Reactions of Aromatic Molecules
Understanding Ortho, Para, and Meta Directors
Last updated: May 28th, 2026 |
Ortho-, Para– and Meta– Directors
- In Electrophilic Aromatic Substitution (EAS), some substituents on benzene will direct the electrophile E to the ortho– (1,2)and para– (1,4) positions. These are called, “ortho, para- directors”.
- Another class of substituents avoids directing the electrophile E to those positions, with the result that the meta- product (1,3) is major. These are called, “meta- directors”.
- The ultimate factor which determines whether a substituent is ortho-, para- or meta- directing is how well it stabilizes an adjacent carbocation.
- Substituents that have a lone pair on the atom adjacent to the aromatic ring will be ortho-, para- directors since this can form a new pi-bond with an adjacent carbocation.
- Substituents that pull electron density away from adjacent carbocations (e.g. CF3, NO2)will avoid directing E to the ortho- or para- position with the result that the meta- product forms instead.
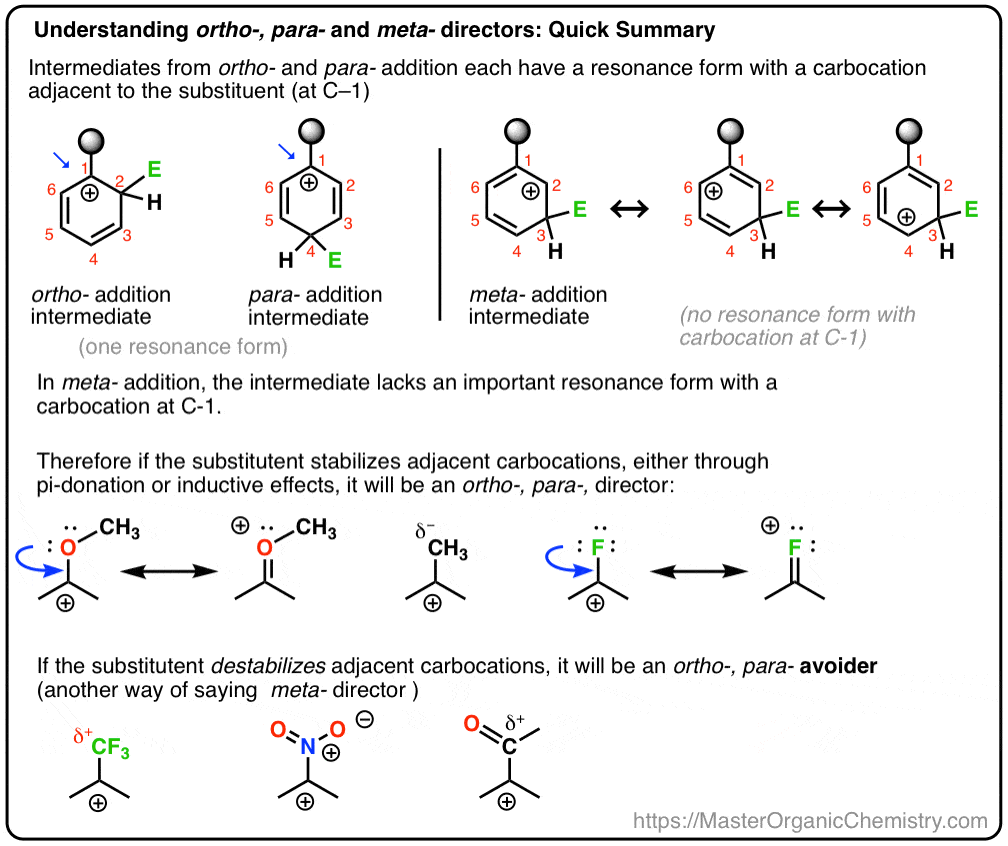
Table Of Contents
- Carbocations Are Stabilized By Adjacent Atoms With Lone Pairs
- Case Study #1: An ortho-,para- Director (OCH3)
- Yes, Oxygen Is Electronegative. But Donation Of Its Lone Pair More Than Makes Up For It!
- The Reaction-Energy Diagram For An Ortho Para Director
- Case Study #2: A meta- director (CF3).
- What We Call “meta– Directors” Are More Like “ortho-, para– Avoiders”
- The Reaction Energy Diagram For A meta- Director
- So Why Are Halogens Ortho-, Para- Directors?
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Carbocations Are Stabilized By Adjacent Atoms With Lone Pairs
In the previous post we introduced ortho- ,para- and meta- directors in electrophilic aromatic substitution. Previous to that we covered the mechanism of electrophilic aromatic substitution, and showed that the mechanism proceeds through a carbocation intermediate.
Today, we’re going to tie those two concepts together, and try to show that whether a substituent is an ortho-, para- or meta- director depends on how the substituent affects the stability of the carbocation intermediate.
And we’ll also see why an alternative name for meta- director might be, “ortho-, para- avoider”. : – )
Let’s think back to which factors increase the stability of carbocations. [See, “3 Factors that Stabilize Carbocations“]
Carbocations are electron-poor species with six electrons in their valence shell. So, any substituent which can donate electron density toward the carbocation will be stabilizing. This includes:
- electron-releasing substituents that can act as “sigma-donors” (e.g. many alkyl groups, which donate electron density through inductive effects [Note 1])
- adjacent atoms with lone pairs that can act as “pi donors” to the carbocation
- adjacent C–C pi bonds that can stabilize the carbocation through charge delocalization (“resonance stabilization”, in other words).
And factors which destabilize carbocations?
- any substituent that removes electron density from a carbocation, such as strong electron-withdrawing groups.
So here’s a quick quiz. Given everything written above, which of the two carbocations below will be more stable?
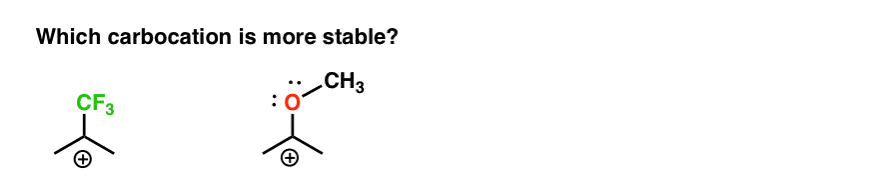
If you can answer this question well, you are most of the way towards understanding the difference between ortho-, para- and meta- directors.
Let’s examine two case studies. We’ll look at a generic electrophilic aromatic substitution reaction of benzene with an ortho-, para- director (methoxybenzene) and examine the intermediates that are obtained from attack at the ortho, meta, and para positions. Then we’ll perform the same exercise with a meta– director (trifluoromethylbenzene).
In the process, we’ll hope to understand why methoxybenzene favors ortho- and para– products while trifluoromethylbenzene favors the meta- product.
