Organometallics
Organocuprates (Gilman Reagents): How They’re Made
Last updated: May 7th, 2026 |
How Gilman Reagents (Organocuprates) Are Made
- Gilman reagents (organocuprates, often written as “R2CuLi” are not made the same way as Grignard or organolithium reagents.
- Instead of being made from alkyl halides, they are made through the reaction of an alkyllithium with a copper salt (e.g. CuBr)
- The use of two equivalents of alkyllithium (R-Li) relative to copper (CuX) is important for forming the organocuprate
- If only one equivalent is used, the result is an “organocopper” reagent (R-Cu) which is a poor nucleophile.
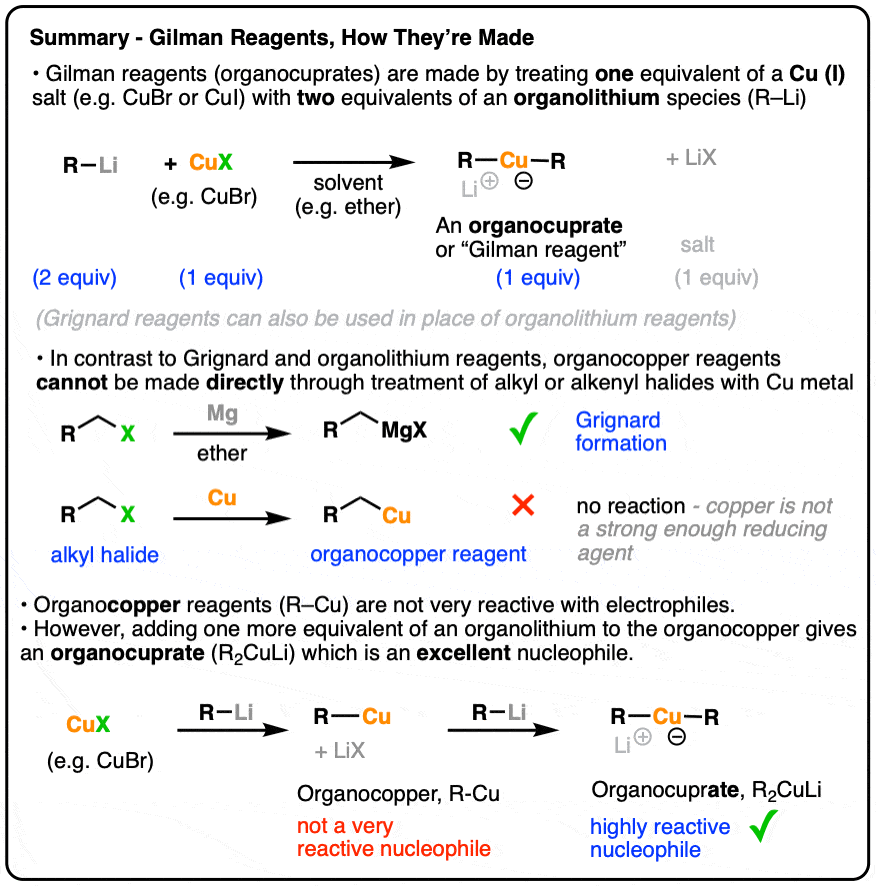
Table of Contents
- Organometallics: What About The Rest Of The Periodic Table?
- Can We Make Other Organometallics The Same Way We Make Grignards (And Organolithiums)?
- Formation of Organometallics From Alkyl Halides Involves Reduction
- Direct Formation of Organometallics From Cu Is Much More Difficult Than From Mg or Li
- A Work-Around: Transmetallation
- OrganoCOPPER reagents: Way Less Reactive Than Grignards
- OrganoCUPRATES: The Much-More-Reactive Cousins of OrganoCOPPER Reagents
- Summary: Organocuprates
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Organometallics: What About The Rest Of The Periodic Table?
In this whole series on organometallics so far has covered exactly TWO metals: lithium and magnesium (with a very brief head-nod to sodium and the comparatively useless Wurzt Wurtz reaction.)
There are dozens of other metals on the periodic table. What about organometallic compounds of them ?
Great question! (That’s also the first thing you say when someone asks you a question you want to dodge.)
Much as we’d love to, we just don’t have enough time to get into the full glory of organometallic chemistry in an introductory course. (If you’re curious, may I suggest The Organometallic Reader?)
In this post and the next we’ll give perfunctory treatment of some organometallic compounds of copper, and perhaps in a later post have something cranky to say about palladium, and call it a series.
Let’s get started.
