Substitution Reactions
The SN1 Mechanism
Last updated: May 28th, 2026 |
The SN1 Reaction Mechanism
- There are two important classes of nucleophilic substitution mechanisms – the SN1 and SN2 mechanisms (See article – Two Types of Substitution Reactions)
- The SN1 mechanism is distinct from the SN2 in three distinct ways.
- The reaction is fastest for tertiary alkyl halides and slowest for primary (and methyl) halides
- The rate law is unimolecular – it is only dependent on the concentration of substrate (i.e. alkyl halide) and not the nucleophile
- Alkyl halides with a chiral center at the “alpha-carbon” will give a product that provides a mixture of retention of configuration and inversion of configuration. [Note 2] Sometimes this is described as “racemization” .
- The best explanation for how this reaction works is that it begins with a (rate-determining) loss of a leaving group to give a carbocation, which can then undergo attack by a weak nucleophile at either face, resulting in the loss of stereochemistry.
- The SN1 reaction is sometimes accompanied by carbocation rearrangements. (See article – Substitution With Rearrangement)
Table of Contents
- Stereochemistry Of The SN1 Reaction: A Mixture of Retention and Inversion is Observed
- The Rate Law Of The SN1 Reaction Is First-Order Overall
- The Reaction Rate Increases With Substitution At Carbon (Tertiary >> Secondary > Primary)
- The Stepwise Reaction Mechanism of the SN1 Reaction
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Stereochemistry Of The SN1 Reaction: A Mixture of Retention and Inversion is Observed
If we start with an enantiomerically pure product, (that is, one enantiomer), these reactions tend to result in a mixture of products where the stereochemistry is the same as the starting material (retention) or opposite (inversion). In other words, some degree of racemization will take place (See post: What Is A Racemic Mixture?)
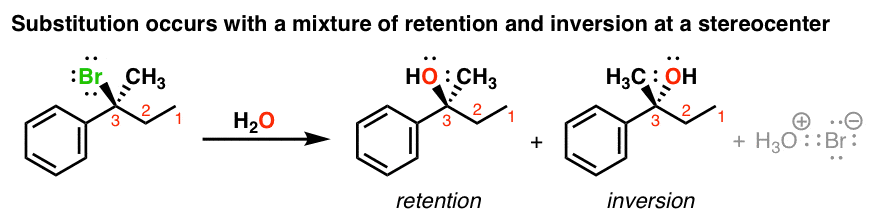
Compare this to the SN2, which always results in inversion of stereochemistry! Clearly something different must be going on here.
2. The Rate Law Of The SN1 Reaction Is First-Order Overall
We can also measure the rate law of these reactions. When we do so, we notice that the rate is only dependent on the concentration of the substrate, but not on the concentration of nucleophile.
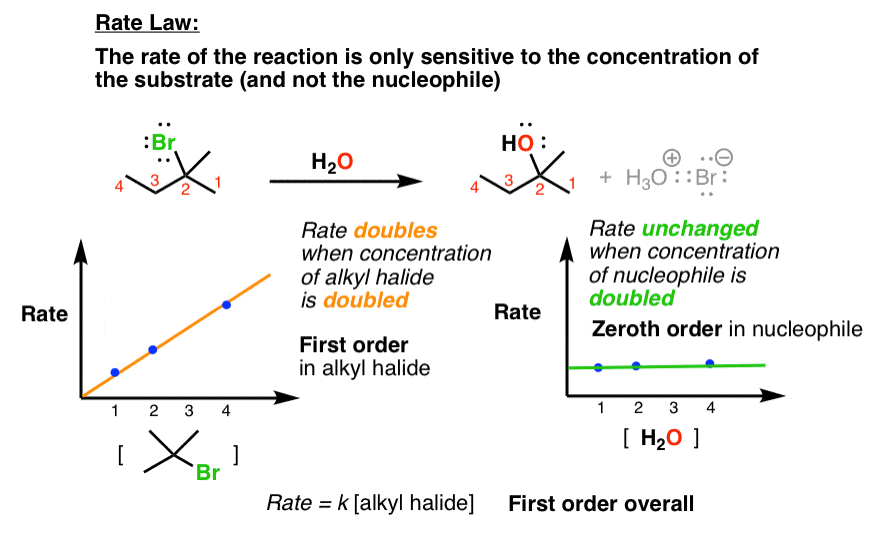
Weird. Remember that the SN2 depends on both. Why might this reaction only depend on the concentration of substrate?
