Organometallics
Formation of Grignard and Organolithium Reagents
Last updated: May 28th, 2026 |
Formation or Grignard Reagents and Organolithium Reagents From Alkyl and Alkenyl Halides
In the last post we introduced the concept of organometallic compounds – molecules where carbon is bound to a less electronegative atom such as Li, Mg, Cu, and many other metals. We said that carbon in these molecules tends to be electron-rich and thus have nucleophilic character, in contrast to functional groups such as alkyl halides, aldehydes, ketones, and epoxides where carbon has electrophilic character. If you need a refresher on what I meant by nucleophilic and electrophilic, read that post first.
In this post we’ll talk about how certain types of organometallic compounds are made – specifically organolithium and Grignard reagents.
Quick summary:
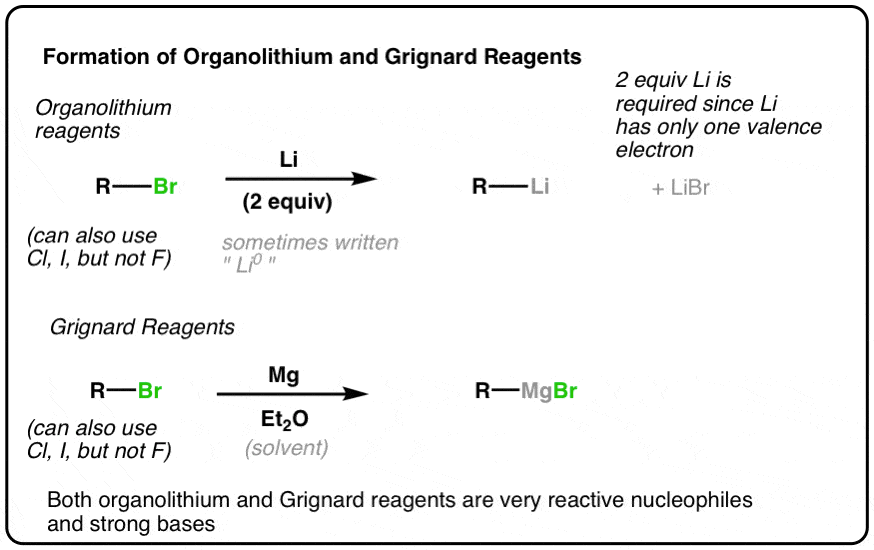
Let’s start with organolithium reagents because they’re the simplest.
Table of Contents
- Organolithium Reagents As Carbanions (The Conjugate Bases Of R-H)
- Making Carbanions Through Reduction, Not Deprotonation
- Formation of Organolithium Reagents From Alkyl Halides
- Formation of Grignard Reagents: The Mechanism
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Organolithium Reagents As Carbanions
If you look closely, you can approximate the structure of an organolithium reagent (R-Li) as “R(–)” , with lithium as the positive counter-ion: in other words, a carbon bearing a negative charge (we call these species “carbanions”). If you think back to earlier lessons on acids and bases, this structure might look familiar – it’s the conjugate base of a species R-H.
Using butane to stand in for “R-H” here, we get:
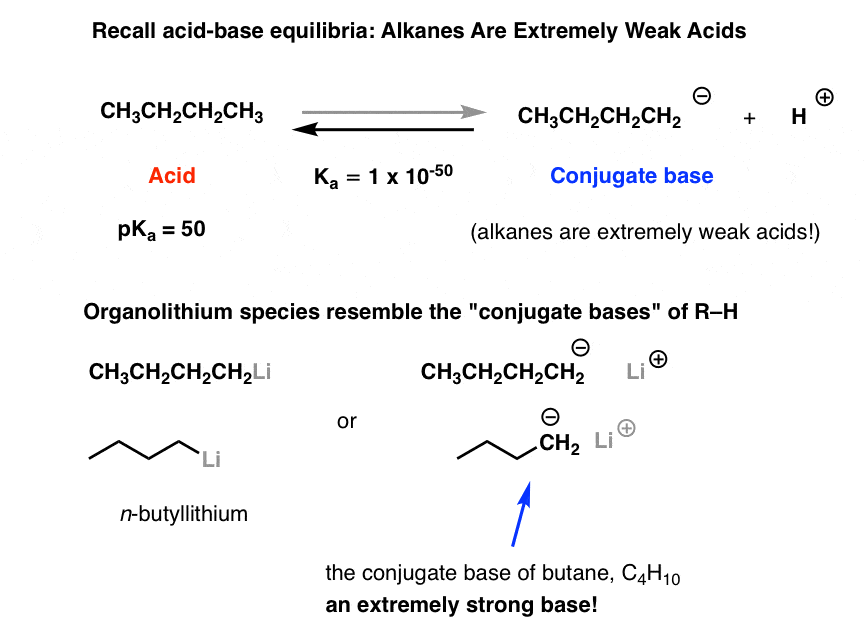
Since conjugate bases are made through deprotonation, we might naively think that we could make organolithium species by taking an organic molecule and just adding a super-strong base to rip off the proton.
