Alcohols, Epoxides and Ethers
The Williamson Ether Synthesis
Last updated: May 28th, 2026 |
The Williamson Ether Synthesis
- In the Williamson Ether Synthesis, an alkyl halide (or sulfonate, such as a tosylate or mesylate) undergoes nucleophilic substitution (SN2) by an alkoxide to give an ether.
- Being an SN2 reaction, best results are obtained with primary alkyl halides or methyl halides. Tertiary alkyl halides give elimination instead of ethers. Secondary alkyl halides will give a mixture of elimination and substitution.
- The alkoxide RO– can be those of methyl, primary, secondary, or tertiary alcohols.
- The reaction is often run with a mixture of the alkoxide and its parent alcohol (e.g. NaOEt/EtOH or CH3ONa/CH3OH). Alternatively, a strong base may be added to the alcohol to give the alkoxide. Sodium hydride (NaH) or potassium hydride (KH) are popular choices.
- When an alkoxide and alkyl halide are present on the same molecule, an intramolecular reaction may result to give a new ring. This works best for 5- and 6-membered rings.
- When planning the synthesis of ethers using the Williamson, take care to select the best starting materials for an SN2 reaction. Avoid planning a Williamson involving a tertiary alkyl halide or a phenyl halide!
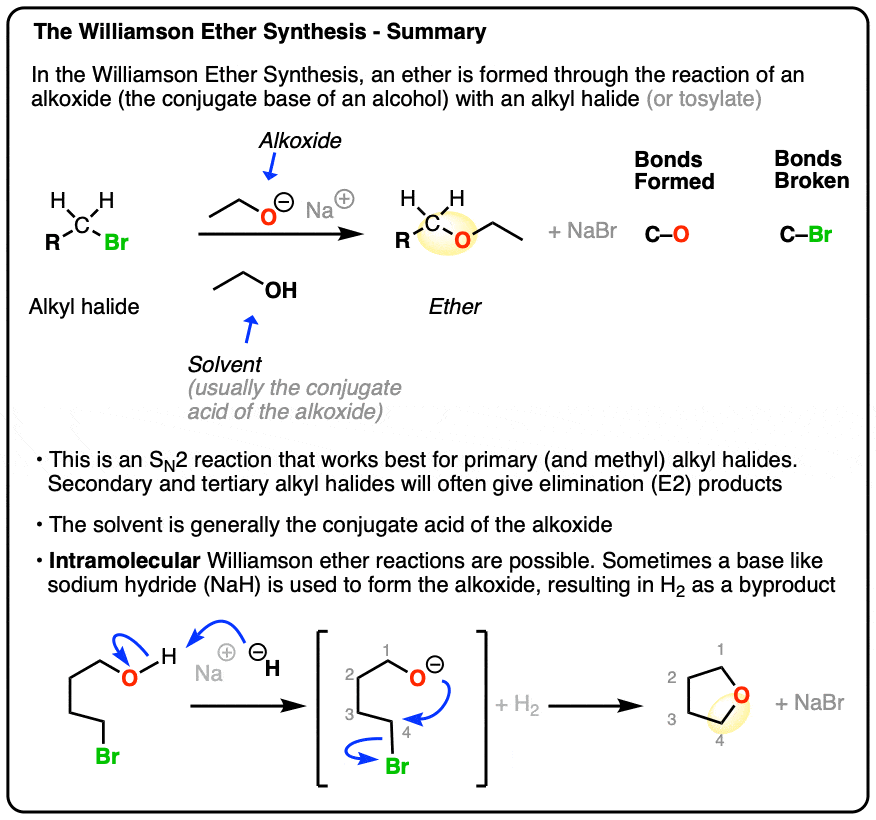
Table of Contents
-
- The Williamson Ether Synthesis
- Mechanism of the Williamson Ether Synthesis is SN2
- Primary and Methyl Alkyl Halides Work Best
- Solvent Choice In The Williamson
- Intramolecular Williamson Ether Syntheses
- Planning Ether Synthesis via the Williamson
- Summary
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. The Williamson Ether Synthesis
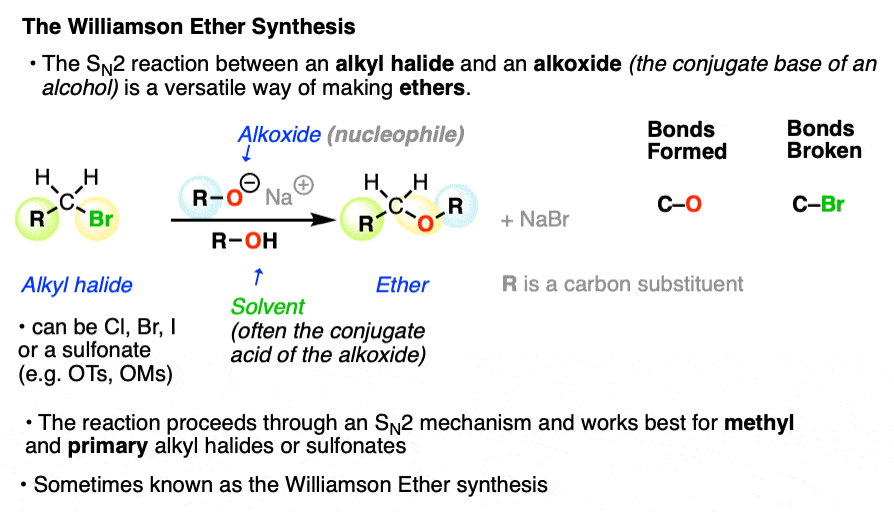
One of the simplest and most versatile ways for making ethers is the SN2 reaction between an alkoxide (RO–, the conjugate base of an alcohol) and an alkyl halide.
Although this is a very old reaction – the first report was in 1850! – it just hasn’t been surpassed. It works well for making a variety of ethers and is known as the Williamson Ether Synthesis.
SN2 reactions between neutral alcohols and alkyl halides are generally quite slow. Since the conjugate base of any species is a better nucleophile, the reaction is sped up considerably (Note 1) by employing an alkoxide instead of a neutral alcohol. [See article: What Makes A Good Nucleophile?]
A common way to do the Williamson is to simply use the alkoxide nucleophile with its parent alcohol as solvent (indeed, that’s how it was done in 1850!)
For example, the classic way to make diethyl ether is to treat the ethyl halide (the chloride, bromide, or iodide all work, but not the fluoride) with the ethoxide ion in ethanol.
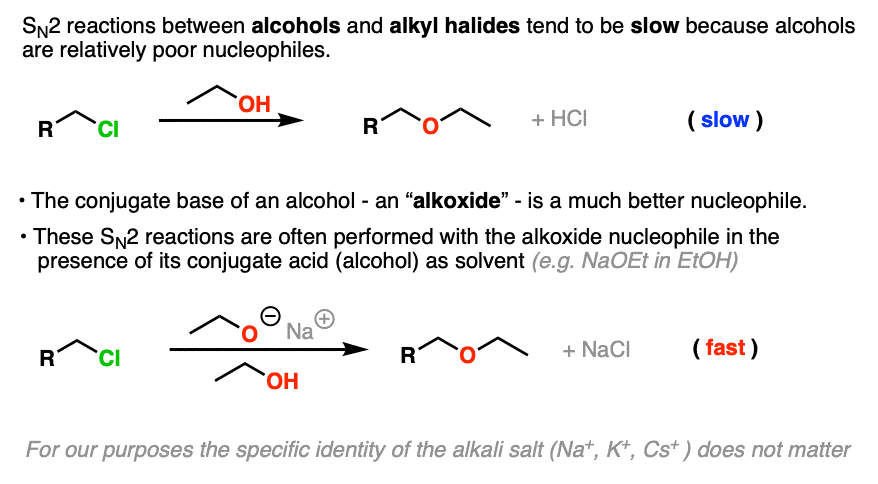
For our purposes the identity of the leaving group (iodide, bromide, chloride, tosylate (OTs), mesylate (OMs) ) does not really matter, although it is important to know their relative leaving group abilities (See article: What Makes A Good Leaving Group).
Likewise the identity of the alkali metal salt (Li+ , Na+, K+, etc.) does not matter much for our purposes and we will use these metal salts interchangeably.