3. The Reaction Rate Increases With Substitution At Carbon
When we subtly change the types of substrates (e.g. alkyl halides) we use in these reactions, we find that tertiary substrates (for instance, t-butyl bromide) are considerably faster than secondary alkyl bromides, which are in turn faster than primary [Note 1]
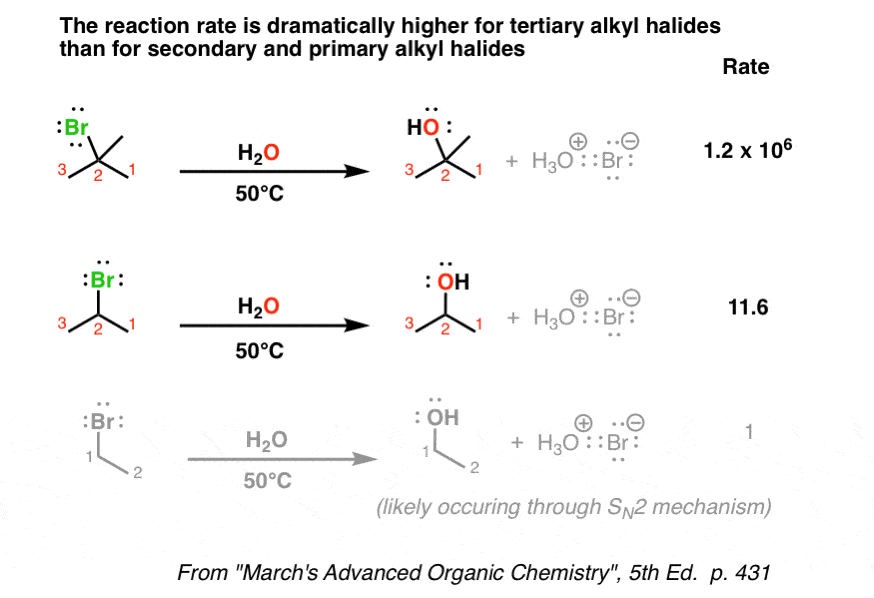
Compare that to the case for SN2, where primary was faster than secondary and tertiary hardly reacted at all. Mysterious!
4. The Stepwise Reaction Mechanism of the SN1 Reaction
The best hypothesis we have for this reaction is a stepwise mechanism.
- In the first step, the leaving group leaves, forming a carbocation.
- In the second, a nucleophile attacks the carbocation, forming the new product.
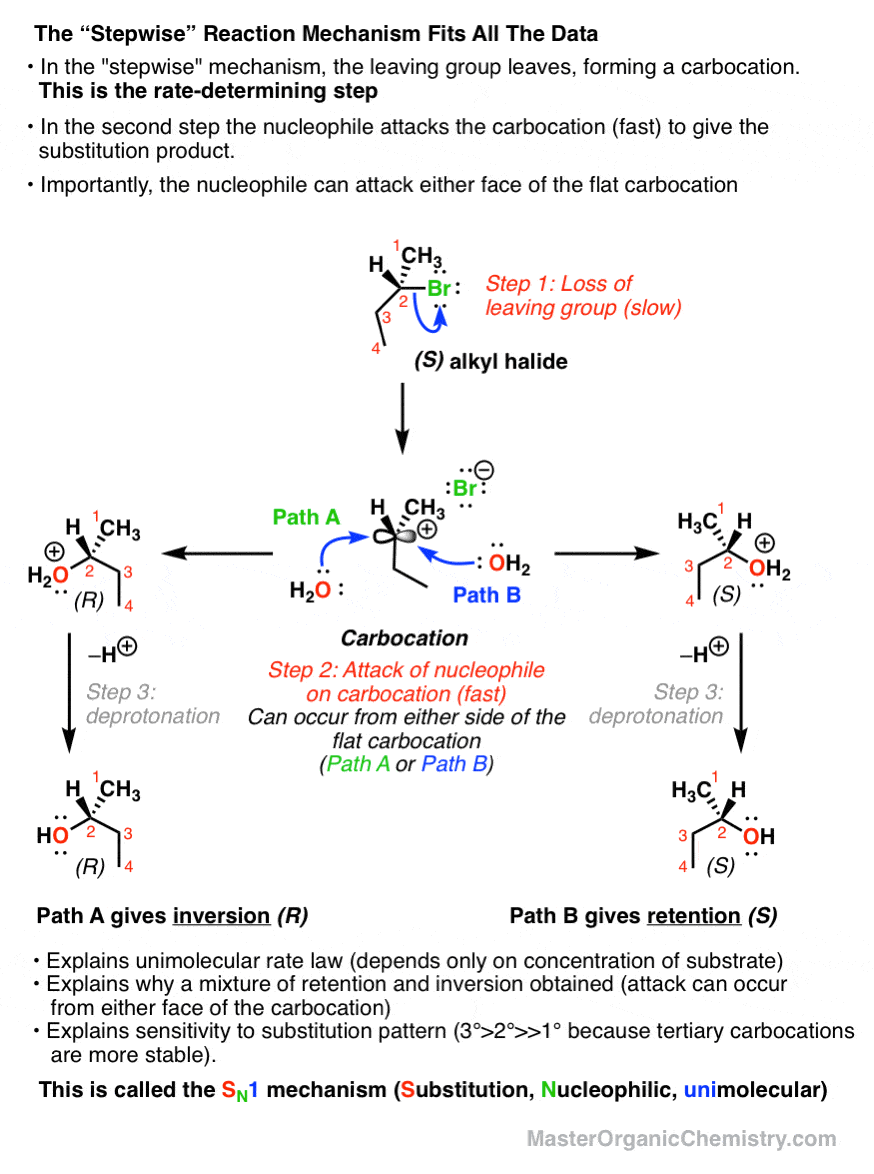
This explains all of our observations nicely. First of all, the slow step should be formation of the (unstable) carbocation – which only depends on the substrate, not the nucleophile.
Furthermore, since the stability of carbocations depends tremendously on substitution pattern (tertiary carbocations are more stable than secondary, which are more stable than primary) this also conveniently explains the dependence of the reaction rate on substitution pattern (See post: Carbocation Stability)
Any factor which stabilizes the carbocation, increases the rate at which the leaving group can leave.
It also helps us understand the stereochemistry. Since the electrophile is flat, attack could occur from either face; which means that we obtain a mixture of retention and inversion products.
This is therefore called the SN1 mechanism – Substitution, Nucleophilic, Unimolecular – to contrast with the SN2 (Substitution, Nucleophilic, Bimolecular).
It all seems to work if you’ve got a good leaving group present (like a halogen). But what if you don’t have a good leaving group? In the next post we’ll talk about how to make a poor leaving group into a good one.
Next Post: The Conjugate Acid Is A Better Leaving Group
Notes
Note 1. – the primary alkyl halide shown here is certainly reacting solely through an SN2 mechanism.
Note 2. Athough it’s often said that the SN1 proceeds with “racemization” of stereocenters, in practice a 50/50 split of stereocenters may not be obtained due to “ion pairing” effects.
In other words, the leaving group could leave, but not fully dissociate from the vicinity of the carbocation, which could block a nucleophile from attacking the electrophile from that face. For that reason it’s a little bit more correct to say that it proceeds with a “mixture of retention and inversion” rather than “racemization”.