2. Case Study #1: An ortho-,para- Director (OCH3)
Electrophilic aromatic substitution of methoxybenzene tends to give ortho- and para- products with very little meta- . Here’s what happens in nitration, for example:
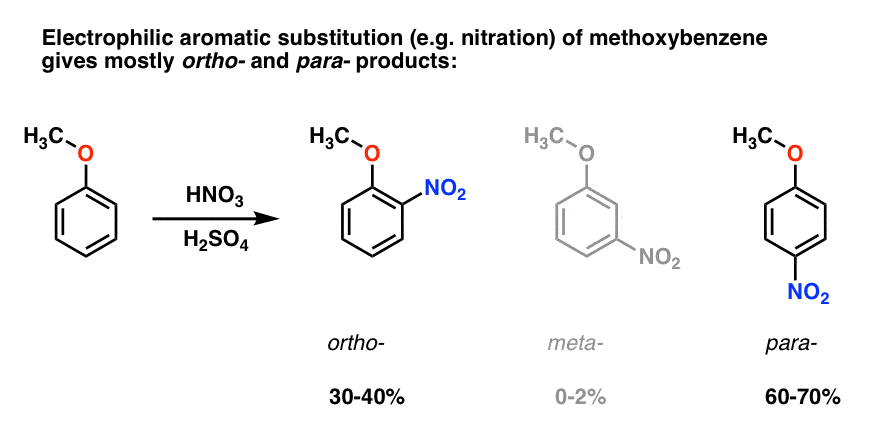
The para- product dominates (60-70%), with ortho- close behind (30-40%) and only a trace of the meta– .
Why?
3. Yes, Oxygen Is Electronegative. But Donation Of Its Lone Pair More Than Makes Up For It!
Here’s the pathway leading to the ortho- product. We break a C–C pi bond and form a new C–E bond (where E is intended to represent a generic electrophile), generating a resonance-stabilized carbocation:
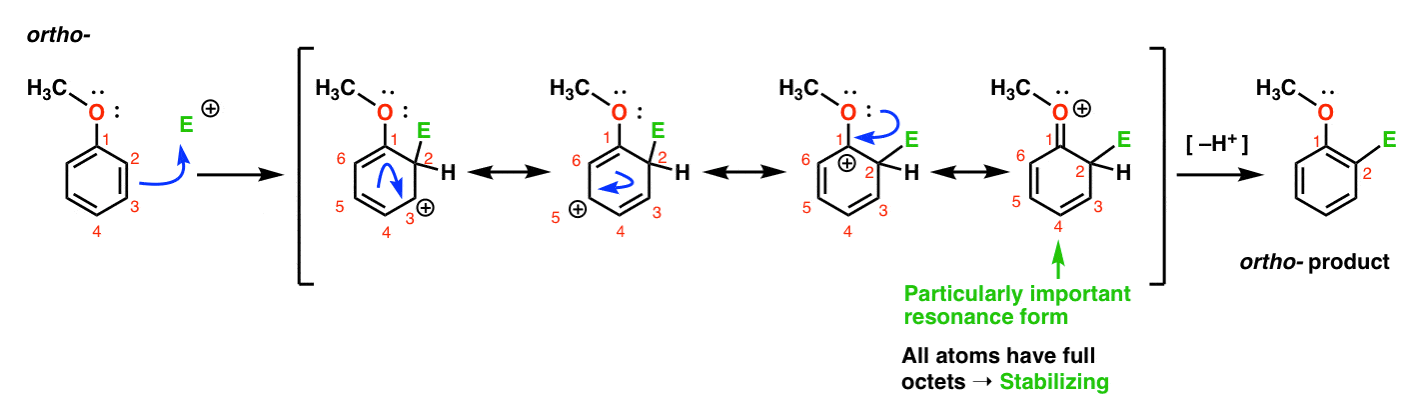
If you look closely, you should note that we can form four resonance forms . Three of them bear full carbocations (on C1, C3, and C5), but it is the the fourth one – the resonance form where there is a C=O pi bond – which is particularly important. Why? In this resonance form, all of the carbon atoms have a full octet of electrons. That’s because the oxygen directly bonded to the ring can donate a lone pair to the adjacent carbocation, forming a pi bond. This is an example of pi donation.
Here’s the pathway for attack of the electrophile at the meta- position. Note the difference!
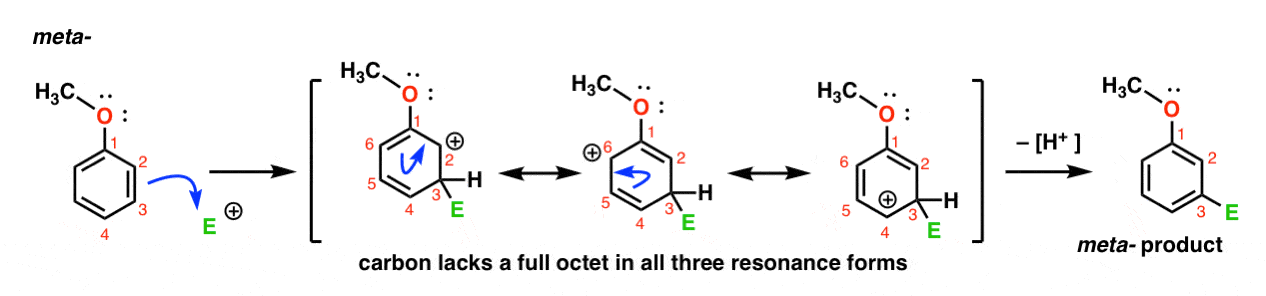
Attack of the electrophile at C-3 results in a carbocation which can be delocalized via resonance to C2, C4, and C6. However, there’s no way to “move” the carbocation to C1, which means that there’s no reasonable resonance form where all atoms have full octets.
This makes the meta- carbocation intermediate much less stable than the ortho- carbocation intermediate.