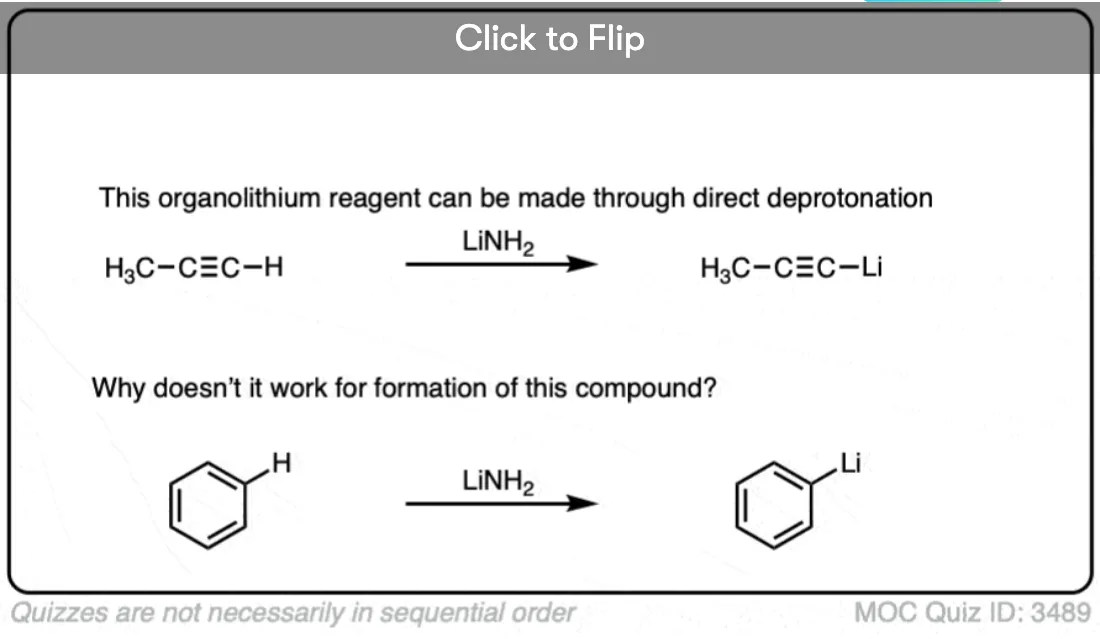
If you’ve covered alkynes, you’ve seen that that process actually works pretty well in the case of terminal alkynes. They are quite acidic species, having a pKa of 25 or so. Their unusually high acidity is due to the considerable s-character on the carbon (meaning that the lone pair is held closely to the nucleus).
Trouble is, as we move towards alkenes and alkanes, the direct deprotonation approach doesn’t work so well. That’s because… well, it’s hard to find any species more basic than a deprotonated alkane! Just as the only thing sharp enough to cut a diamond is another diamond, about the only thing basic enough to deprotonate an alkane is… another deprotonated alkane.
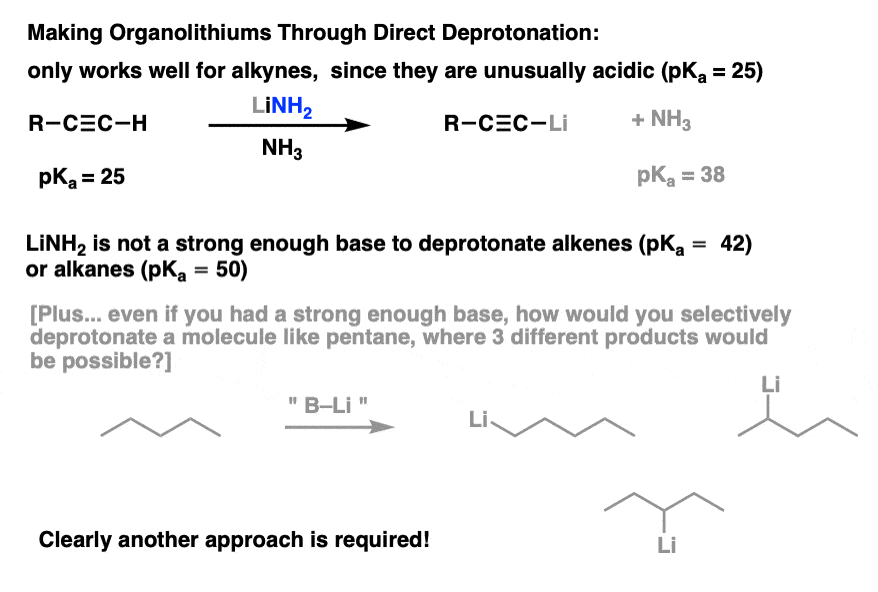
This isn’t a practical approach to make organolithiums for several reasons – primarily, the fact that there are often many C-H bonds and it’s hard to selectively remove just one. [For instance, if you tried to make 1-pentyllithium by deprotonating pentane, you could potentially end up with multiple different isomers].