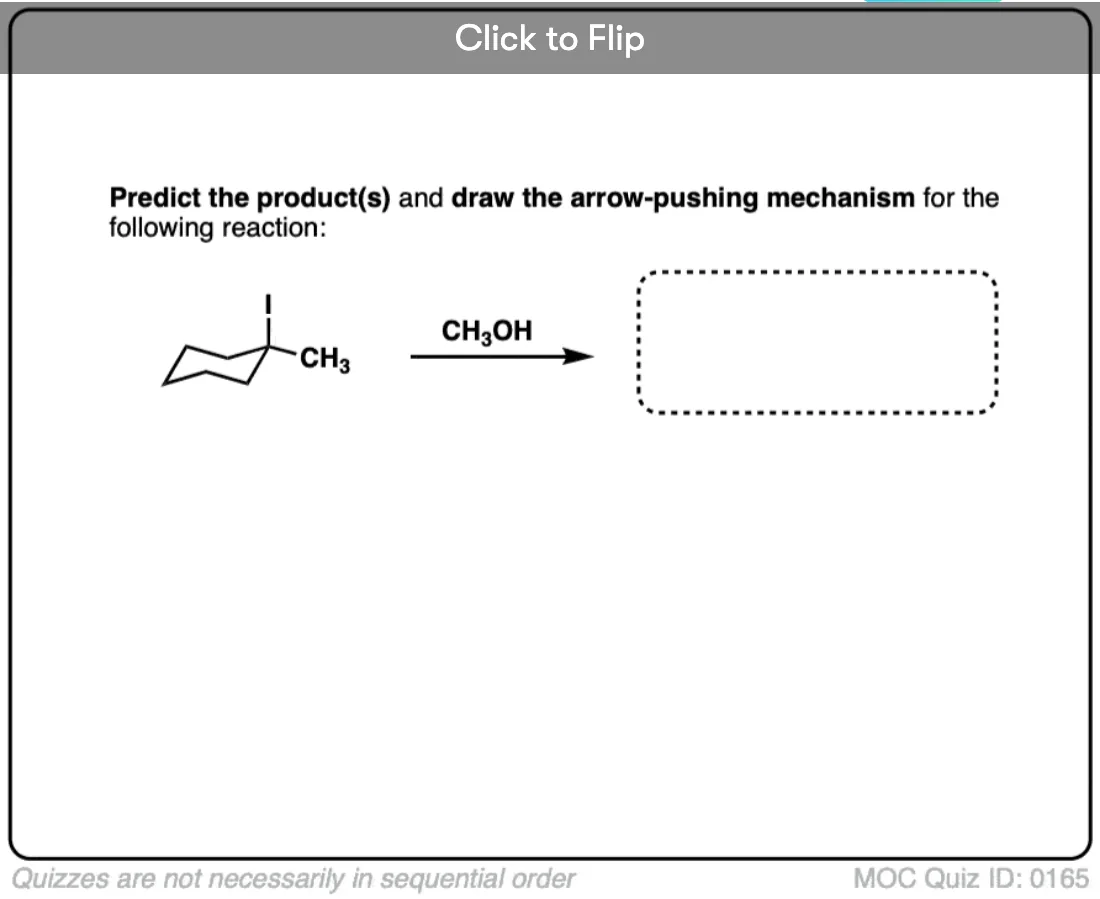
Quiz Yourself!
 Click to Flip
Click to Flip

Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
-
56. Mechanism of substitution at a saturated carbon atom. Part V. Hydrolysis of tert.-butyl chloride.
Edward D. Hughes.
J. Chem. Soc. 1935, 235
DOI: 10.1039/JR9350000255
Original study where the hydrolysis of t-butyl chloride was found to be first-order in alkyl halide and zero order in base, giving rise to the mechanism we now know as SN1. - Mechanism of substitution at a saturated carbon atom. Part IX. The rôle of the solvent in the first-order hydrolysis of alkyl halides
Leslie C. Bateman and Edward D. Hughes
J. Chem. Soc. 1937, 1187-1192
DOI: 10.1039/JR9370001187
In the hydrolysis of alkyl bromides by water in formic acid, the relative rates at 100° are MeBr 1.00, EtBr 1.71, iPrBr 44.7, and tBuBr ca. 10^8. - Reaction kinetics and the Walden inversion. Part I. Homogeneous hydrolysis and alcoholysis of β-n-octyl halides
Edward D. Hughes, Christopher K. Ingold and Standish Masterman
J. Chem. Soc. 1937, 1196-1201
DOI: 10.1039/JR9370001196 - Reaction kinetics and the Walden inversion. Part IV. Action of silver salts in hydroxylic solvents on β-n-octyl bromide and α-phenylethyl chloride
Edward D. Hughes, Christopher K. Ingold and Standish Masterman
J. Chem. Soc., 1937, 1236-1243
DOI: 10.1039/JR9370001236
These two papers examine reactions of 2-octyl halides in an attempt to see if pure SN1 or SN2 pathways on the same substrate can be favored simply by varying the reaction conditions. - The Correlation of Solvolysis Rates
Ernest Grunwald and S. Winstein
Journal of the American Chemical Society 1948, 70 (2), 846-854
DOI: 1021/ja01182a117
This is a very important paper, discussing the ‘Grunwald-Winstein equation’ for the first time. This equation is an extension of the Hammett equation, taking solvent effects (i.e. ‘ionizing power’) into consideration. - The Reactivity of Bridgehead Compounds of Adamantane
Paul von R. Schleyer and Robert D. Nicholas
Journal of the American Chemical Society 1961, 83 (12), 2700-2707
DOI: 1021/ja01473a024
Bridgehead carbocations are generally quite unstable since they cannot achieve the planar geometry necessary for good hyperconjugative stabilization. Somewhat surprisingly, in this paper it is found that the SN1 reaction of 1-bromoadamantane proceeds only about 1000 times slower than that of t-butyl bromide, albeit (of course) only with retention of configuration. - The Common Basis of Intramolecular Rearrangements. VI.1 Reactions of Neopentyl Iodide
Frank C. Whitmore, E. L. Wittle, and A. H. Popkin
Journal of the American Chemical Society 1939, 61 (6), 1586-1590
DOI: 1021/ja01875a073
An early paper demonstrating that SN1 reactions can be induced by reaction of an alkyl halide with silver salts. In this case, the neopentyl cation quickly rearranges to the significantly more stable t-amyl cation, and those products are obtained. - Mechanism of substitution at a saturated carbon atom. Part XXIX. The rôle of steric hindrance. (Section D) the mechanism of the reaction of neopentyl bromide with aqueous ethyl alcohol
I. Dostrovsky and E. D. Hughes
J. Chem. Soc., 1946, 166-169
DOI: 10.1039/JR9460000166