Finally, here is the reaction leading to the para- product:
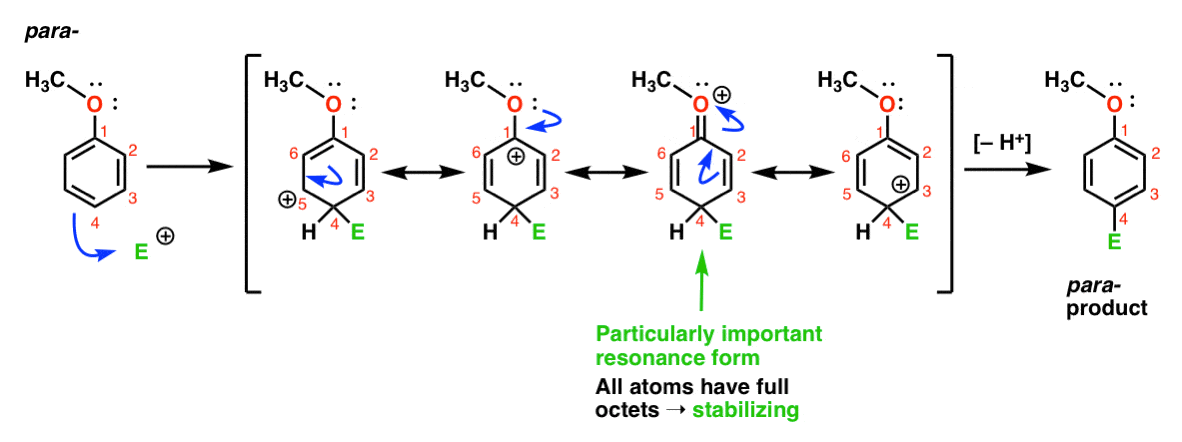
Here we can again draw resonance forms with carbocations on C1, C3, and C5, as well as a fourth resonance form where the attached oxygen atom donates an electron pair to the carbocation on C1, resulting in a full octet at carbon. This is a situation essentially the same as the ortho- intermediate.
So, by analysis of the resonance forms (and mark my word, you may well be asked to do the same on a test in the near future) we can see that the intermediate carbocations resulting from ortho- and para– addition are considerably more stable than the intermediate from meta– addition.
This explains is why ortho- and para- products dominate, and the meta- product is minor. [Note 2]
It might be helpful to visualize this by drawing a reaction energy diagram.
4. The Reaction-Energy Diagram For An Ortho Para Director
The transition state leading to the ortho- and para- addition products is much lower in energy than the meta– .
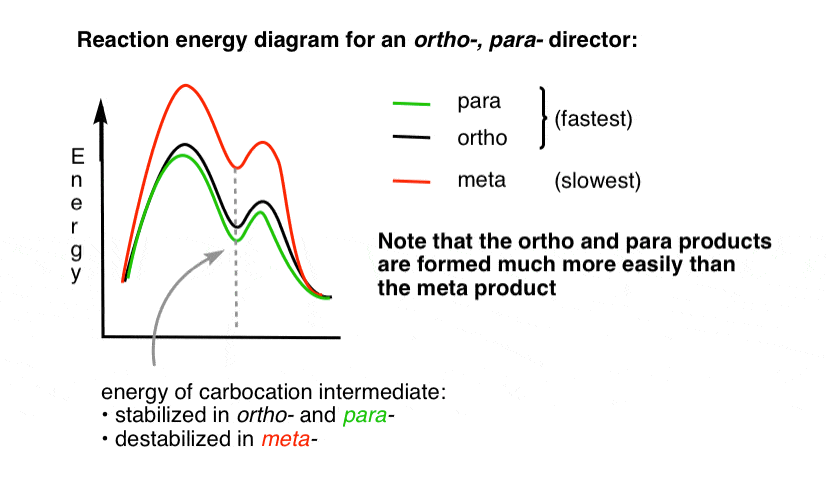
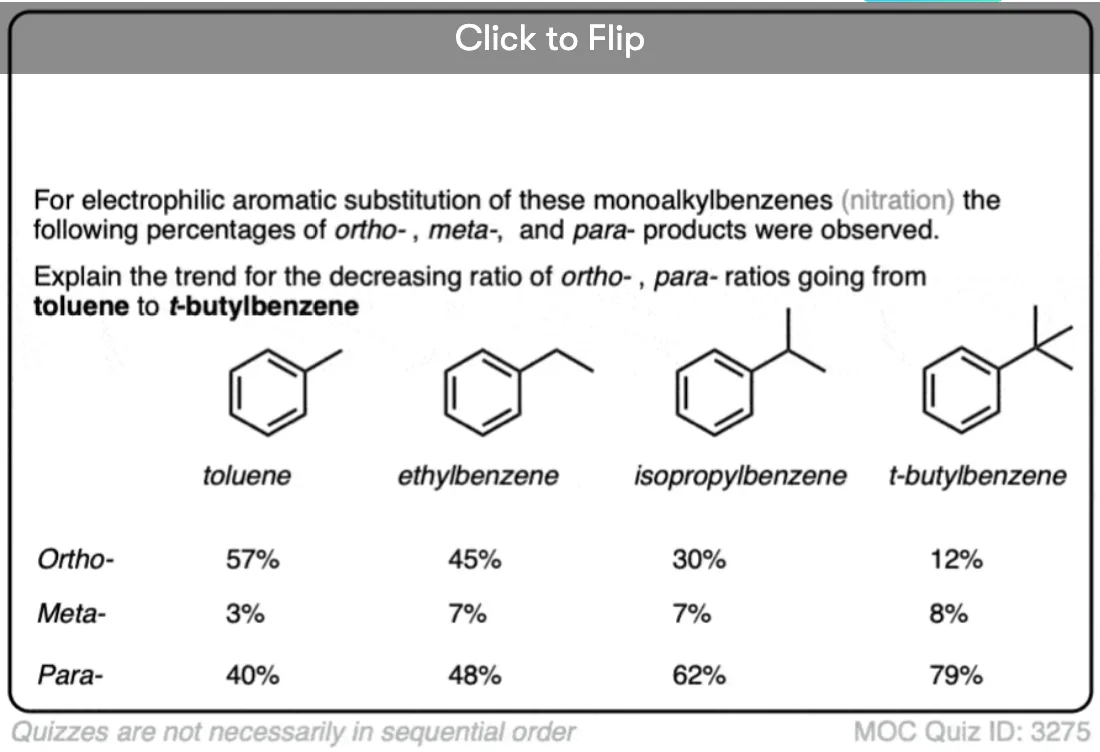
One question – why do you think that para- might be favored (e.g. 60%-70% yield for the para– product of nitration, above, versus 30-40% for ortho-) even though there are two ortho- positions and only one para- ?
Think about that for a minute, and we’ll address it below.
5. Case Study #2: A meta- director (CF3).
So what about CF3? Why does it give meta- products? Let’s do the same type of analysis.
Here’s the pathway leading to the ortho- product. As with the ortho- intermediate above, we can draw three resonance forms placing the carbocation on C1, C3, and C5, respectively.