2. Can We Make Other Organometallics The Same Way We Make Grignards And Organolithiums?
First, recall what we’ve said so far about how to make organometallic compounds: basically, take an organohalide, add magnesium or lithium, and stir. (See post: Synthesis of organolithium and Grignard reagents)
You might wonder: is it this easy for making other organometallics?
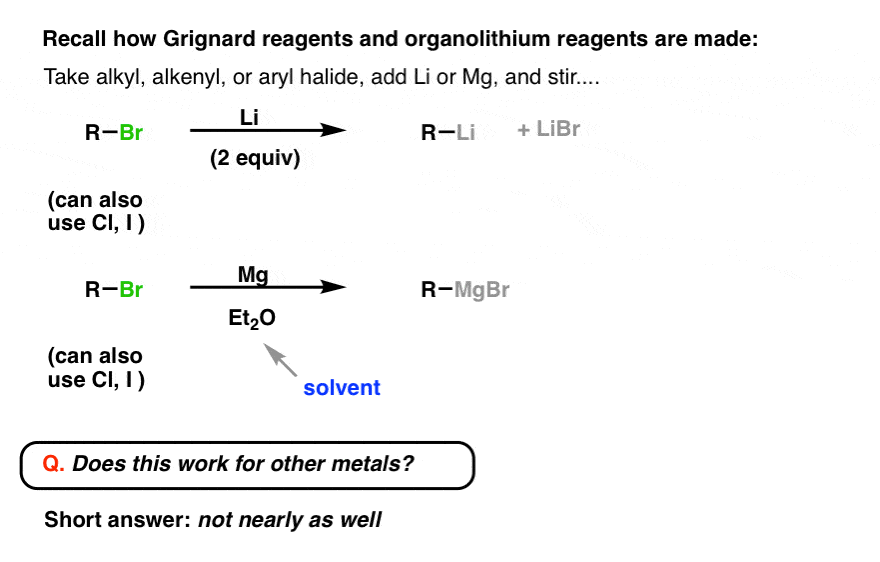
The short answer is: for most metals, it doesn’t work nearly as well.
3. Formation of Organometallics From Alkyl Halides Involves Reduction
Remember that formation of organometallic compounds from organohalides is a reduction reaction. Here it’s illustrated for the synthesis of organolithium compounds.
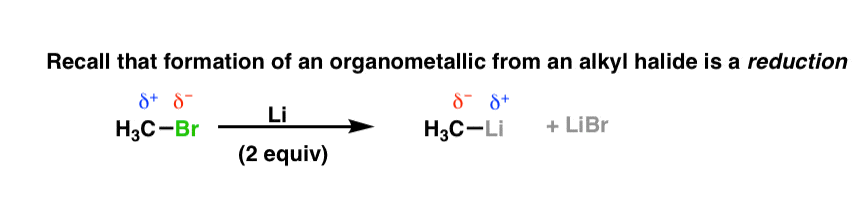
Notice how the polarity on the carbon changed from positive to negative? Not that we normally do such things, but if you keep track of the carbon oxidation state, you’d see that it changed from (–2) to (–4). (See post: Calculating the oxidation state of carbon)
This works well for lithium and magnesium because those metals are so easily oxidized. We can quantify this by looking at a table of oxidation potentials.
See how lithium, sodium, and magnesium are near the top of this table of oxidation potentials for metals? They’re easily oxidized – which means they are extremely strong reductants.

[note that these voltages are in aqueous solution, so take these numbers with a grain of salt for reactions performed in organic solvents].
As we move from lithium down the table to metals like nickel and copper, we should expect the reduction reaction should become progressively more difficult. And it is!
4. Direct Formation Of Organometallics From Metals Like Cu Is Much More Difficult Than For Mg or Li
In other words, direct formation of an organometallic from the organohalide and that metal [a process we call ‘insertion’ , or “oxidative addition”, FYI] is less favoured.
Look at copper for instance: the “direct reduction” doesn’t work nearly as well as it does for Li and Mg.
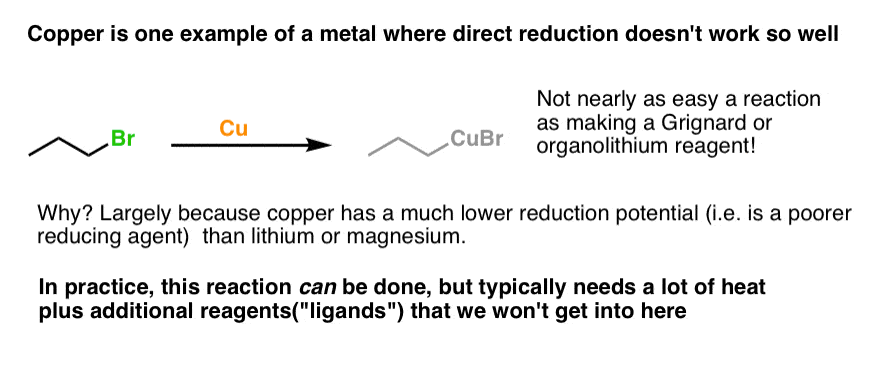
I don’t mean to imply that it can’t be done, but it generally requires heat and the addition of extra reagents that influence the oxidation potential of the metal (these are called ligands) in order to get this reaction to go.
