Elimination Reactions
Elimination (E1) Reactions With Rearrangements
Last updated: May 28th, 2026 |
Elimination Reactions (E1) That Occur With Rearrangements – Hydride Or Alkyl Shifts
Where there are carbocations (see last post), rearrangement reactions are never far behind. Our old friends have come back for a short visit in this chapter on elimination reactions.
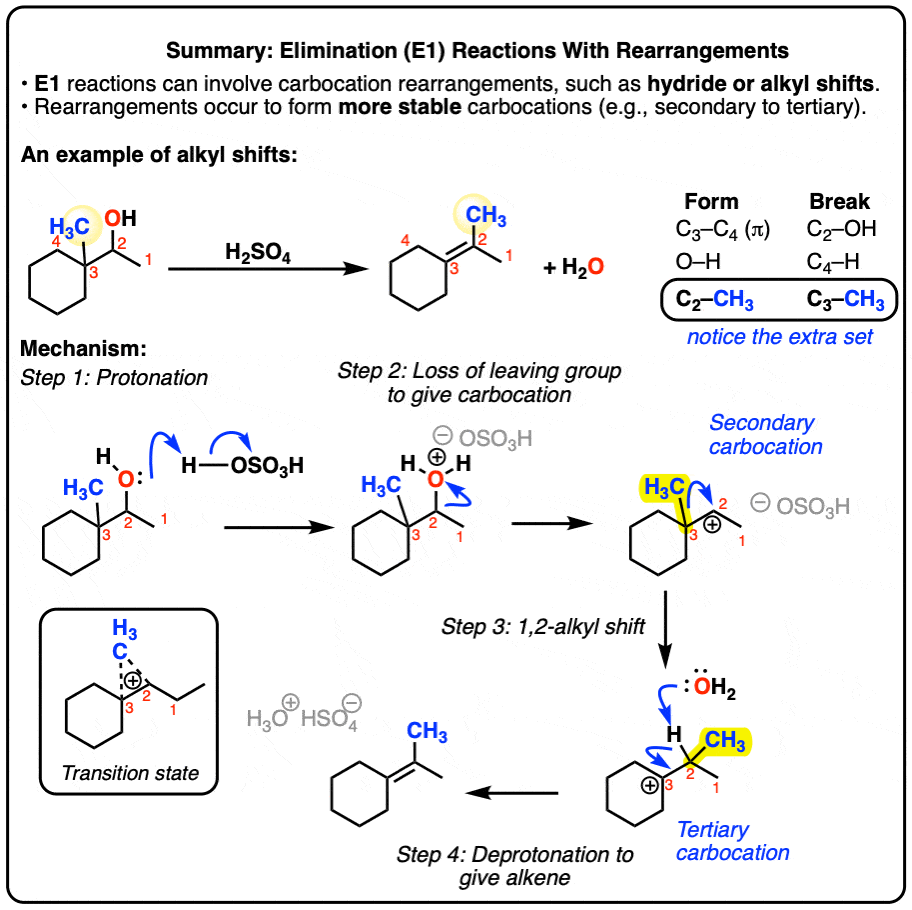
Table of Contents
- What’s Weird About This Elimination Reaction?
- Elimination (E1) With Hydride Shift
- Elimination (E1) With Alkyl Shift
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. What’s Weird About This Elimination Reaction?
One last (weird) reaction to show you with regard to elimination reactions. Can you see what’s weird about it?
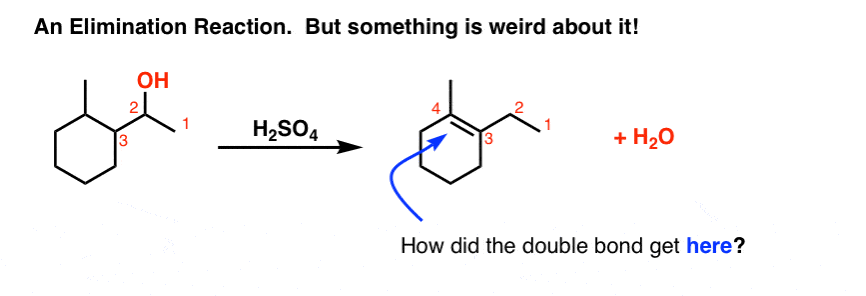
How did that double bond get over there? Normally when elimination occurs, we remove a hydrogen from the carbon adjacent to the leaving group. But here, something extra has taken place.