This is an example of an SN1 reaction with rearrangement. Neopentyl bromide in aqueous ethyl alcohol gives t-amyl alcohol (and t-amyl ethyl ether). - Mechanism of substitution at a saturated carbon atom. Part XXXV. Effect of temperature on the competition between unimolecular solvolytic and non-solvolytic substitutions of di-p-tolylmethyl chloride. Activation in the fast step of unimolecular non-solvolytic substitution
Audrey R. Hawdon, E. D. Hughes and C. K. Ingold
J. Chem. Soc., 1952, 2499-2503
DOI: 10.1039/JR9520002499
It is possible to run SN1 reactions in the presence of added nucleophile, such as in the hydrolysis of benzyl chlorides in the presence of added sodium azide. The separate rates of formation of the carbocation and production of the azide can thus be measured. - Methanolysis of Optically Active Hydrogen 2,4-Dimethylhexyl-4-phthalate
von E. Doering and Harold H. Zeiss
Journal of the American Chemical Society 1953, 75 (19), 4733-4738
DOI: 10.1021/ja01115a035
An early example of an SN1 reaction without full racemization. Prof. Doering proposes a mechanism in the paper, interesting read. - Quaternary stereocentres via an enantioconvergent catalytic SN1 reaction
Wendlandt, A.E., Vangal, P. & Jacobsen, E.N.
Nature 556, 447–451 (2018)
DOI: 1038/s41586-018-0042-1
This is a rare example of an asymmetric SN1 reaction – normally the SN1 reaction is taught as giving achiral products, but in this particular case it is possible to induce chirality because the carbocation is so highly stabilized (tertiary, benzylic, and propargylic).
If after nucleophilic attack, the carbon is not a chiral centre, then wont it just give us a retained configuration product?
For something like t-butyl bromide going to t-butyl alcohol, there is no possibility of forming stereoisomers, so the question of retention or inversion becomes a non-issue.
I’m still learning about electrophilic sub/elim reactions, and your website is a blessing! I’ve got one question regarding the fast method to determine a reaction is SN1 or SN2. Let’s say that Br- is the leaving group. It’s said that if a nucleophilic substitution reaction has an acid (in this case HBr) as its product, then it’s a SN1 reaction. On the other hand, if a nucleophilic substitution reaction has a anion (in this case Br-), then it’s a SN2 reaction. I’m sure this method is useful and heuristic, but whether it be SN1 or SN2, shouldn’t the leaving group Br- not able to form HBr (as it’s a weak conjugate base)? I assume you wrote H3O+ Br- to show this, but I might be wrong. Would you please give some clarification on this? Thank you!
You’ll likely only ever see HBr if you are performing substitution reactions on alcohols. The purpose is to turn the poor leaving group OH into the good leaving group H2O, and then, depending on whether the substrate is primary (SN2) tertiary (SN1) or secondary (ask your instructor, answers vary!) you will obtain the resulting substitution product.
Hey James, I recently came across this paper: http://dx.doi.org/10.1021/ed086p519
Does this mean that the SN1 mechanism is not applicable for secondary alkyl halides at all? If so, please make a post explaining why (I’m still a student, and don’t really understand the paper)
That is a very interesting paper.
