formation of 1,3 di-nitrobenzene?
It would be in no way different from the mechanism for the nitration of benzene.
Superb explanation.
Wonderful! I was able to get the concepts right.