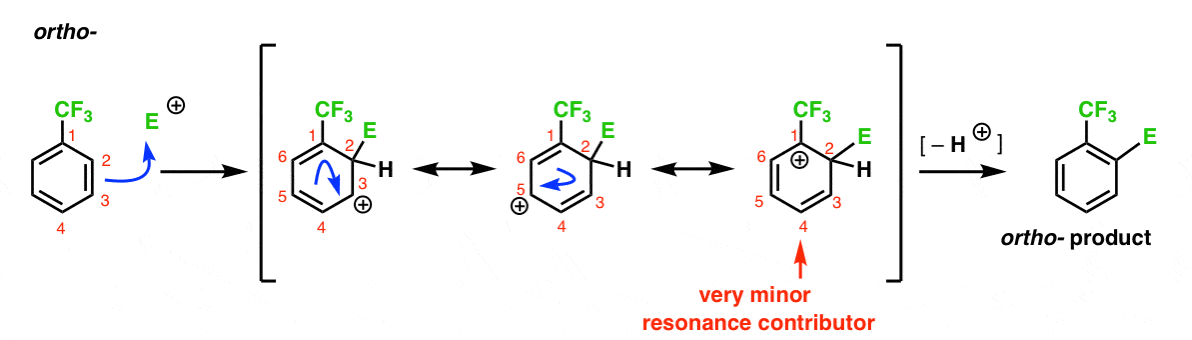
So what’s different in this case?
What’s different is the presence of a strong electron withdrawing group (CF3) at C1, and this completely changes the ballgame.
As we’ve said, carbocations are destabilized by neighbors which withdraw electron-density. Therefore we would expect this to be a very minor resonance contributor, with the result that the positive charge is only delocalized over C3 and C5.
[Note: I’m avoiding saying that this is a “less stable” resonance form, because I want you to remember that resonance forms don’t really exist. What does exist is the resonance “hybrid”, which is built up of contributions from each of the resonance forms. You won’t be struck down by lightning if you refer to “more stable” or “less stable” resonance forms in casual conversation, so long as you always keep the “imaginary” nature of resonance forms in mind].
We’ve noted that CF3 is a meta- director. It’s reasonable to think that there’s something about the meta– that makes it particularly stable. By analyzing the intermediate for the meta– product below, can you see why?
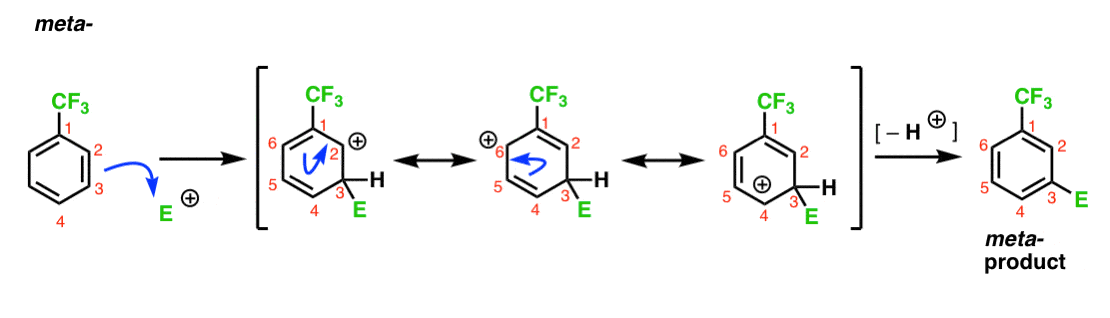
Hmmm. There’s no intermediate where a carbon has a full octet. There are no particularly stable resonance forms.
It’s kind of “meh”, isn’t it?
6. What We Call “meta– Directors” Are More Like “ortho-, para– Avoiders”
On the other hand, there aren’t any particularly unstable resonance forms either. Since the positive charge is localized on C2, C4, and C6, a situation where the positive charge is directly adjacent to the electron-withdrawing CF3 group is avoided. And the positive charge is thus delocalized nicely throughout the ring, unlike in the ortho- situation, above.
So it’s a “less bad” intermediate than the ortho-.
Finally, the para- intermediate has the same problem as the ortho- ; an intermediate with a positive charge on C1, adjacent to the CF3.
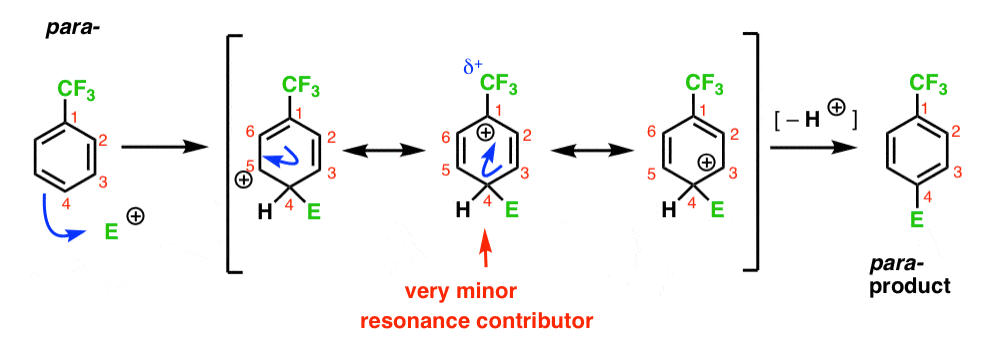
This results in only two important resonance forms, which leads to a less delocalized (and therefore less stable) carbocation intermediate.
Bottom line: the meta carbocation intermediate is more stable than either the ortho– or the para- intermediates.
But it’s not because the substituent itself has any stabilizing effect on the meta– intermediate. [And as we’ve seen, all meta- directing substituents are deactivating, meaning that they decrease the rate of reaction relative to H].
I’d argue that it’s more helpful to think of the meta- as less unstable than the ortho- and para– .
In this way, it’s not so much that CF3 is a meta- director, so much that it is an “ortho– , para– avoider. “
7. The Reaction Energy Diagram For A meta- Director
Here’s a sketch of the reaction energy diagram:
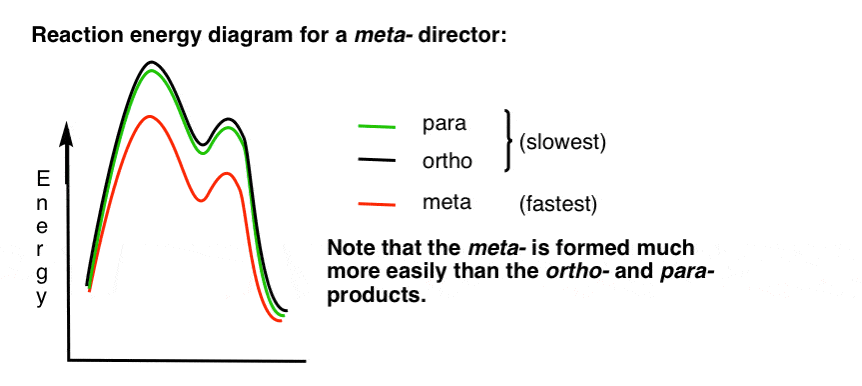
Alright.
Back to our original question. So which carbocation is more stable?