I would not say that the SN1 doesn’t apply to secondary alkyl halides *at all* but let’s just say they are much less important than they appear to be.
Let’s just say that the whole teaching of SN1/SN2/E1/E2 is pretty muddled, and has been this way for quite some time. If you take introductory organic you would come away thinking that organic chemists spend a great deal of their time trying to plan out SN1/Sn2/e1/e2 reactions whereas in reality the ratio of published SN2 and E2 reactions vs SN1 and E1 reactions is at least 100:1. Even then the SN2 is best done on primary substrates to avoid side reactions.
It’s a mess. I’m glad someone has gone back and looked through the original Ingold/Hughes papers, because they are the basis for a lot of what is taught.
Can kinetics provide all the necessary details about nucleophilic substitution reactions?
I’m not sure what details you mean by “all necessary details”, but even a limited reading of that would neglect issues like stereochemistry, etc. So the answer would be no.
Awesome and helpful content- specific typo is under #3. types of substrates***
Thanks!
Not seeing it. Anyone?
Thanks for the very helpful site!
I have a question about the first step in an SN1 reaction – the leaving group leaves. What causes the leaving group to want to leave? Is this action set in motion by heat, light or some other energy source?
Thanks for the help.
It’s helpful to think of bonds as balls and springs. At any given moment the bond between the carbon and the leaving group is oscillating like two balls connected by a spring, and the bond length is alternating between being shorter than normal and longer than normal. The average is the “bond length”.
Now imagine the energy as that bond extends to its longest length. The carbon will have a partial positive charge (delta plus) and the leaving group will have a partial negative charge (delta minus). If the carbon is able to stabilize positive charge very well (e.g. a tertiary carbocation) and the leaving group is able to stabilize negative charge very well (e.g. a weak base) then it’s not hard to imagine the bond length getting longer and longer until it goes beyond the length of what we’d consider a normal bond and can be considered a carbocation in the vicinity of a leaving group. If a nucleophilic solvent is around (like CH3OH) it may then “trap” that free carbocation, resulting in a substitution reaction.
conversely if we have a situation like CH3CH2OH, where we have a bad leaving group (OH) connected to a carbon poorly able to stabilize positive charge (primary) then the energetic barrier for that bond length getting long enough to lead to dissociation is just too high.
In sn1 rexn most usually racemic mixture is formed in product but it isnot 100% racemized,why?
The reason is that the resulting carbocation and the leaving group can form what is known as a “tight ion pair”. In a tight ion pair the leaving group has not completely dissociated, and this will block the face of the carbocation that was connected to the leaving group.
In order for full racemization to occur there needs to be complete dissociation such that either face of the carbocation can be attacked with equal probability.
Would you please write a textbook – I would gladly buy it!
No plans as yet, but thanks for the vote of confidence!
I can’t tell you how brilliantly every topic is explained. This is great. Your analogies and phrasing are the only way I understand organic chem. Thanks a ton
Glad to hear it Anisha!
I got it. thank you
In the paragraph under “final note”, I believe “In other words, the leaving group could leave, but not fully dissociate from the vicinity of the carbocation, which could block a nucleophile from attacking the nucleophile from that face” should read “block a nucleophile from attacking the electrophile” instead.Also,In the paragraph under “So what could be going on here?”, I believe “Since the nucleophile is flat, attack could occur from either face; which means that we obtain a mixture of retention and inversion products” should read “Since the electrophile is flat” instead.
sir plz tell me the percentage of inverted and retented prodct in ion pair mechanism…???
It would depend on a variety of factors, such as temperature, solvent, the identity of the leaving group and the substrate. Highly variable!
EXTREMELY helpful! The content is very straightforward and understandable! MUCH appreciated! This website will be used for the whole semester!!
yes … very helpful but organic chemistry is very intrusting chemistry than other branches of chemistry <3
You’ve created an awesome website here. I haven’t even used your reaction guide much yet, but I still like being a member simply because I feel like you totally deserve my money for all the help I’m getting from the blog. (It’s taught me more in a few days than my book has all year!)
Anyway, just wanted to point out that on your starred note at the bottom, I think you meant that the primary alkyl halide is certainly reacting through an SN2 mechanism (not an SN1).
Yes, you’re right. Thanks for the catch – and glad you find it useful!
Tremendously useful site, your efforts are highly appreciative, Kindly correct the spelling of Carbocation in the fourth line after mechanism.
thank you.