2. Elimination (E1) With Rearrangement: Hydride Shift
Let’s look at all the bonds that form and the bonds that break so we can track down exactly what happens:
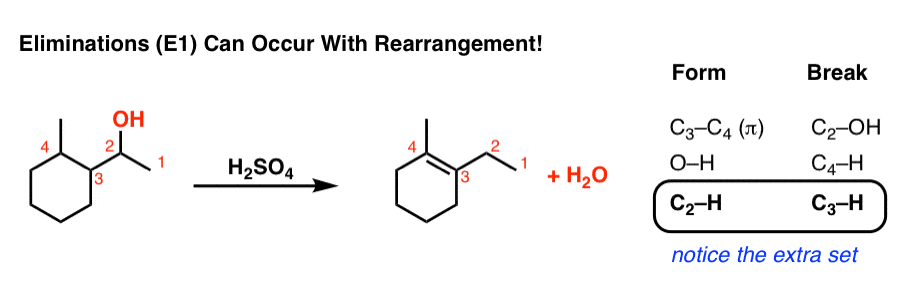
Notice how it differs from a typical elimination reaction? Sure, we’re forming C-C (π), and breaking C-H and C-OH, but we have an extra C-H that forms and an extra C-H that breaks.
This is a sure sign of a rearrangement step!
So what’s going on here?
Well, we start by protonating the alcohol. This allows for water to leave in the next step, which is going to form a carbocation.
Here’s the thing: the carbocation is secondary, and we’re adjacent to a tertiary carbon. So if the hydrogen (and its pair of electrons) were to migrate from C3 in our example to C-2, we’d now have a tertiary carbocation, which is more stable.
Then, a base (water in this example) could remove C-H, forming the more substituted alkene (the Zaitsev product in this case). And that’s how the alkene ends up there.
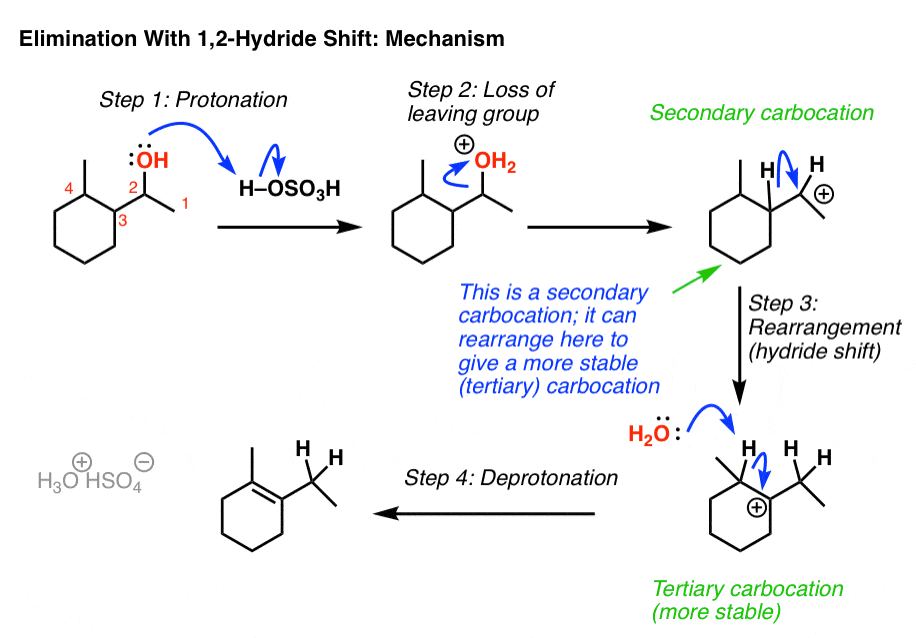
OK. So that’s one mystery solved.