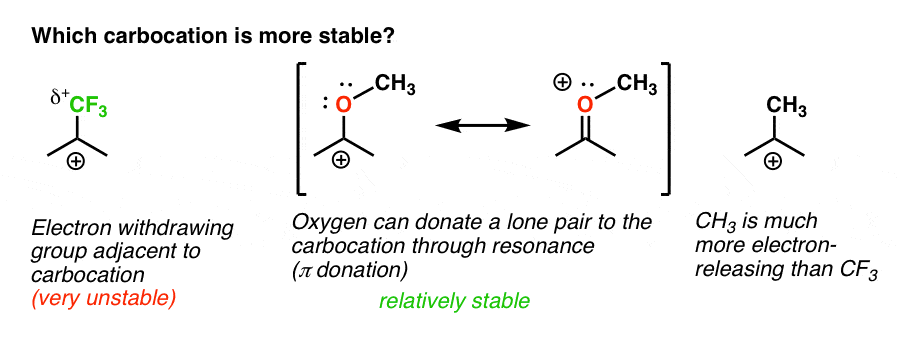
Clearly, the carbocation with an adjacent oxygen bearing a lone pair is far more stable than a carbocation adjacent to an electron-withdrawing group like CF3.
And that comprises the difference between an ortho-, para- director like OCH3 and a meta- director like CF3.
It’s worth noting that most alkyl groups (such as CH3 ) lack a lone pair, but are still ortho-, para- directors. This is consistent with everything we’ve learned before about how alkyl groups are generally stabilizing for carbocations- recall that carbocation stability generally increases with substitution [primary (least stable) < secondary < tertiary (most stable). ]
So about those para– products…
Why are para- products produced at a greater rate than ortho- ?
The answer is steric hindrance.
Attack at the para position is less encumbered by the substituent than attack at the ortho- positions are.
8. So Why Are Halogens Ortho-, Para- Directors?
This leaves us with the somewhat tricky example of halogens.
Why are fluorine, chlorine, bromine, and iodine ortho-, para- directors even though they are deactivating groups?