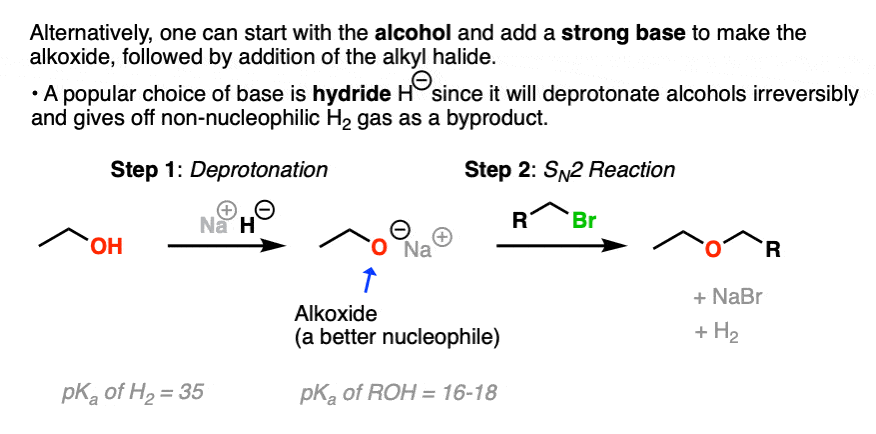
Since it’s not always an option to use the alcohol as solvent, another option is to generate the alkoxide by using a strong base that will irreversibly deprotonate the alcohol.
A popular choice is the hydride ion H– , which is the conjugate base of hydrogen gas (pKa about 35) and is also a poor nucleophile to boot. Sodium hydride (NaH) or potassium hydride (KH) can be added to the starting alcohol to generate the alkoxide. The hydrogen gas byproduct then bubbles out of solution into the atmosphere.
2. The Williamson Ether Synthesis Proceeds Through an SN2 Mechanism
The Williamson ether synthesis is a substitution reaction, where a bond is formed and broken on the same carbon atom. In this substitution reaction, a new C-O bond is formed, and a bond is broken between the carbon and the leaving group (LG) which is typically a halide or sulfonate.
It proceeds through an SN2 mechanism (nucleophilic substitution, bimolecular) where the nucleophile approaches the carbon atom from the backside of the carbon-leaving group bond. (See article: The SN2 Mechanism)
A pair of electrons from the nucleophile are donated into the sigma* (antibonding) orbital of the C-leaving group bond.
This requires that the nucleophile actually makes its way to the orbital on the backside of the carbon! For this reason the SN2 is fastest for methyl and primary alkyl halides, and does not occur on tertiary alkyl halides due to the fact that nucleophiles can’t make their way through the tangled thicket of alkyl groups on the backside. (See article: Steric Hindrance Is Like A Fat Goalie) .
Substitution reactions of alkoxides with secondary alkyl halides can occur, but often occur with significant elimination through the E2 pathway.
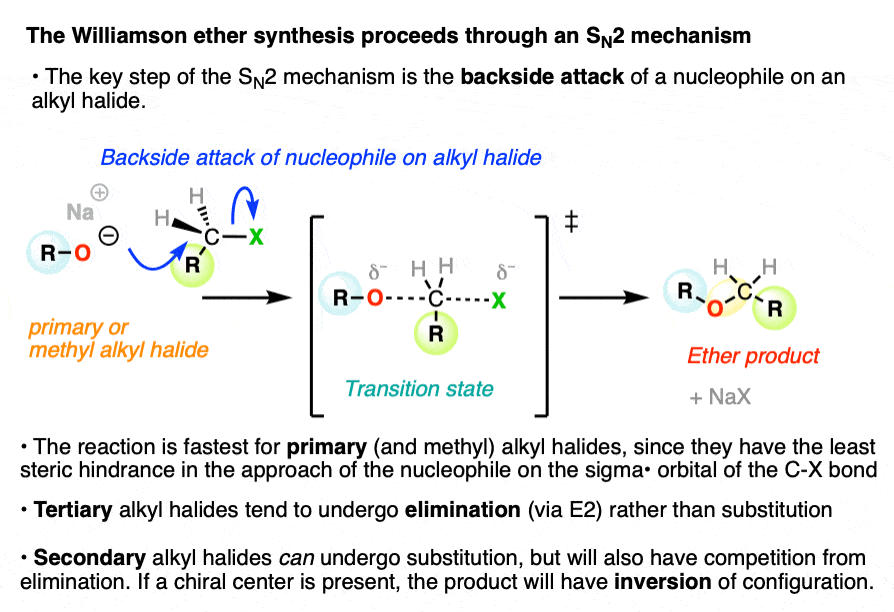
In the transition state of the SN2 there is a five-coordinate geometry about the carbon with partial bonds to the nucleophile and to the leaving group.
As the carbon-nucleophile bond strengthens and the carbon-leaving group bond weakens, the geometry of the carbon becomes inverted, like the proverbial umbrella in a strong wind. This is generally not noticeable unless the alkyl halide carbon is a chiral center.
3. Primary and Methyl Alkyl Halides Work Best
The Williamson works best for primary and methyl alkyl halides. Let’s look at some examples:
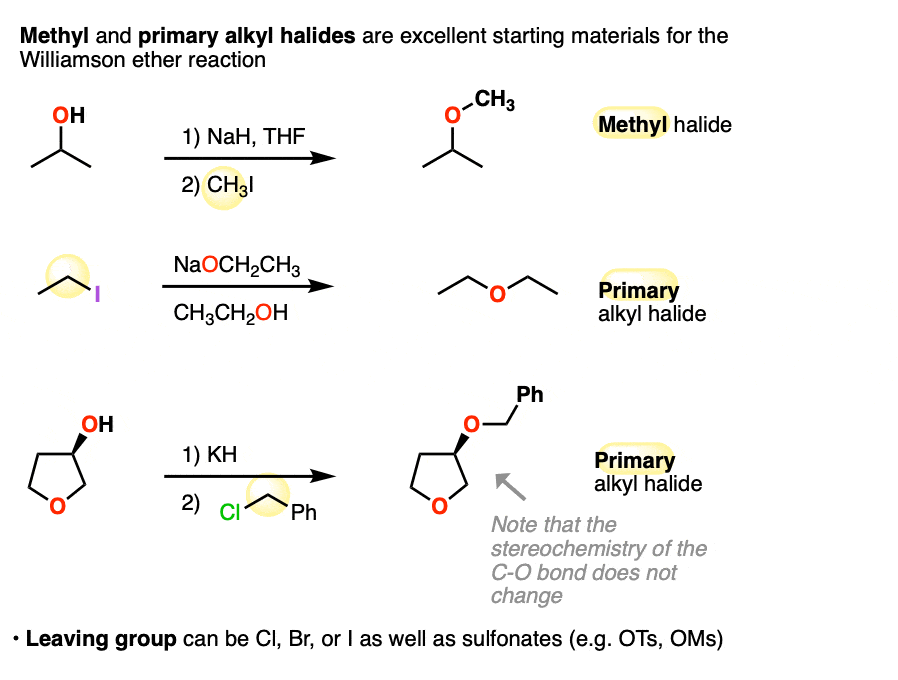
Note that the third example the stereochemistry of the C-O bond is unaffected. That’s because it’s only the geometry of the electrophile (i.e. the alkyl halide) in the SN2 that becomes inverted.
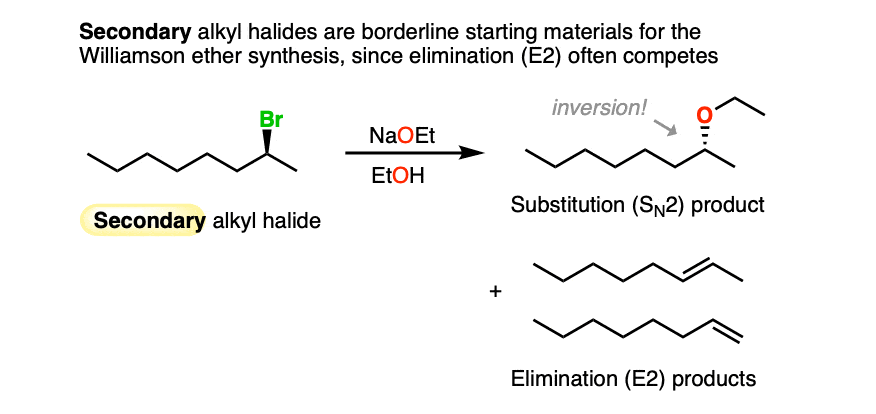
If the alkyl halide carbon is chiral, inversion can occur, as it does for this secondary alkyl halide below.
Since alkoxides are strong bases, there will be significant competition between SN2 and E2 reactions with secondary alkyl halides. Alkenes and ethers are generally obtained as mixtures. [Note 2].