5. A Work-Around: Transmetallation
Let’s say we really need to make an organometallic compound of copper. Can we get around this difficulty we generally experience in direct reduction? Yes – there’s a workaround.
What we can do instead is to start with a pre-made organometallic (such as an organolithium or Grignard reagent) where the reduction has already occurred. We can then add a copper (I) salt such as CuBr or CuCl.
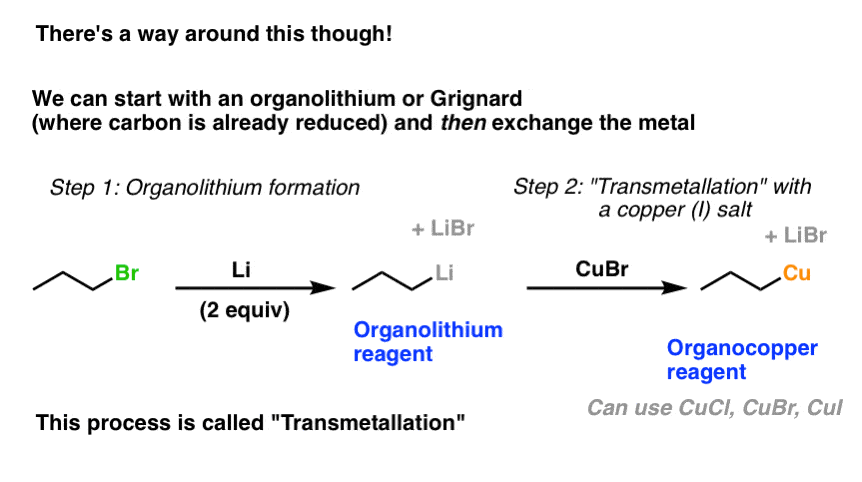
The result is displacement of the halide at copper by the carbon bound to lithium, and we form an organocopper reagent (plus a lithium salt). [You might wonder: why does this work at all? Note 1. ]
Yahoo for shortcuts!!
(Although not shown, this also works for Grignard reagents)
Now that we have a way to make organocopper reagents, the next question is: so what? What can we do with them?
6. Organocopper Reagents: WAY Less Reactive Than Grignards
In comparison to Grignard and organolithium reagents which are violently destroyed by water (and sometimes air) organocopper reagents are fairly sedate. For instance, there are organocopper reagents that you can leave out in the air without incident, like 1-hexynylcopper.
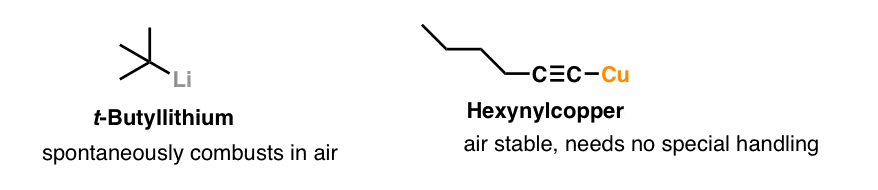
Also, organocopper reagents don’t add to carbonyls or epoxides like organolithium reagents or Grignard reagents do.

While organocopper reagents are interesting, it turns out that they have a chemical “cousin” that is even more versatile, reactive, and useful, and for that reason we will from this point further focus on these species: “organocuprate” reagents.
7. Organocuprates: Far More Reactive “Cousins” Of Organocopper Reagents
In the 1940’s Iowa chemist Henry Gilman discovered that adding one further equivalent of an organolithium reagent to an organocopper compound resulted in an “organocuprate” reagent, with two Cu–C bonds and is also comprised of a positive counter-ion (lithium in this case).
[Note that “-ate” at the end. Notice how many species that end in the name “ate” [e.g. sulfate, nitrate, tosylate] are negatively charged?]