The answer should come as no surprise if you’ve been following along, but we’re going to leave this until the next post, because this one is long enough already.
Next post: Why are halogens ortho-, para- directors?
Notes
Note 1. Or hyperconjugation, which most textbooks (with the notable exception of Maitland Jones) generally avoid.
Note 2. It’s more correct to say that ortho- and para- products dominate because the transition states leading to these products are lower in energy, rather than the energies of the intermediates themselves. After all, it’s the energy of the transition states which determines the activation barrier, and therefore the reaction rate.
Note 3. Useful table with yields of meta– products for various substituted benzenes:
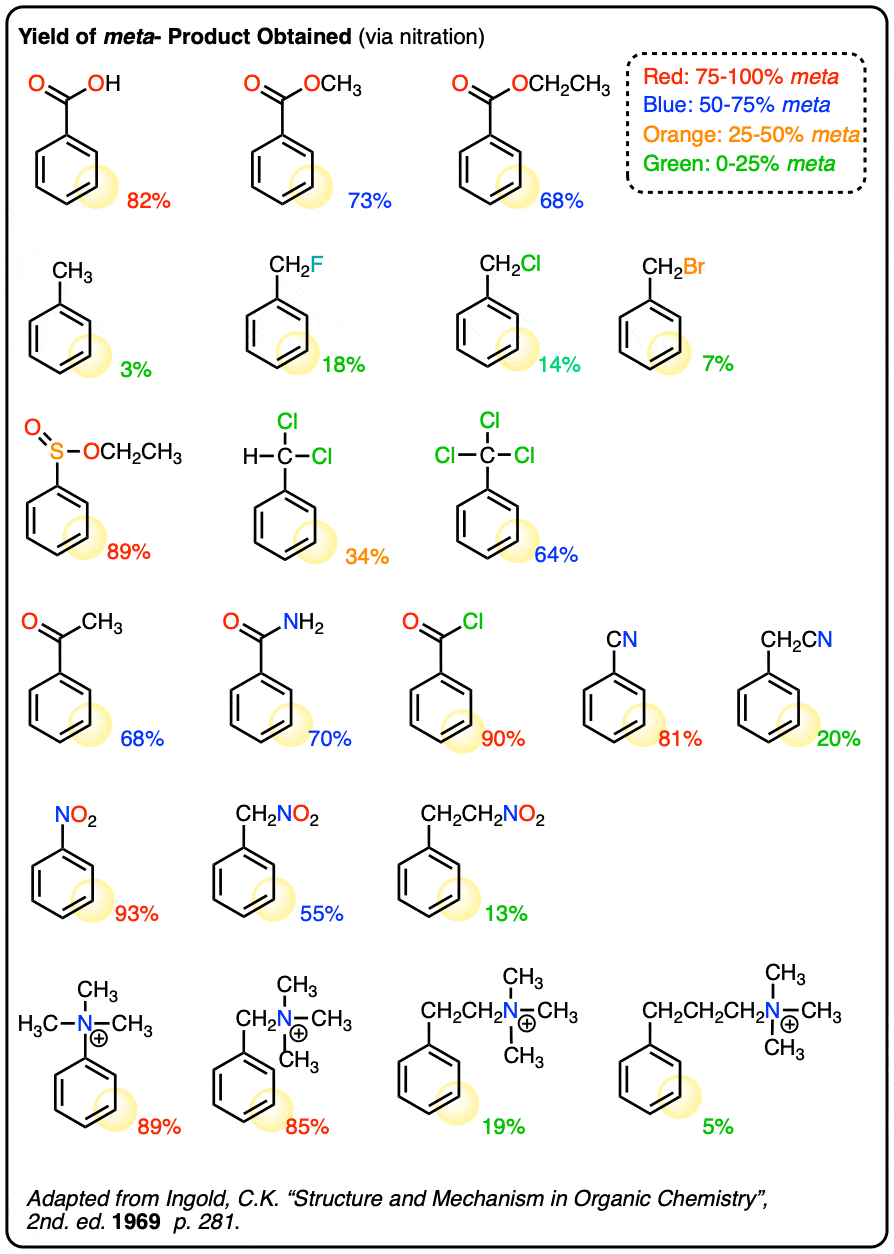
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
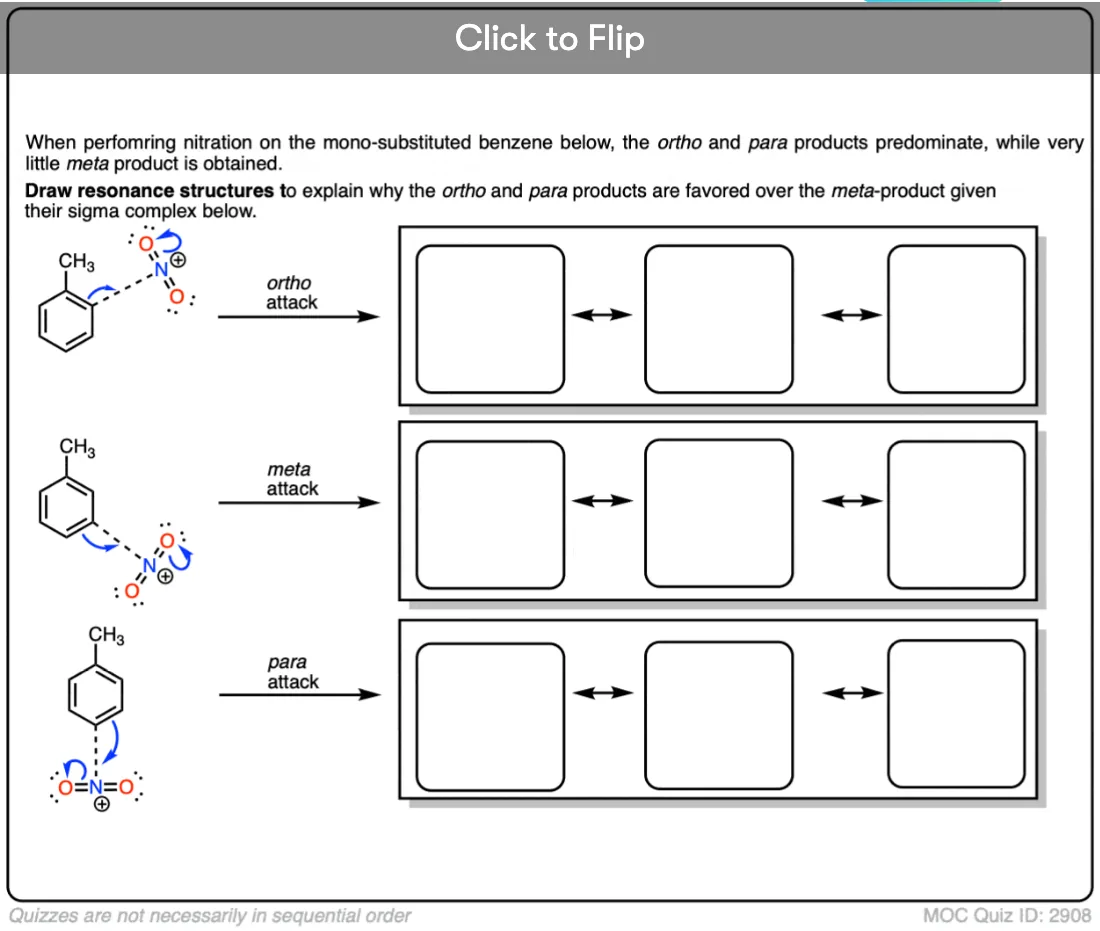
Become a MOC member to see the clickable quiz with answers on the back.
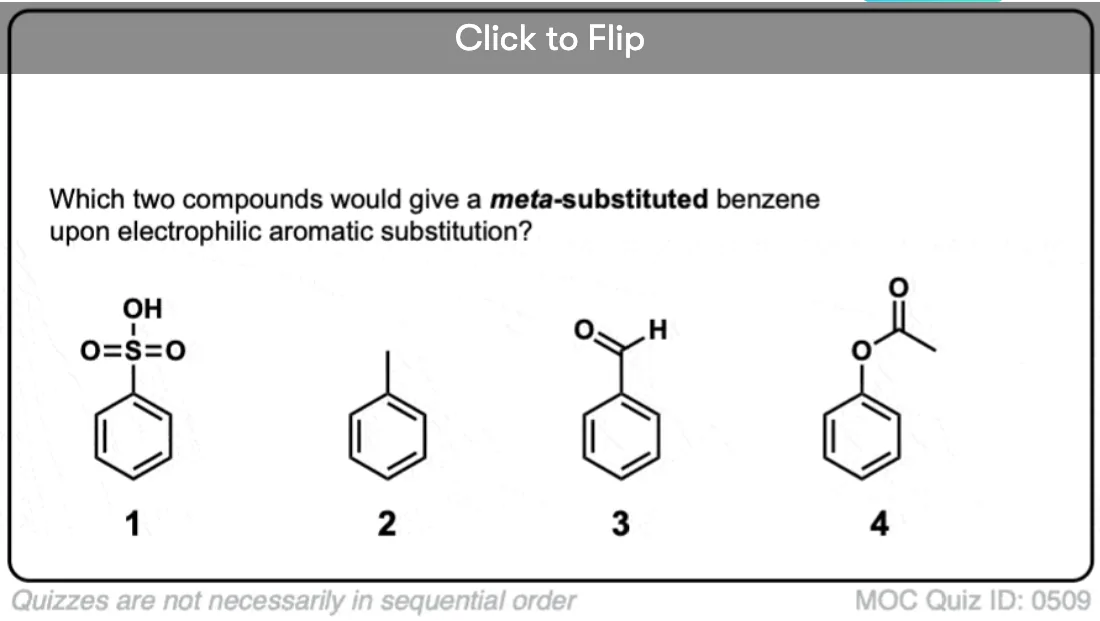
Become a MOC member to see the clickable quiz with answers on the back.
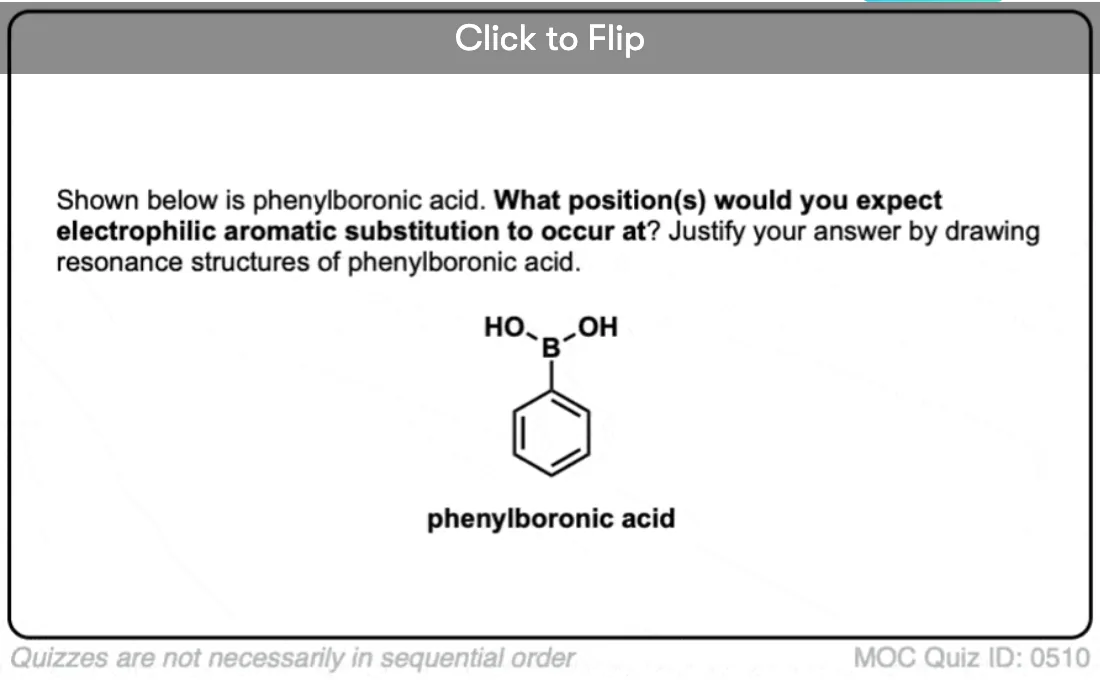
Become a MOC member to see the clickable quiz with answers on the back.