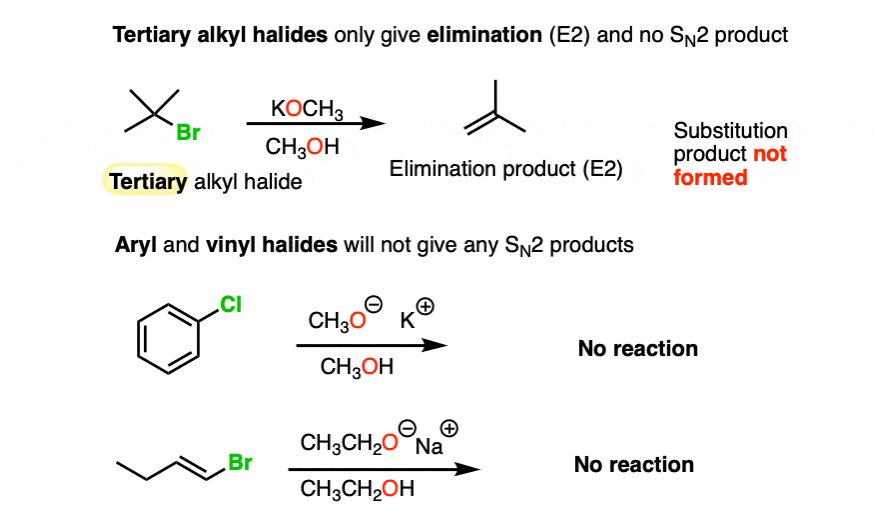
With tertiary alkyl halides, the Williamson ether reaction fails completely, and only alkenes are obtained.
Since SN2 and E2 reactions generally do not occur with sp2 hybridized carbons, another case where Williamson reactions fail is with aryl and alkenyl halides.
4. Choice of Solvent For Ether Formation
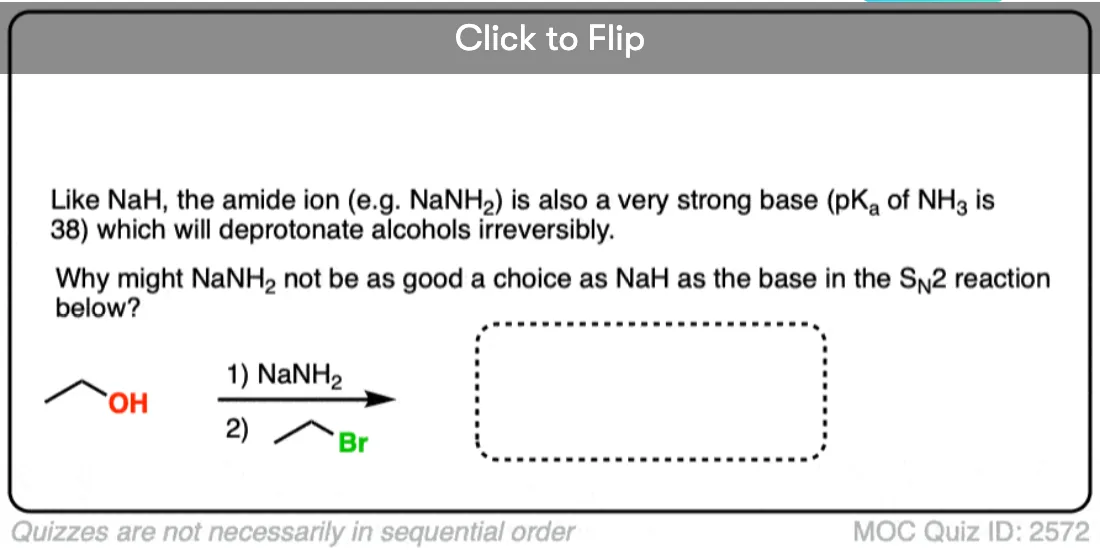
A common choice of solvent for the Williamson is to use the parent alcohol of the alkoxide, such as ethanol when using sodium ethoxide.
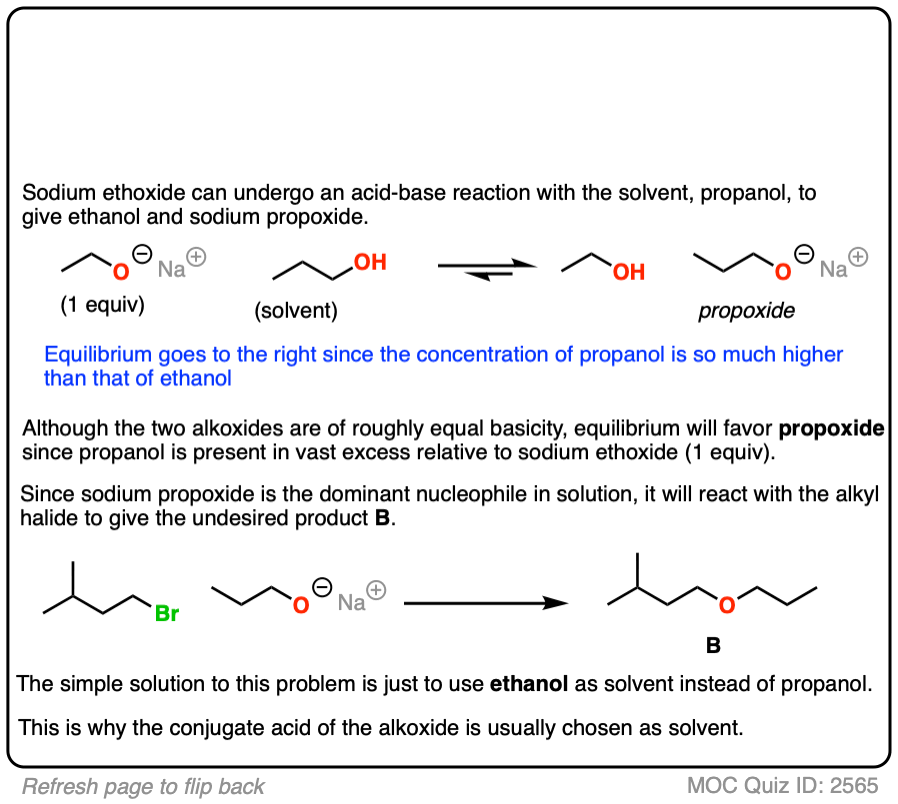
It is generally a bad idea to use an alcoholic solvent that is not the conjugate acid of the alkoxide, as discussed in the quiz below:
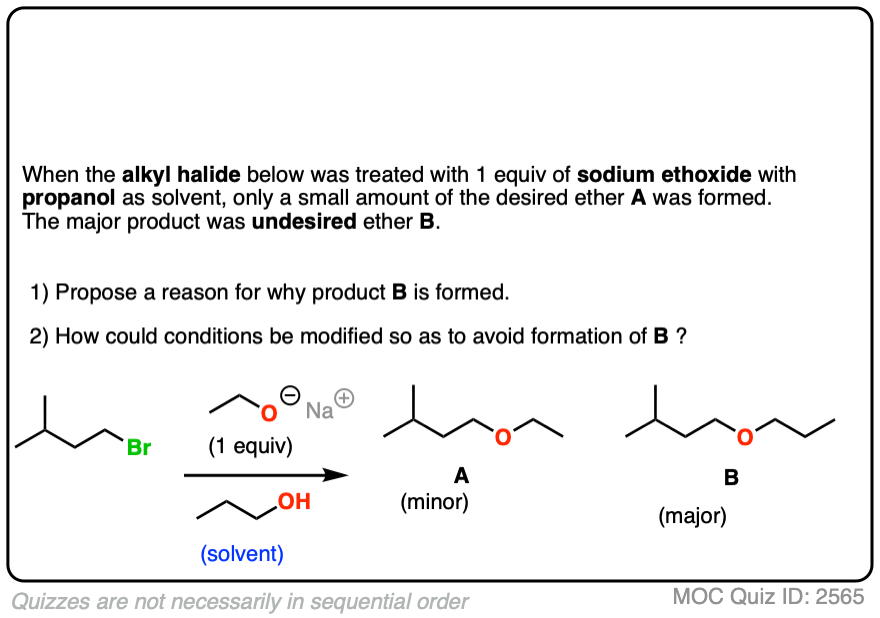 Click to Flip
Click to Flip
When using sodium hydride (NaH) or potassium hydride (KH) a common choice of solvent is ethers such as tetrahydrofuran (THF), diethyl ether, or polar aprotic solvents such as dimethylsulfoxide (DMSO).