3. Elimination (E1) With Rearrangement: Alkyl Shift
You might remember that these types of rearrangements can occur in SN1 reactions too. And if you read that post, you might recall that in addition to shifts of hydrogen (“hydride”, because there’s a pair of electrons attached) we can also have alkyl shifts. Here’s a final example.
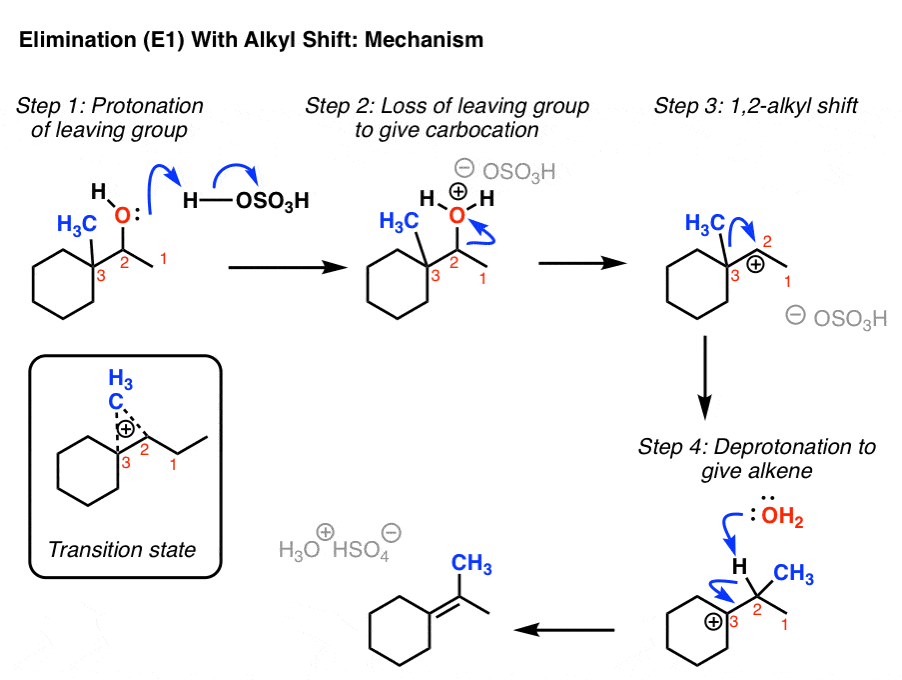
This pretty much does it for elimination reactions.
In the next series of posts, let’s go though one of the biggest questions students struggle with.
Now that we’ve gone through substitution and elimination reactions, HOW DO WE DECIDE WHICH ONE IS GOING TO OCCUR IN EACH SITUATION?
Great question. That’s next.
Next Series, post 1: SN1/SN2/E1/E2 Decision (1) – The Substrate
Notes
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
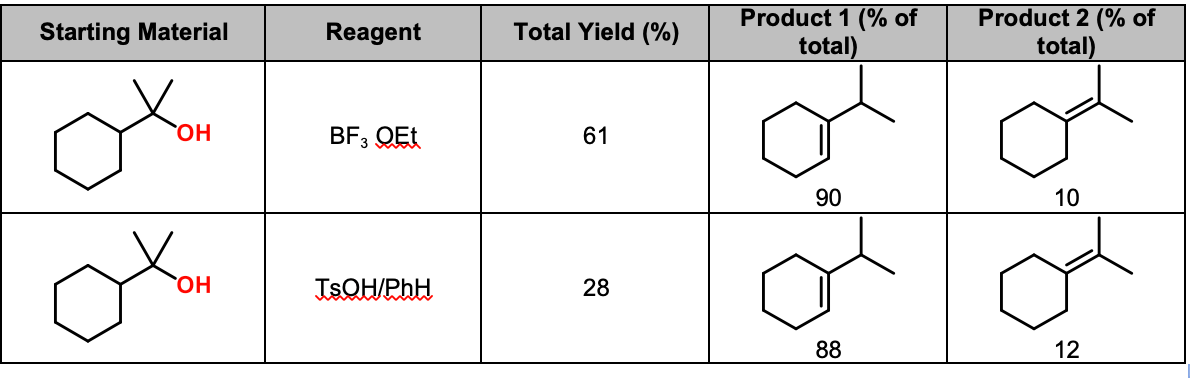
- Check this paper out for some very clean, classic examples of dehydration with alkyl shift. The authors take 1-cyclohexyl-1-methylethanol and treat it with either TsOH/benzene or BF3•OEt2. You might think that they’d get the tetrasubstituted olefin, but the dominant product is the trisubstituted alkene (90:10). Reason is greater acidity of the axial C-H bonds which are aligned with the intermediate carbocation.