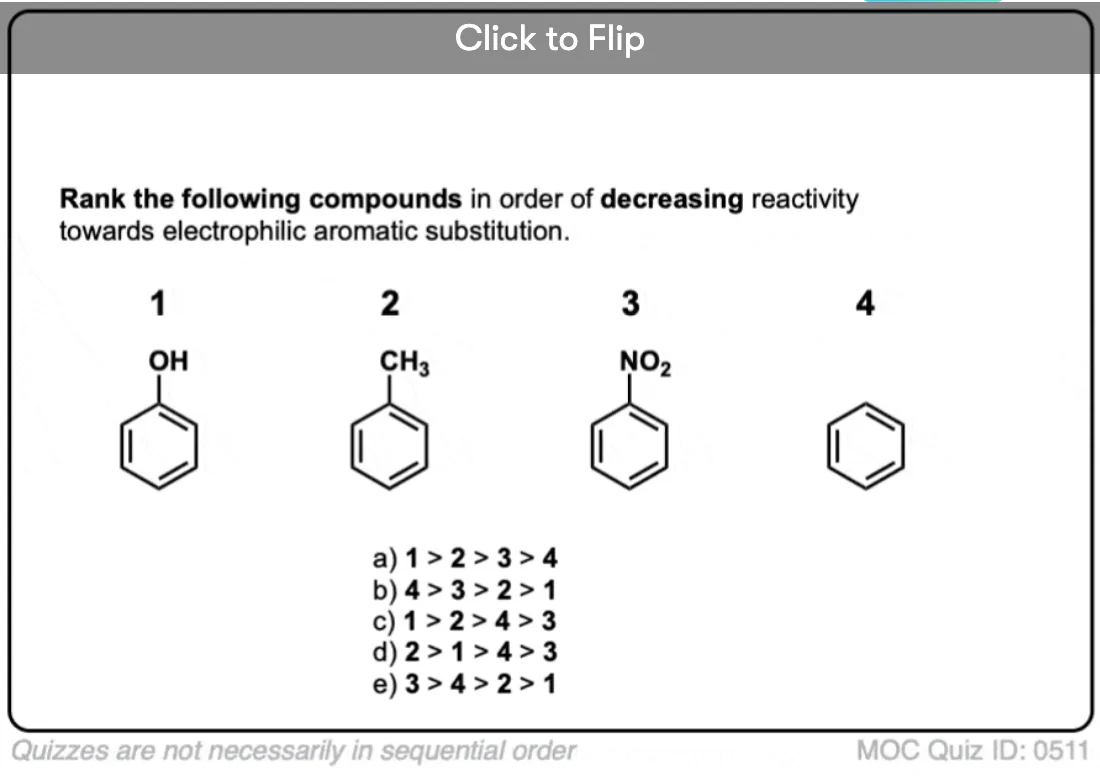
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- A. F. Holleman, Die direkte Einführung von Substituenten in den Benzolkern
Rec. Trav. Chim. Pays-Bas 1910, 12, 455-456
DOI: 10.1002/recl.19100291205
A.F Holleman from 1910 said that ortho–para orientation is associated with activation and meta orientation with deactivation. - —The nature of the alternating effect in carbon chains. Part XXIII. Anomalous orientation by halogens, and its bearing on the problem of the ortho–para ratio, in aromatic substitution
Christopher Kelk Ingold and Charles Cyril Norrey Vass
J. Chem. Soc. 1928, 417-425
DOI: 10.1039/JR9280000417
This paper discusses directing effects in 1,2-dihalobenzenes. - Some observations concerning steric hindrance and the effects of substituents on the ortho : para ratio in aromatic substitution
P. B. D. de la Mare
J. Chem. Soc. 1949, 2871-2874
DOI: 10.1039/JR9490002871
In this paper, de la Mare discusses various factors that may account for a predominance of para over ortho selectivity, after dividing the yield of ortho product by 2 (since there are 2 ortho positions but only 1 para position in monosubstituted benzenes). - Volume effects of alkyl groups in aromatic compounds. Part V. The monosulphonation of p-cymene
R. J. W. Le Fèvre
J. Chem. Soc. 1934, 1501-1502
DOI: 10.1039/JR9340001501
In p-cymene, the major product obtained upon electrophilic sulfonation is the 2-product (ortho to the methyl group), likely due to sterics. - Effects of Alkyl Groups in Electrophilic Additions and Substitutions
COHN, H., HUGHES, E., JONES, M. and PEELING, M. G.
Nature 1952, 169, 291
DOI: 1038/169291a0
This paper has data comparing the nitration of t-butylbenzene and toluene. T-butylbenzene is much more p-directing than toluene (79.5% para for t-butylbenzene vs. 40% para for toluene), which is likely due to sterics (ortho approach is blocked by the bulkier t-butyl group). - Distribution of Isomers in the Mononitration of Ethyl- and Isopropylbenzene. Further Evidence for a Steric Effect in Isomer Distribution
Herbert C. Brown and W. Hallam Bonner
Journal of the American Chemical Society 1954, 76 (2), 605-606
DOI: 10.1021/ja01631a084
Table II in this paper illustrates that the ortho product obtained from nitration of monoalkylbenzenes decreases as the alkyl group gets larger (e.g. t-butylbenzene yields very little ortho product upon nitration compared to toluene). - —The nature of the alternating effect in carbon chains. Part XXII. An attempt further to define the probable mechanism of orientation in aromatic substitution
Christopher Kelk Ingold and Florence Ruth Shaw
J. Chem. Soc. 1927, 2918-2926
DOI: 10.1039/JR9270002918
An early paper by the influential Physical Organic Chemist, Prof. C. K. Ingold, stating that halogenobenzenes are inductively electron-withdrawing but simultaneously resonance-stabilizing. - The Anomalous Reactivity of Fluorobenzene in Electrophilic Aromatic Substitution and Related Phenomena
Joel Rosenthal and David I. Schuster
Journal of Chemical Education 2003, 80 (6), 679
DOI: 1021/ed080p679
A very interesting paper, suitable for curious undergrads, and discusses something that most practicing organic chemists will know empirically – fluorobenzene is almost as reactive as benzene in EAS or Friedel-Crafts reactions, which is counterintuitive when one considers electronic effects! - Organic chemistry
N. Haworth, C. K. Ingold, T. A. Henry
Ann. Rep. Prog. Chem. 1926, 23, 74-185
DOI: 10.1039/AR9262300074
An early paper discussing directing effects in EAS. Pp. 136 and 137 contain figures that depict the flow of electrons. Pg. 140 discusses meta-direction, which occurs for the ‘opposite’ reasons as o/p-direction by alkyl groups.