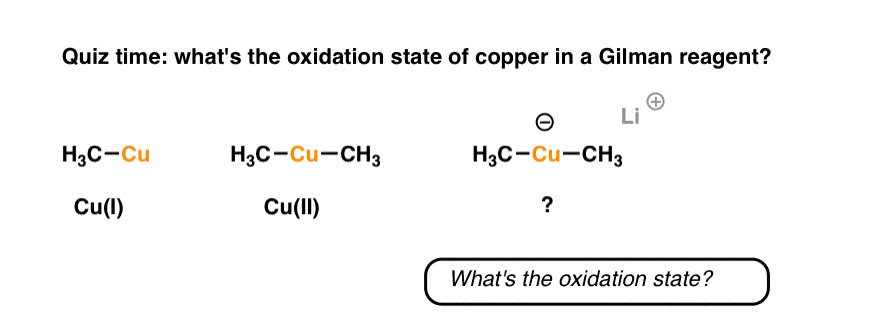
Organocuprates, with the general formula R2CuLi , have the same general pattern of reactivity as organocopper reagents, but are much more reactive. These compounds are commonly referred to as “Gilman reagents” in ol’ H.G.’s honour. [Why are they more reactive? Note 2].
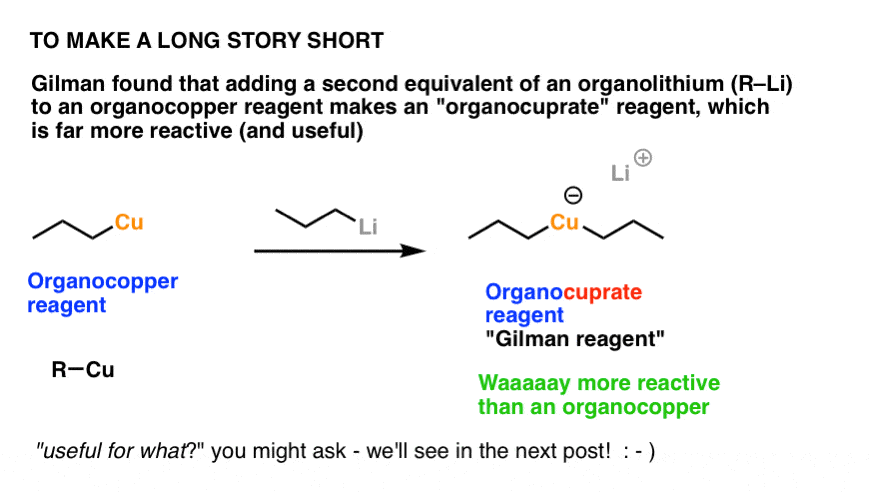
Just like organocopper reagents (and in contrast to Grignards) organocuprates do not generally add to aldehydes, ketones, or esters.
However, as we’ll see in the next post, they do participate in substitution and “conjugate addition” reactions – reactions that Grignards and organolithiums reagents typically don’t do. In this way (as we’ll see) they have a somewhat complimentary function.
8. Summary: Synthesis of Organocuprates
Let’s sum up. We saw that we generally don’t make organocopper reagents directly, but via organolithium or Grignard reagents. However, there’s an even more reactive “cousin” of organocopper reagents – “organocuprates” – that we can also make if we use a 2:1 ratio of organolithium to copper.
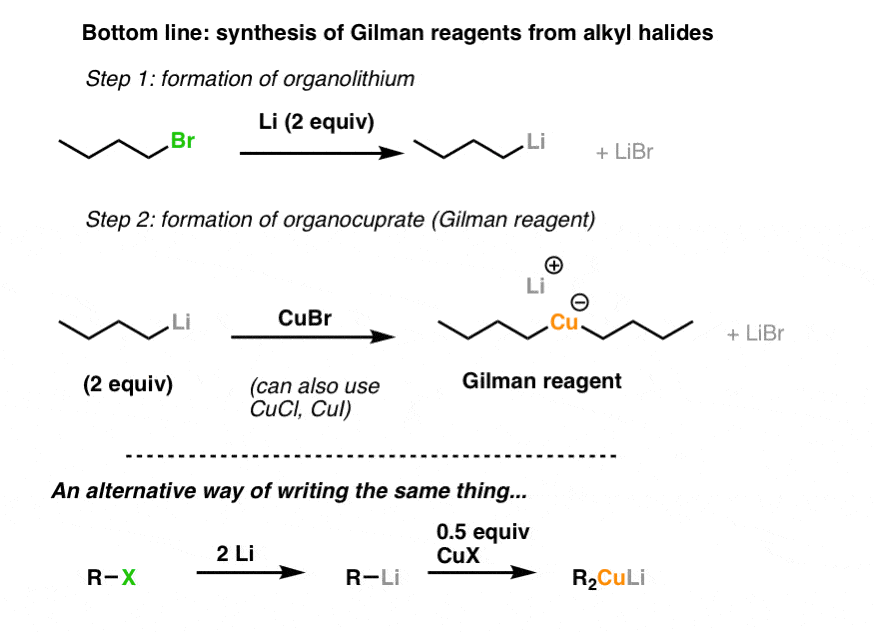
Note again: you need two equivalents of organolithium for every equivalent of copper.