BF3·OEt2 Promotes Fast, Mild, Clean and Regioselective Dehydration of Tertiary Alcohols.
Posner, G. H.; Shulman-Roskes, E. M.; Oh, C. H.; Carry, J.-C.; Green, J. V.; Clark, A. B.; Dai, H.; Anjeh, T. E. N.
Tetrahedron Lett. 1991, 32 (45), 6489–6492.
DOI: 10.1016/0040-4039(91)80200-P
- Neighboring hydrogen, isotope effect, and conformation in solvolysis of 3-methyl-2-butyl p-toluenesulfonate
S. Winstein, J. Takahashi
Tetrahedron 1958, 2 (3-4), 316-321
DOI: 10.1016/0040-4020(58)88053-9
3-methyl-2-butyl-tosylate is an example of a system that rearranges readily under solvolysis, which is illustrated in Table 3. - Mechanisms of elimination reactions. XIII. Effect of base, solvent, and structure on product ratios in elimination reactions of some secondary tosylates
Irving N. Feit and William H. Saunders
Journal of the American Chemical Society 1970, 92 (6), 1630-1634
DOI: 1021/ja00709a035
Towards the end, this paper states, “An interesting sidelight of the E1 reactions is that the olefins resulting from hydride shift with 2-methyl-3-pentyl and 3-methyl-2-butyl tosylates, 2-methyl- 1 -pentene, and 2-methyl-l-butene, respectively, are found in increasing amounts along the solvent series n-BuOH <s-BuOH < t-BuOH.” - REARRANGEMENT STUDIES WITH C14: IX. THE FORMOLYSIS OF METHYL-C14-ISOPROPYLCARBINYL p-TOLUENESULPHONATE
J. Finlayson and C. C. Lee
Can. J. Chem. 1960, 38, 787-792
DOI: 10.1139/v60-114
Another study of the same system from Ref. 1, this uses C14 labeling to study the course of the rearrangements – see pg. 700. - Über die Pinakolinumlagerung cyclischer Verbindungen
Hans Meerwein, Walter Unkel
Lieb. Ann. Chem. 1910, 376 (2), 152-163
DOI: 10.1002/jlac.19103760203
This paper by Hans Meerwein, an early pioneer in the study of carbocations and acid-catalyzed rearrangements, is on pinacol and semipinacolic rearrangements. In this paper, he demonstrates that 2,2-dimethylcyclohexanol is converted by acid into a mixture of isopropylidenecyclopentane and 1,2-dimethylcyclohexene. - Über Ringveränderungen bei der Wasserabspaltung aus alicyclischen Alkoholen
Hans Meerwein
Lieb. Ann. Chem. 1918, 417 (2-3), 255-257
DOI: 10.1002/jlac.19184170205 - The Common Basis of Intramolecular Rearrangements. II.1 The Dehydration of Di-tert-butylcarbinol and the Conversion of the Resulting Nonenes to Trimethylethylene and Isobutylene
Frank C. Whitmore and E. E. Stahly
Journal of the American Chemical Society 1933, 55 (10), 4153-4157
DOI: 1021/ja01337a042
Prof. F. C. Whitmore is mentioned in reviews on the history of carbocation chemistry, as he was the first person to suggest that carbocations be represented with an ‘open sextet’ of electrons and draw them as such. - Mechanism of substitution at a saturated carbon atom. Part XXIX. The rôle of steric hindrance. (Section D) the mechanism of the reaction of neopentyl bromide with aqueous ethyl alcohol
I. Dostrovsky and E. D. Hughes
J. Chem. Soc., 1946, 166-169
DOI: 10.1039/JR9460000166
Under the conditions used here (aqueous ethanol with NaOH), about 36% of rearranged olefin (trimethylethylene) was obtained from neopentyl bromide. Likely higher yields of olefin would be possible if acidic conditions are employed, which favor formation of carbocations. - Lanostane to Cucurbitane Transformations.