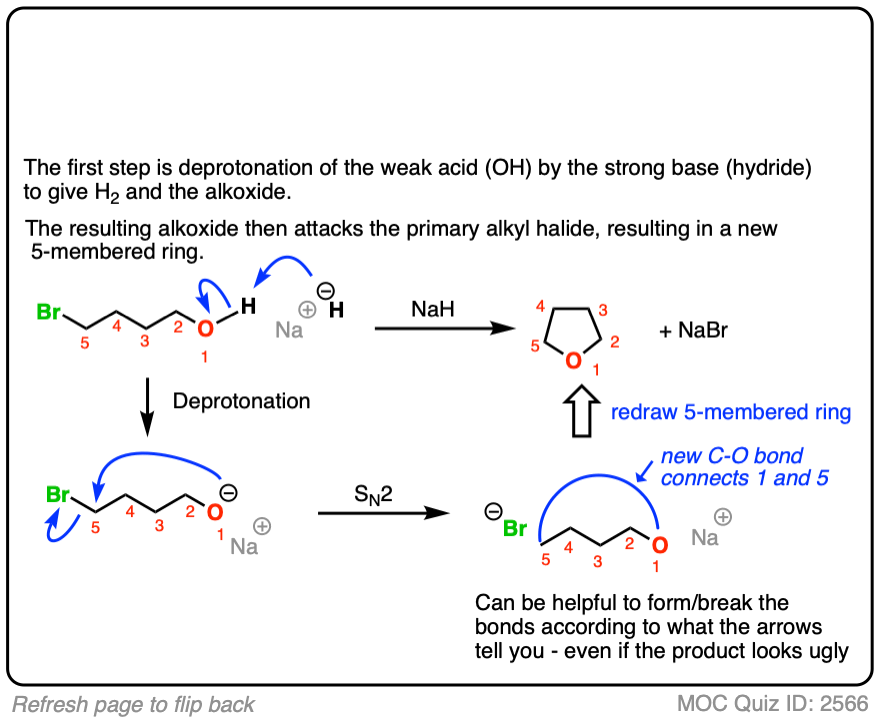
5. Intramolecular Williamson Reactions
If an alcohol and alkyl halide are present on the same molecule, there is the potential for an intramolecular Williamson ether reaction to occur.
This will result in a new ring.
Ring formation is best for 5- and 6-membered rings. [Note 3 ]
The mechanism for the intramolecular Williamson reaction is identical to that for a normal SN2; it just looks weird the first time you see it.
See if you can draw the mechanism:
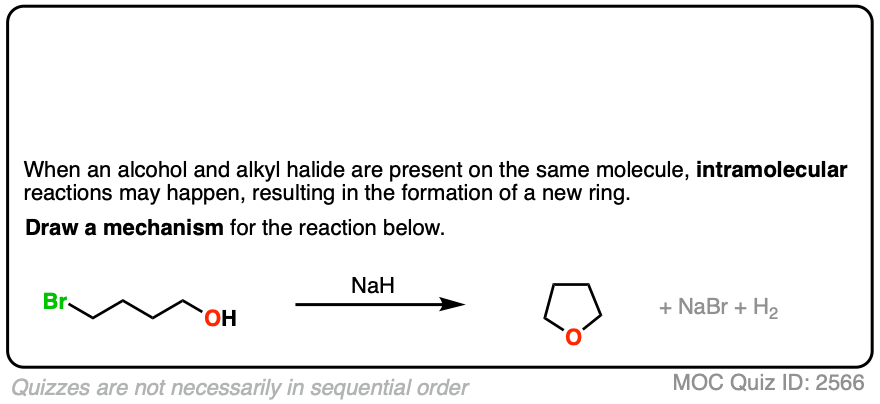 Click to Flip
Click to Flip
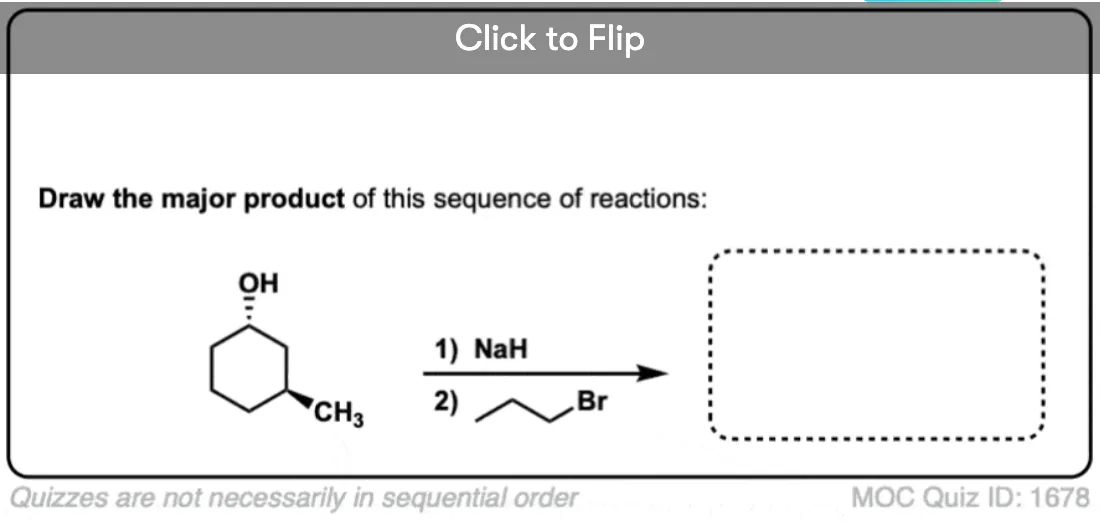
It’s also valuable to be able to work backwards from a final product to propose plausible starting materials. See if you can draw a feasible starting material that will result in the Williamson product below:
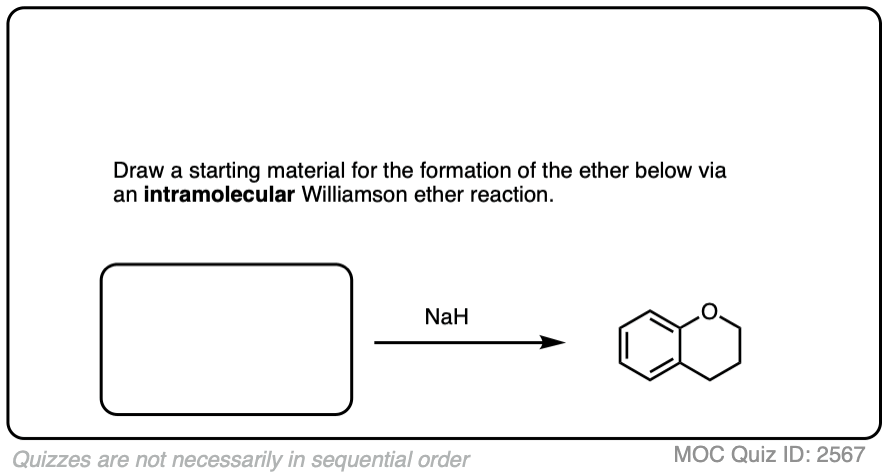 Click to Flip
Click to Flip
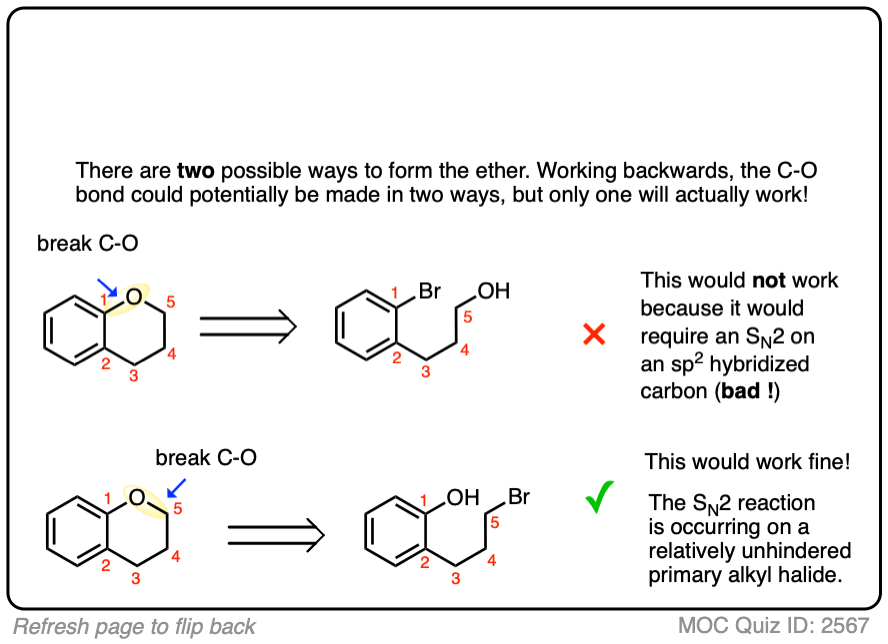
6. Planning Syntheses of Ethers via The Williamson
All right. Given all we have said about the Williamson, let’s see some examples of using this reaction to plan some syntheses of ethers.