Also note that you can use various different Cu(I) salts, as well as Grignard reagents.
So what’s so great about Gilman reagents, and what can they be used for?
We’ll talk about that in the next post. See you next time!
Next Post: Reactions Of Gilman Reagents
Notes
Bonus question for today. What’s the oxidation state of Cu in Gilman reagents?
Note 1. Why does transmetallation from lithium (or magnesium) to copper work? It’s a good question. In a nutshell, the carbon-copper bond is stronger than the C-Li or C-Mg bond, and that provides the driving force.
Why is the C-Cu bond stronger than C-Li ? That’s a difficult question to answer succinctly, but I’ll try.
You can think of bonding as having two components:
- an electrostatic component, where there is attraction between opposite charges (such as in salts)
- the overlap of molecular orbitals, resulting in shared electron density between atoms.
Bonding between a carbanion R– with Li+ is almost purely ionic (note the electronegativity difference of about 1.5) meaning that a significant portion of the bonding interaction is due to electrostatic interactions.
In contrast, bonding between C and Cu is considerably more covalent. There’s less of an electronegativity difference (Cu 1.9 vs. C 2.5) and the bonding is better described by metal-carbon bond overlap.
[waves hands] Generally, carbon forms stronger bonds through orbital overlap than via electrostatic interactions. This is largely because carbon is significantly more polarizable (the negative charge is “squishy”, or “diffuse” – we often use the shorthand “soft” to describe this behaviour) than a generally non-polarizable (“hard”) atom like lithium or oxygen. Likewise copper is also a fairly “soft” atom.
If you want to get super technical, for C-Cu the covalent term in the Klopman equation is large, and the coulombic term is small.
Note 2. Organocuprate reaagents are more reactive than organocopper reagents because after transferring R(-), the result will be a neutral organocopper reagent. After organocopper reagents transfer R(-) the result will be an electron-deficient copper ion.
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- The Preparation of Methylcopper and some Observations on the Decomposition of Organocopper Compounds
Henry Gilman, Reuben G. Jones, and L. A. Woods.
The Journal of Organic Chemistry 1952 17 (12), 1630-1634
DOI: 10.1021/jo50012a009
One of the first papers by Prof. Henry Gilman (U. of Iowa) on ‘Gilman Reagents’ – diorganocopper compounds. In this case he describes the preparation of dimethylcopper.Over the decades, a lotof people have carried out a lot of work in order to elucidate the true nature of copper intermediates and various copper species in reactions. There is a lot of literature in this area, and this is barely scratching the surface: - The composition of lithium methylcuprates in ether solvents
C. Ashby and John J. Watkins.
Journal of the American Chemical Society 1977 99 (16), 5312-5317
DOI: 10.1021/ja00458a015 - Mechanism of thermal decomposition of n-butyl(tri-n-butylphosphine) copper(I)
George M. Whitesides, Erwin R. Stedronsky, Charles P. Casey, and Joseph San Filippo Jr.
Journal of the American Chemical Society 1970 92 (5), 1426-1427
DOI: 10.1021/ja00708a067 - The Chemistry of Carbanions. XII. The Role of Copper in the Conjugate Addition of Organometallic Reagents1
Herbert O. House, William L. Respess, and George M. Whitesides.
The Journal of Organic Chemistry 1966 31 (10), 3128-3141
DOI: 10.1021/jo01348a012 - Autocatalytic decomposition of alkylcopper(I) species. Electron spin resonance spectrum of binuclear copper(O) intermediates
Jay K. Kochi, Keisuke Wada, and Masuhiko Tamura.
Journal of the American Chemical Society 1970 92 (22), 6656-6658
DOI: 10.1021/ja00725a055 - Chemistry of carbanions. XVII. Addition of methyl organometallic reagents to cyclohexenone derivatives
Herbert O. House and William F. Fischer Jr.
The Journal of Organic Chemistry 1968 33 (3), 949-956
DOI: 10.1021/jo01267a004
[quote]
Also note that you can use various different Cu(I) salts, as well as Grignard reagents
[/quote]
Does this statement mean that Grignard reagents can also be used in the formation of organocuprates? Does the magnesium participate as the cation in this particular case? If so, what would the handling requirements be for a Grignard reagent-derived organocuprate?