Thank you for great explanations! In this article, the reason why some substituents “direct” the electrophile to certain position is explained by resonance. However, since resonance is an imaginary concept that doesn’t actually take place in actual interaction of molecules, I have been trying to explain every resonance-induced stability with MO theory.
Well… it just didn’t work this time. I tried to explain the stability with idea based on pi system that the longer the conjugation, the more stable it is, and it could explain ortho and meta position (ortho has 6 atoms in pi system, where meta has two cases of 5 atoms in pi system) but it couldn’t explain the case of para position (one case of 4 atoms, one case of 5 atoms). And I wasn’t even sure that if using the pi system was the right approach(also having many different ways of conjugation that has different number of atoms in the system just felt so wrong).
So I went on to molecular calculation using MolCalc, and got the result that matches our expectations, but I still couldn’t explain it with MO theory.
Even if it is just what Schrödinger equation and Hückel method says, there still has to be the reason for that… and I just can’t find it but I’m dying to find it. Could you help?
Hi, I would highly recommend Ian Fleming’s “Frontier Orbitals and Organic Chemical Reactions” for a discussion of MO theory. Mostly covers pericyclic reactions, but it also does a frontier orbital treatment of electrophilic aromatic substitution.
https://archive.org/details/frontierorbitals0000flem
Is (-NHCOPh) an Ortho/Para-directing group like (-NHCOCH3) ?
Will be obliged if you can give some explanation.
Yes. Same effect. Lone pair on nitrogen can stabilize an adjacent carbocation.
thank you for your nice explanation. but I have a question. why you said ‘most’ alkyl groups not all of them lack a lone pair?
A carbanion would have a lone pair.
*xylen :)
thanks for the article
why ortho-zailen and para-zailen tend to change into meta-zailen in 80 degrees Celsius in presence of HCL and AlCl3
but in 0 degree Celsius it doesn’t happen
how it happens?
depends on HCL or something else?
Sir it is very beautiful explanation i understood complete concept of ortho , meta and para . Suprb explanation. Thankyou very much sir for .
On the fourth resonance structure for the para addition of any E+ to the benzene ring with OCH3, wouldn’t there be a second pair of electrons on the Oxygen after the arrows push them from the double bond up to the Oxygen?
Yes, you are correct. I will put this in the corrections queue.
okay I just found your page explaining this exact problem never mind! Thank you!
if you have a benzene ring with two substituents ( lets say OCH3 and an ethyl) that are both ortho/para directors which are on the 1,3 carbons and you react it with Br2/FeBr3, would you expect to have major products be with the Br on the ortho and meta positions of the higher ranked directing and activating substituent (OCH3) or would the major products be the Br on the ortho and meta positions for both substituents?
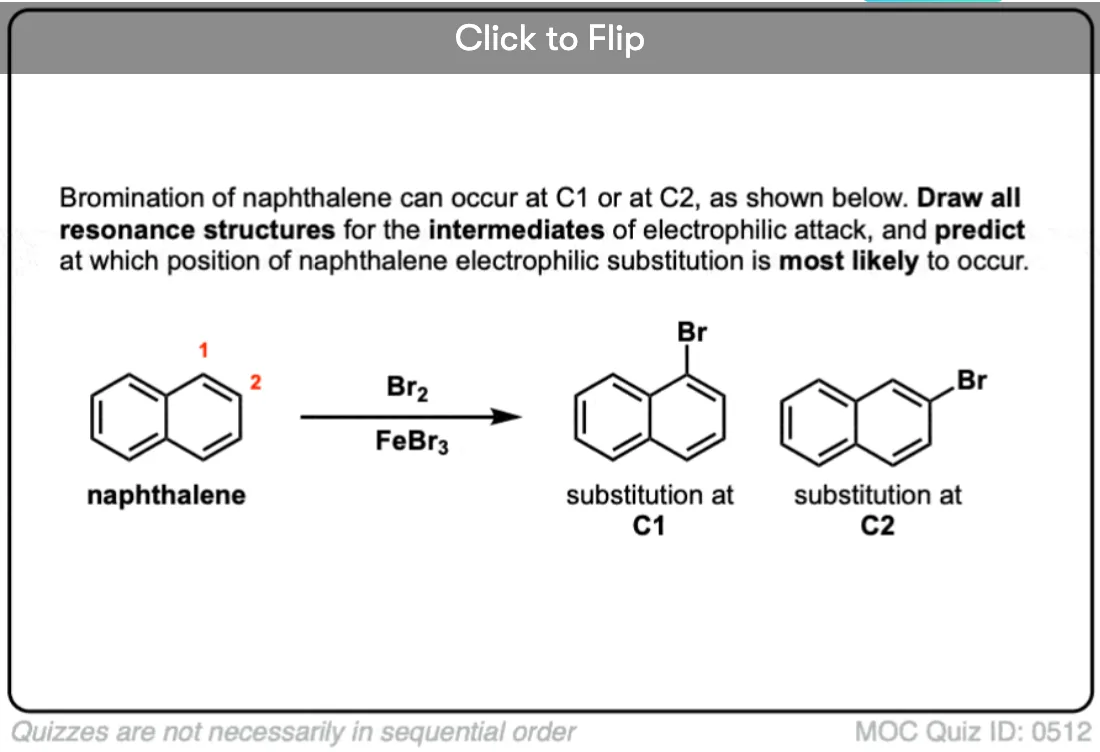
How about larger aromatic systems, such as naphthalene and anthracene. Do the directing effects/rules still hold?
It can be much more complicated. For naphthalene, for instance, there are two possible products, and it turns out that substitution on the 1-position is kinetically favored whereas substitution on the 2-position will occur under equilibrating (thermodynamic) conditions. In anthracene, the central aromatic ring (9- and 10- positions) is the easiest to break (and can even undergo diels-alder reactions).
I had doubt about these all directors but now my mind is clear thank you for explaining it in a simple language
Glad to hear it Rutuja – thanks for leaving the comment.
Beautiful explanations and illustrations! Thanks James!
Hey, glad you liked it Dave. Hope it was useful.