With any ether there are two potential sites where a new C-O bond could be formed, which gives you two alkyl halide + alkoxide combinations to choose from.
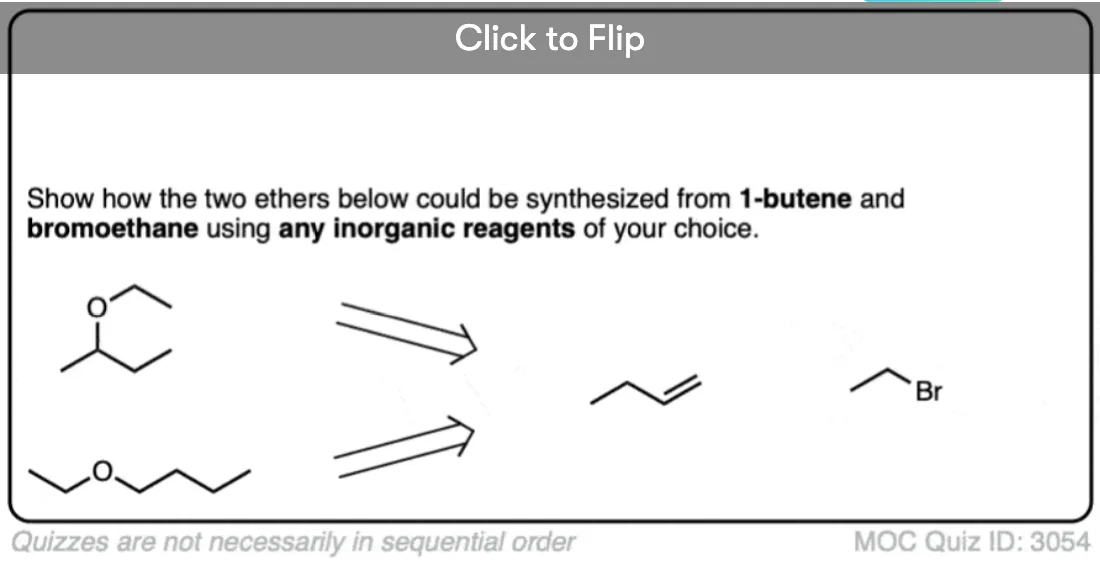
Let’s start with a fairly easy example. See if you can come up with reasonable starting materials for the synthesis of propyl methyl ether via the Williamson reaction.
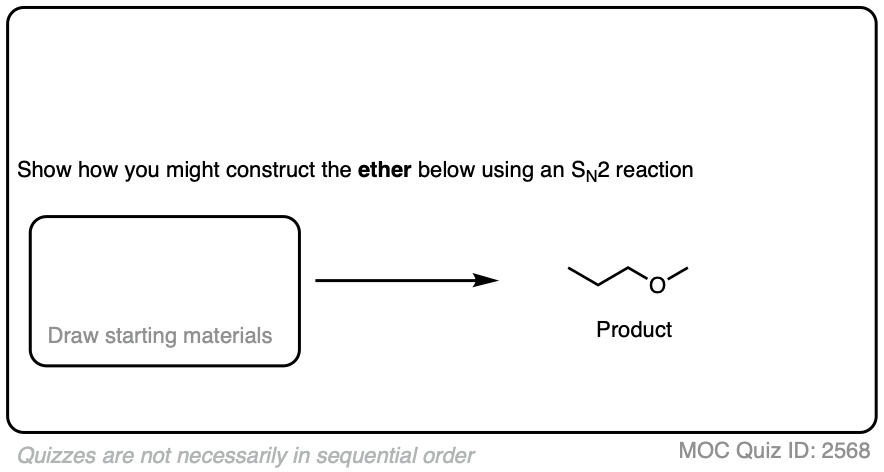 Click to Flip
Click to Flip
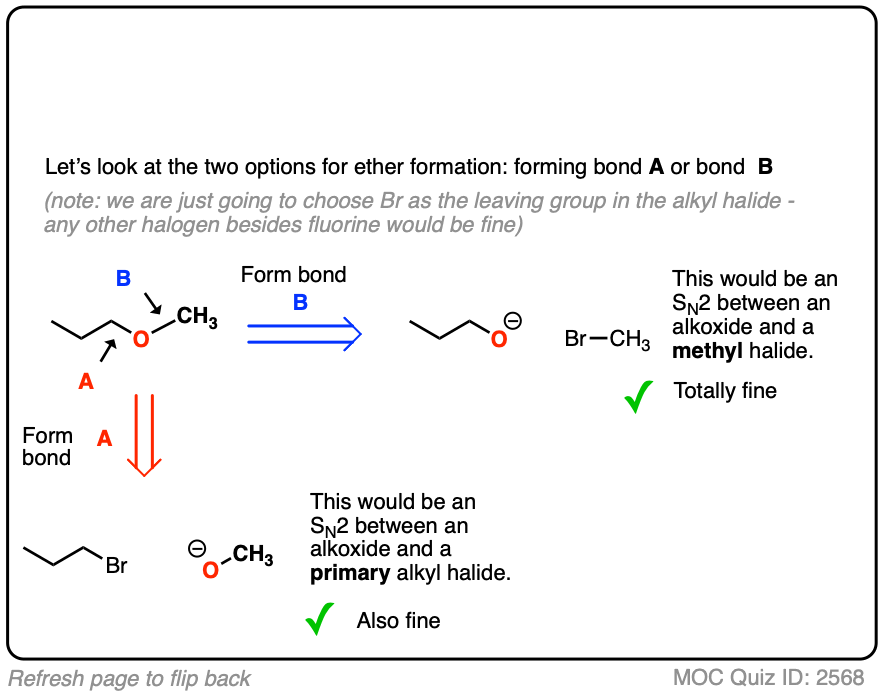
Note that for our purposes it doesn’t matter whether you choose Cl, Br, I or another good leaving group for your alkyl halide/sulfonate, so long that it isn’t F.
With propyl methyl ether, there are actually two good choices for building the ether. You can either use propyl halide (primary) with methoxide, or methyl halide (methyl) with propoxide. Both of these SN2 reactions should work well.
Since elimination is absolutely impossible on methyl, I’d give a slight preference to using the propoxide/CH3Br combination, but either one would work.