Edwards, O. E.; Kolt, R. J. .
Can. J. Chem. 1987, 65 (3), 595–612.
DOI: 10.1139/v87-104
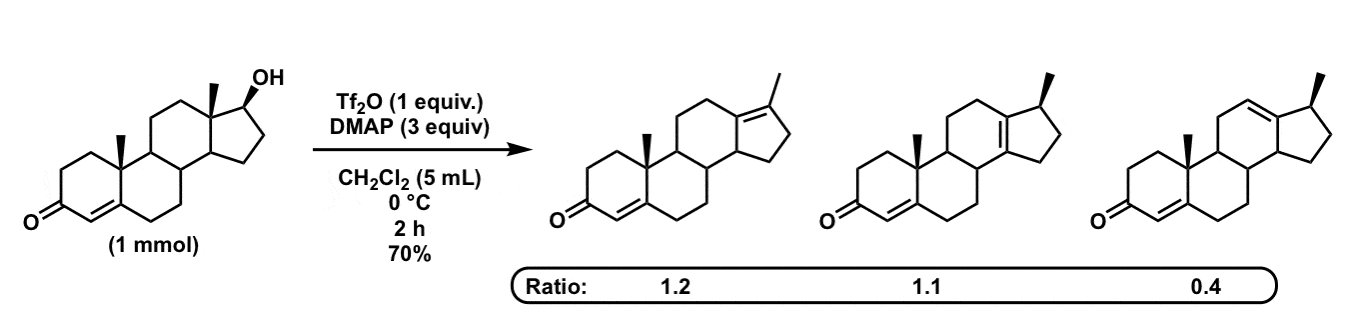
The authors take a very rigid system (the steroid lanostane) containing a tertiary alcohol and observe what happens when it is dehydrated with strong acid (H2SO4 – AcOH – Ac2O, so-called, “Westphalen conditions”). After loss of water, a methyl shift from the adjacent quaternary carbon is observed (NOT a hydride shift, interestingly!) and the authors compare the ratio of alkenes (trisubstituted vs tetrasubstituted). Ratios are greatly affected by subtle electronic effects of remote groups. - A Mild One-Pot Method for Conversion of Various Steroidal Secondary Alcohols into the Corresponding Olefins.
Kumar, R. R.; Haveli, S. D.; Kagan, H. B.
Synlett 2011, 2011 (12), 1709–1712.
DOI: 10.1055/s-0030-1260803
Slightly different steroid system, giving mixture of rearrangement + elimination products.
In neopentylchloride and isopropylchloride .Which one undergo fast E1 elimination Reaction ?
I think you can figure this one out.
Is rearrangement requires energy ?
Any time there is a bond-breaking step, there will be an energy barrier. So, yes.
Can E1 reaction of secondary alkyl halide have carbocation rearrangement as well?
Yes, secondary alkyl halides should be the most common case where you would observe carbocation rearrangements.
Hi , thanks for your great post, i am 16 but i love chemistry and i am going to paticipate in IChO.but a question, if both alkyl and hydrid shift is possible, what will happen?
Hydride shifts, generally, will perform 1,2 shifts much faster than alkyl shifts. First, think about the driving force for a 1,2 shift. The driving force is formation of a less substituted carbocation. If you have a secondary carbocation adjacent to a tertiary carbon, and the tertiary C-H migrates, you obtain a (more stable) tertiary carbocation. However if an alkyl group migrates, you obtain an (equally stable) secondary carbocation.
Exceptions can occur. In the lab, there are cases where 1,2- alkyl shifts will happen preferentially, especially in rigid cyclic systems. In rigid, cyclic systems containing a free carbocation the groups likeliest to migrate are those which have their bonds aligned with the empty p-orbital; axial groups, in other words. See the references at the end of the post for some examples.