Ready for the next one? See if you can come up with a plan for the ether below:
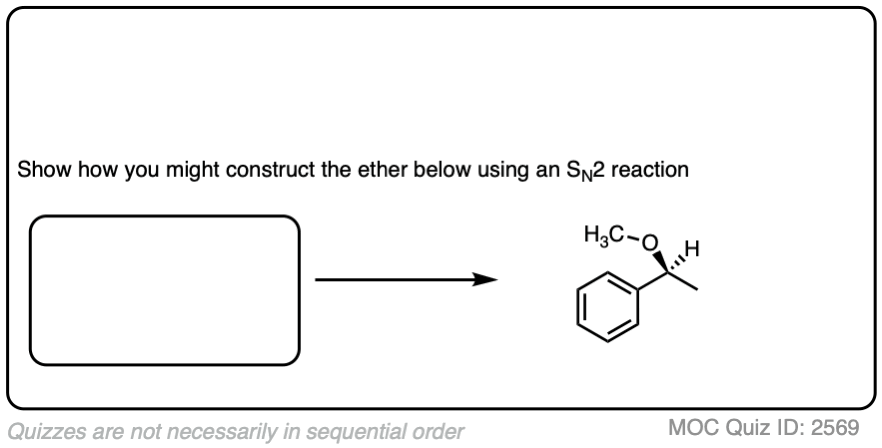 Click to Flip
Click to Flip
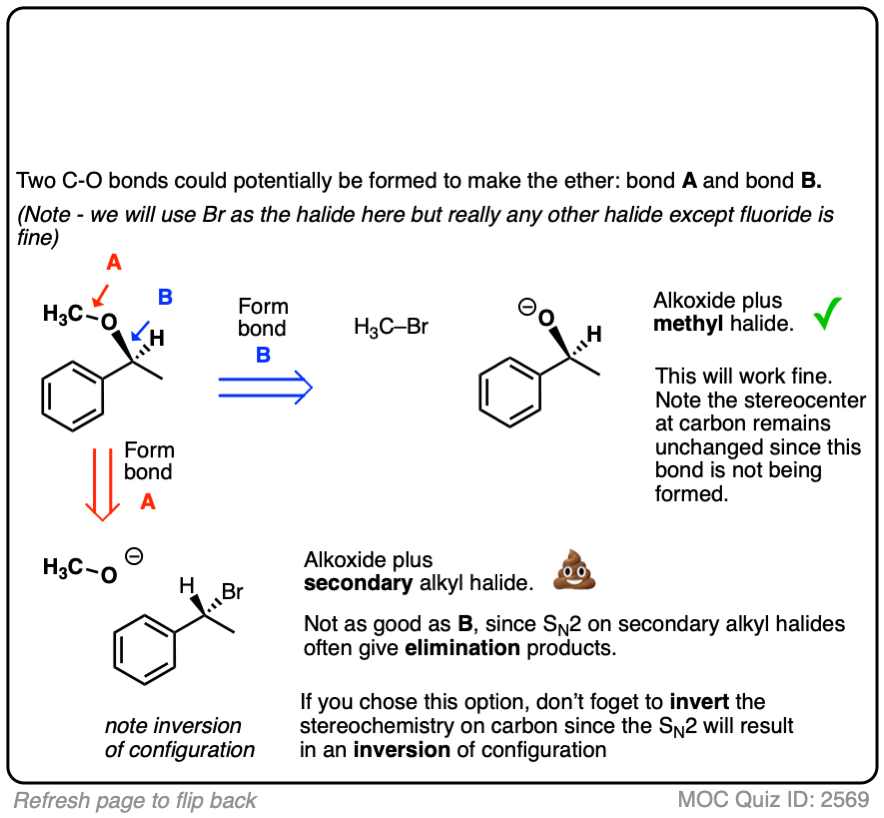
Working backwards gives us two sets of reactants. The one that will work best will be the alkoxide of the secondary alcohol with a methyl halide, since SN2 reactions are fastest on methyl groups and elimination reactions (E2) are impossible and will not compete.
A worse choice would be a secondary alkyl halide with methoxide. Note that since this is an SN2 – which proceeds with inversion of configuration – and the secondary alkyl halide chiral, the starting alkyl halide will have the opposite configuration to that of the starting material.
Let’s try t-butyl ethyl ether next.
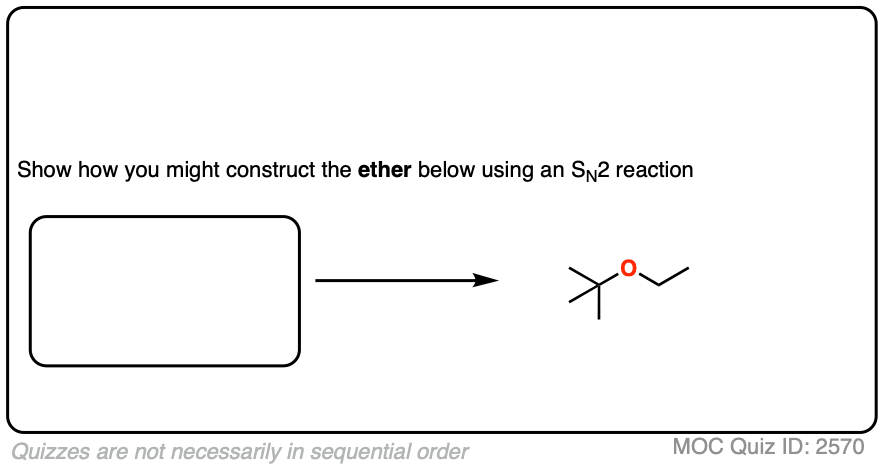 Click to Flip
Click to Flip
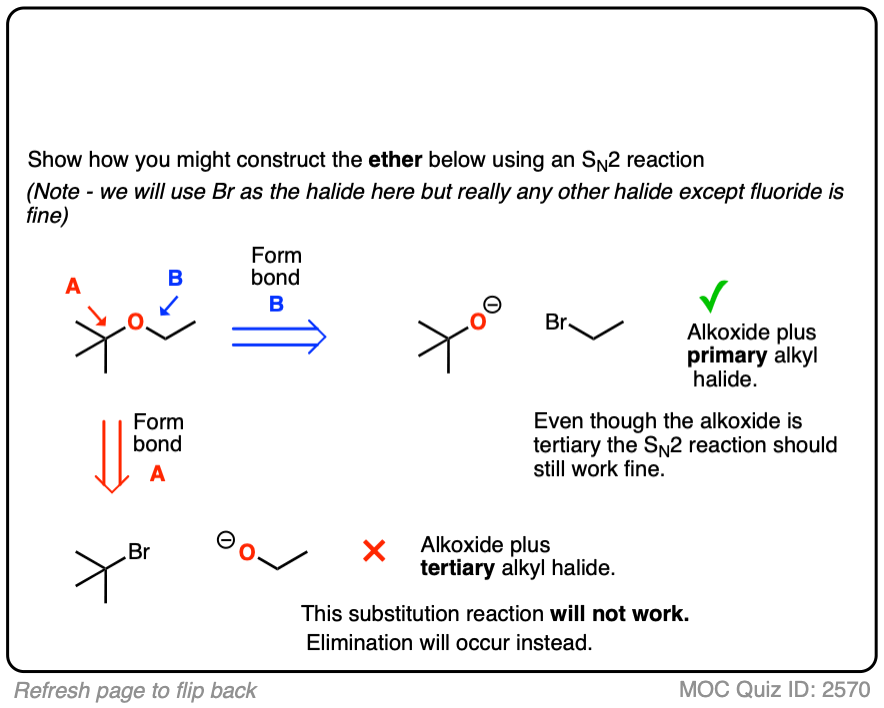
Our two choices of reactants include
- A tertiary alkoxide with a primary alkyl halide
- A tertiary alkyl halide with a primary alkoxide
Hopefully by now it should be clear that the first choice is best, since primary alkyl halides are excellent substrates for SN2 reactions and tertiary alkyl halides are not.
Even though the alkoxide is tertiary, the reaction should still work fairly well. Being bulky, there will be more elimination (E2) than normal, but this could be minimized by keeping the reaction temperature relatively low. [See article: Bulky Bases In Elimination Reactions]
Our final quiz asks how to synthesize phenyl methyl ether.
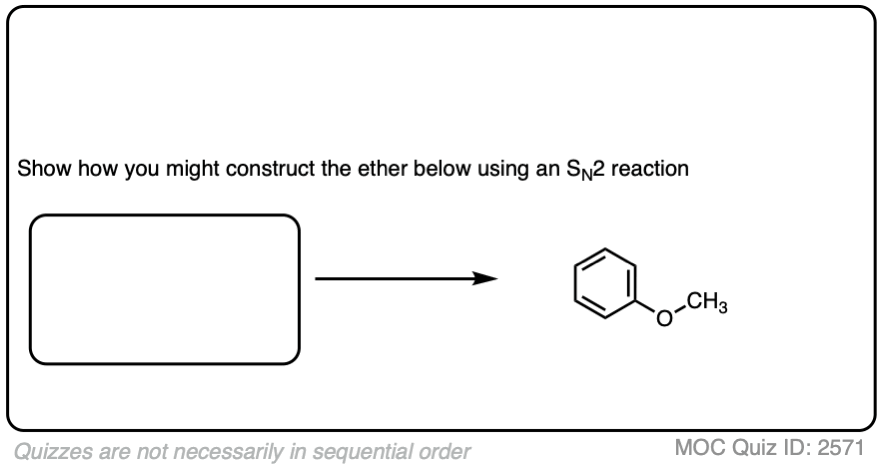 Click to Flip
Click to Flip
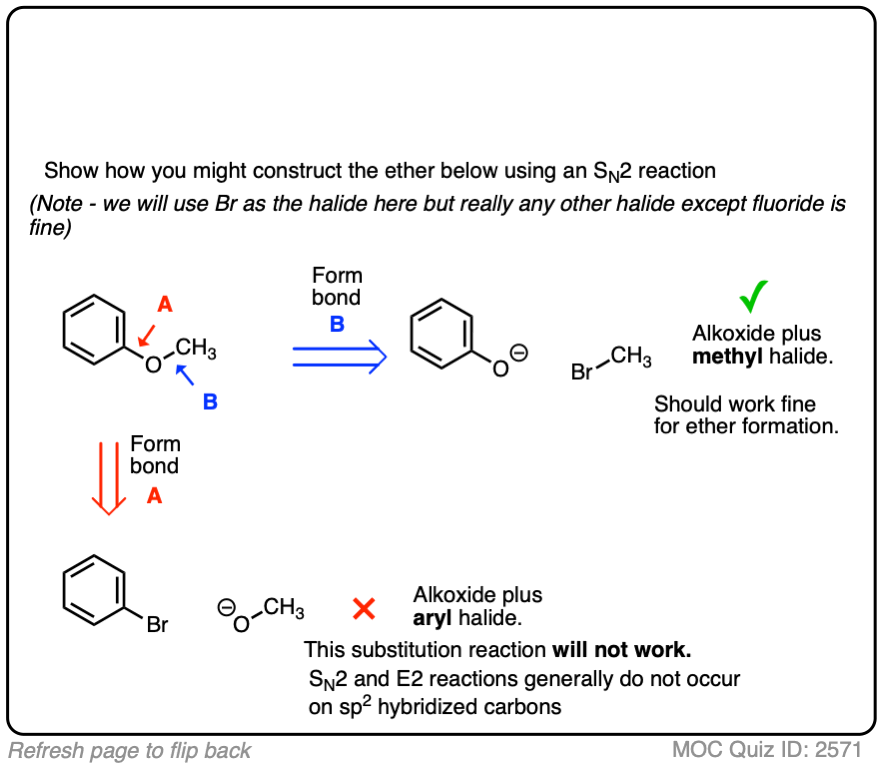
Our choices are:
- A methyl halide with a phenyl alkoxide
- An aryl halide with methoxide
Again, there is a very clear good choice here, as sp2 hybridized carbons (aryl halides) do not undergo SN2 reactions (just try doing a backside attack inside that phenyl ring!). In constrast, the reaction between the phenoxide and CH3Br should work much better without any complication.
7. Summary
So what are the key takeaways here?
If you’ve already learned the SN2 reaction, this should mainly be a refresher.
- SN2 reactions work well for methyl and primary alkyl halides, don’t work for tertiary, alkenyl or aryl halides, and are pretty borderline for secondary alkyl halides.
- Make sure you are familiar with drawing intramolecular examples of this reaction, because instructors tend to love throwing these types of mechanisms at you on exams.
- Get comfortable with planning the synthesis of ethers through Williamson reactions. There will generally be two alkoxide/alkyl halide combinations to choose from. Choose the best SN2 reaction available.
There are situations where the Williamson is not the best choice for ether synthesis and we must resort to other methods. For more, see Ethers From Alkenes, Tertiary Alkyl Halides, and Oxymercuration.
Notes
The contents of a separate article, “The Williamson Ether Synthesis – Planning” has been combined into this article – Sept 2023.
Note 1 – [Background rate of SN2]
Note 2 – [Secondary alkyl halide]
Note 3 – A Final Note About Rates of Ring Formation
I should end with a cautionary note about reactions that lead to ring formation. One question that comes up a lot is, “when do I know when a new ring will form?” [Shortcut spoiler: yes to 5 and 6 (and 3), generally “no” to rings 7 and above]
Great question! This is one of those issues that makes organic chemistry “hard” for the beginner, but “deep” and “interesting” for the lifelong practitioner because there are several key factors that often work in opposite directions.
First of all: Not all rings form at the same rate. That is, the rate at which a ring will form is, to some extent, dependent on the length of the chain.
How does this fit in with what we already know about substitution reactions?
Remember that the rate of a substitution reaction is proportional to the concentration of nucleophile and the concentration of electrophile. But what happens when the nucleophile and electrophile are on the same molecule? For this we use a concept called “effective concentration” which is to say that the reaction rate will be related to how much time the nucleophile spends in the vicinity of the electrophile. There isn’t space to go into this in detail in this post, but let’s use this velcro straps on this shoe as an overly simplistic example.

If the velcro straps are too short, then the nucleophile (strap) can’t reach the electrophile (on the shoe). If the velcro straps are too long (imagine if they were each a foot long, for instance!) , then the shoes will be annoying to put on because of the decreasing likelihood that the nucleophile will be in the vicinity of the electrophile (an example of low “effective concentration”). The rate of formation for very large rings will approach the rate of intermolecular reactions.
Of course molecules are more complicated than belts (or velcro straps) because of the ideal 109° angles of tetrahedral carbons. That creates some additional complexity, notably the issue of ring strain.
For example you are probably aware by this point that 3 and 4 membered rings are quite strained, whereas rings of size 5, 6, and 7 are relatively unstrained. [See article – Ring Strain in Cyclopropane and Cyclobutane]
If you’re just starting out you’re likely unaware that rings of size 8-11 are strained for a very interesting reason (transannular strain) and then rings of size 12 and above are generally unstrained.
For a student in an introductory course, a good rule of thumb is: Formation of 5 and 6 membered rings is fast. Formation of rings of size 7 and above is slow. As for the smaller ring sizes, we’ve seen examples where 3 membered rings form (from halohydrins). Seen less often, but also fast is the formation of 4 membered rings.
This is a vague generalization. “Give me numbers!” you might be saying. My copy of March says the following. Note that this is for a different reaction than the Williamson ether synthesis [formation of cyclic esters through SN2 of carboxylates with alkyl halides], but the trend should hold.
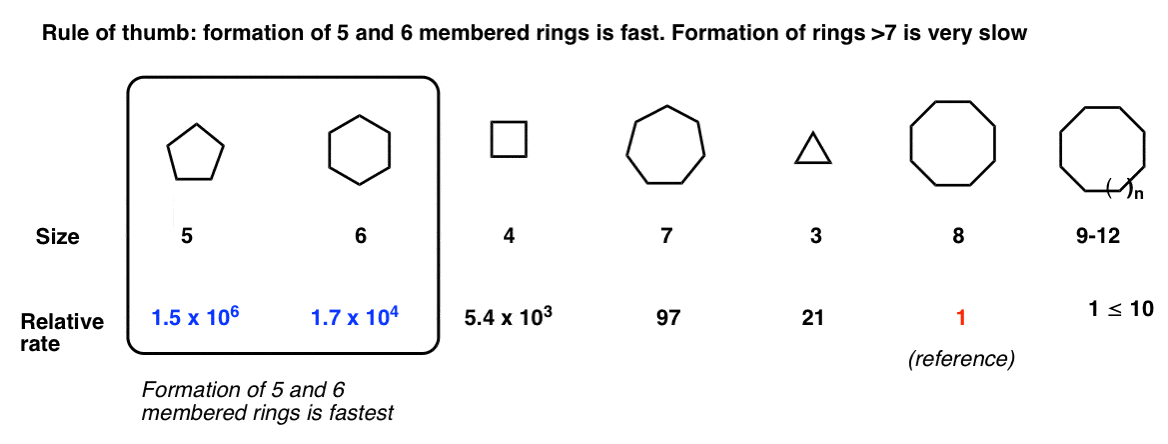
For a more quantitative approach, I suggest you look into this paper on ring closure kinetics.
Quiz Yourself!

Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- XLV. Theory of ætherification.
Alexander Williamson (1850) , The London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science, 37:251, 350-356,
DOI: 080/14786445008646627
The original Williamson paper. When Williamson reported the reaction in 1850 he didn’t know what an SN2 was – scientists didn’t even know what electrons were, for that matter – which again goes to show that the science of organic chemistry developed through a lot of empirical observations first, and the theory developed later. - Equilenin 3-Benzyl Ether
M. Hoehn, Clifford R. Dorn, and Bernard A. Nelson
The Journal of Organic Chemistry 1965 30 (1), 316-316
DOI: DOI: 10.1021/jo01012a520
One of the reactions in this paper is a classic Williamson reaction – protection of the alcohol in dehydroestrone as a benzyl ether, using benzyl chloride. - Total Synthesis of (+)-7-Deoxypancratistatin: A Radical Cyclization Approach
Gary E. Keck, Stanton F. McHardy, and Jerry A. Murry
Journal of the American Chemical Society 1995 117 (27), 7289-7290
DOI: 1021/ja00132a047
In modern organic synthesis, the Williamson reaction is used for the protection of reactive alcohols in a substrate. Common protecting groups include methoxymethyl (MOM) and 2-methoxyethoxymethyl (MEM). MOM protection is employed in this total synthesis by Prof. Keck and coworkers.
For the methyl cyclohexanol example, shouldn’t the dash be inverted into wedge when ether is formed?
I had a similar question to the one above–bulky bases are supposed to be an example of a poor nucleophile but a really strong base, so cause E2 reactions, with Hoffmann elimination when applicable for steric reasons. But the textbook and this site also say that using a tert-butoxide ion is a better bet for making an ether by SN2 reaction than say, a tertiary alkyl halide in which case only elimination product comes.
But then, I guess you could make an ether by SN1 reaction on a tertiary alkyl halide? As you said in a response to another comment. The textbook says the reaction of CH3ONa with (CH3)3C-Br gives exclusively 2-methylpropene. Guess that’s an experimental result.
Source:
https://flic.kr/p/2ogyr3S
https://flic.kr/p/2ogxHyZ
NCERT chemistry class 12
Will reaction of sodium tert-butoxide and chloroethane give ethene (due to E2) or t-butyl ethyl ether (due to SN2) as major product?
Mechanism for Williamson synthesis
You are right that it isn’t in there. Need to fix that. It’s just an SN2.
Thanks for sharing with such useful details.
If the product is water insoluble, you can get rid of excess base simply by pouring the reaction in water; however, my product is highly water soluble, do you have any suggestions how to separate it from the excess base? (I use K2CO3)
Is your product soluble in any organic solvents at all? One way to do it would be to quench the base with saturated NH4Cl solution, and then add equal volumes of brine and n-BuOH. Perform 3 extractions with n-BuOH and your organic molecule should persist in that layer while all the salts will be in the aqueous layer.
Is it possible to use NaOH instead of NaH as a base in the formation reaction of alcohol to Alkoxide?
Sure! There will be an equilibrium between alkoxide and alcohol but will still get the job done. It’s best when the solvent is the conjugate acid of the alkoxide (e.g. EtO- / EtOH).
hello and thank you very much for your notes, I found them really helpful.
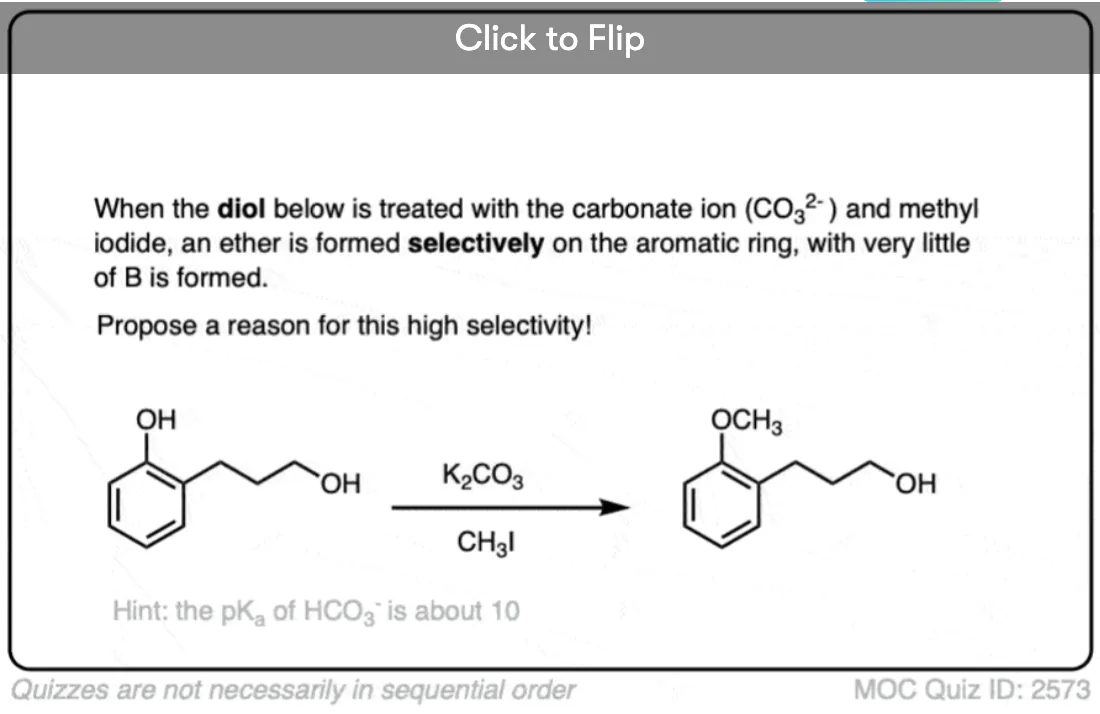
I have a question: in my project I had to form a benzylic ether to protect a phenol. I did it using NaI, K2CO3 and BnBr in DMF at 80oC as literature suggested. Is this a Williamson synthesis and does the ether form through an Sn2? If yes then why NaI and K2CO3 and not a stronger base?
The NaI makes benzyl iodide from benzyl bromide. Benzyl iodide is too unstable to isolate.
The pKa of phenol is 10. The pKa of carbonic acid is about 6. The pKa difference is 4. A good rule of thumb is that a pKa difference of 8 or less will be sufficient to get your conjugate base to participate. So although the acid-base reaction will like far on the carbonate side, there will be enough phenoxide to react with your benzyl iodide.
Can we please tell me if SN1 is possible in Williamson ether synthesis?..Because our Teacher today said it can… And i have learned otherwise 😕..Iam a 12th standard student
*ether formation* is possible via SN1. For example, take a tertiary alkyl halide like t-butyl bromide and dissolve in methanol; you’ll get a new ether, t butyl methyl ether.
However that’s not technically a Williamson; a Williamson involves deprotonating an alcohol to give an alkoxide, and then having that react with an alkyl halide to give a new ether. This proceeds through an SN2 reaction.
Ultimately the name “Williamson” isn’t very important; what’s more important is to realize that ethers can form both through SN1 and SN2 reactions.
Hii, I have a dought. Is that possible of good yield with a primary alcohol and a secondary halide having steric hindered groups ?. I tried with a strong base in polar solvent but yield was very less 6-7% with unreacted SM.
The SN2 with secondary alkyl halides, particularly hindered secondary alkyl halides, is quite poor. Why don’t you switch it around and use a secondary alkoxide with a primary alkyl halide? That would work much better.
Hi James,
According to you, the following sequence :
1) NaH
2) BnBr
is a SN1 or SN2 mechanism ?
Thanks for your answer
Phil
I don’t see the molecules you’re working on, but I assume that NaH is to deprotonate an alcohol, and the BnBr reacts with the resulting alkoxide.
Hi, I learned a lot in this page. However, could you give some more examples about the conjugate acid of the alkoxide while choosing solvent.
Hello! I have a question: why heptanol can not be deprtonated by NaOH?
It can, it’s just that it will be highly reversible. you might have some solubility issues as well as heptanol is on the greasier side of things. Try a phase transfer catalyst. Tetraammonium hydroxide
Outstanding Presentations.
Found your web pages while helping my Daughter find good sources of info for her Organic course.
Concise but not Terse…
Pros, Cons, Comparisons, Rationale for choosing Reactant-Substrates with Reagents to yield preferred products along with related ” Be Aware Of This” notations are on point.
Your presentations perfectly full fill my Golidlocks criteria for selecting a Professors and Teachers.
Not To Little
Not Too Much,
PERFECT Presentation
Tom Jones, D.D.S.
Thank you
Can we use K2CO3 as a base to make the alkoxide ?
Not a great base to use because it’s quite weak. Equilibrium greatly favours the alcohol, not the alkoxide.
It depends also on the irreversibility, the extrusion of CO2 may help; maybe you need then the alkyliodide ….
Excellent stuff! World needs authors like you in chemistry! This is how an information should be conveyed. Short & covered all essential points..
Using DMSO(aprotic solvent) will favour E2 and not Sn2.Instead of Aprotic,protic solvent should be used.
This is not correct. Aprotic solvents favor SN2 over E2, and DMSO is a useful solvent for an SN2 reaction. Think of it this way. A polar PROTIC solvent would hydrogen bond to the nucleophile and hinder its backside attack.
“… after the base does its deprotonation, its conjugate base is still swimming around in solution…”
“it is non-nucloephilic”
“Easily avoided if we we just…”
Please make the appropriate corrections.
But a really great and useful post, actually (well, as usual).
Glad the mistakes are minor this time. Thank you, as always.