Thankfully, another approach has been devised.
2. Making Anions Through Reduction, Not Deprotonation
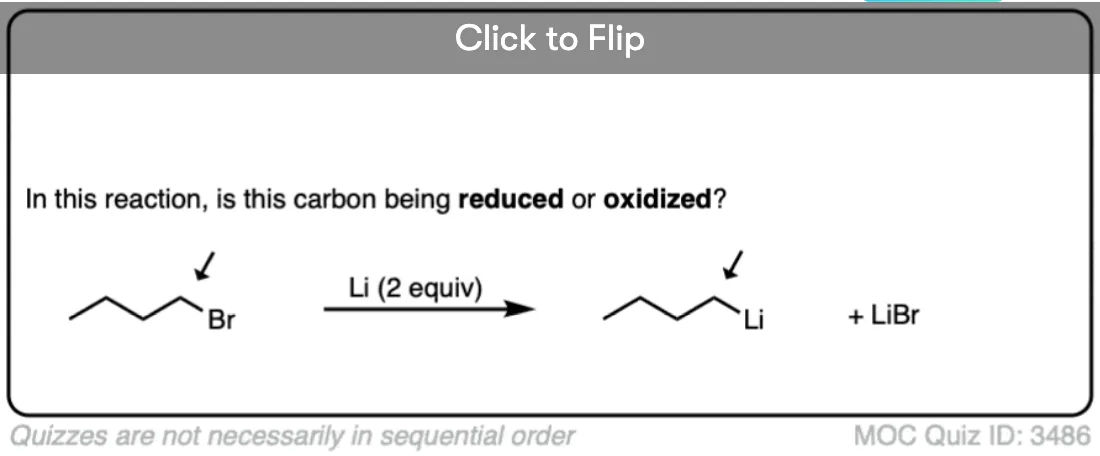
If you look at the reaction below, and count the electrons carefully, you might note that the product has two more electrons than the starting material. In other words, particularly if you remember the OIL RIG mnemonic, reduction has occurred.
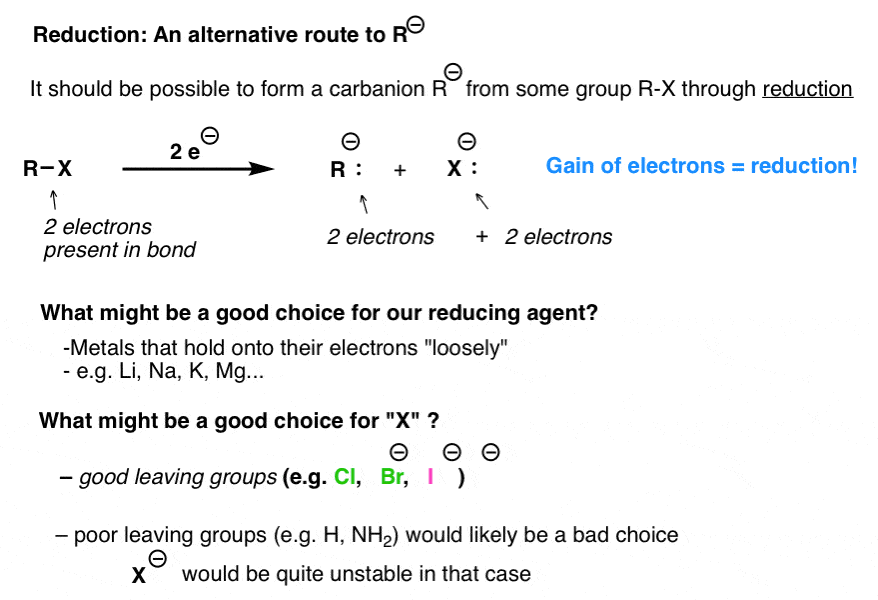
This means that if we were to add some species which was particularly likely to give up its electrons, we might thus be able to effect this transformation.
Can you think of any members of the periodic table which hold onto their electrons particularly loosely? If you said “the far left part of the periodic table” (particularly the alkali and alkaline earths), ding ding ding! you would be correct.
Lithium, in short, would be a great choice as a reductant for this reaction. [We’ll get to the other metals in a minute].
What might be a good choice for X? One factor which would make this reaction easier is if X(–) was a fairly stable species – a good leaving group, in other words. Good candidates for X are halides such as Cl, Br, and I. A bad candidate would be H(–) or some other strongly basic version of R(–) , since we’d be generating another unstable anionic species.
Now let’s get to specifics.
3. Making Organolithium Reagents From Alkyl Halides
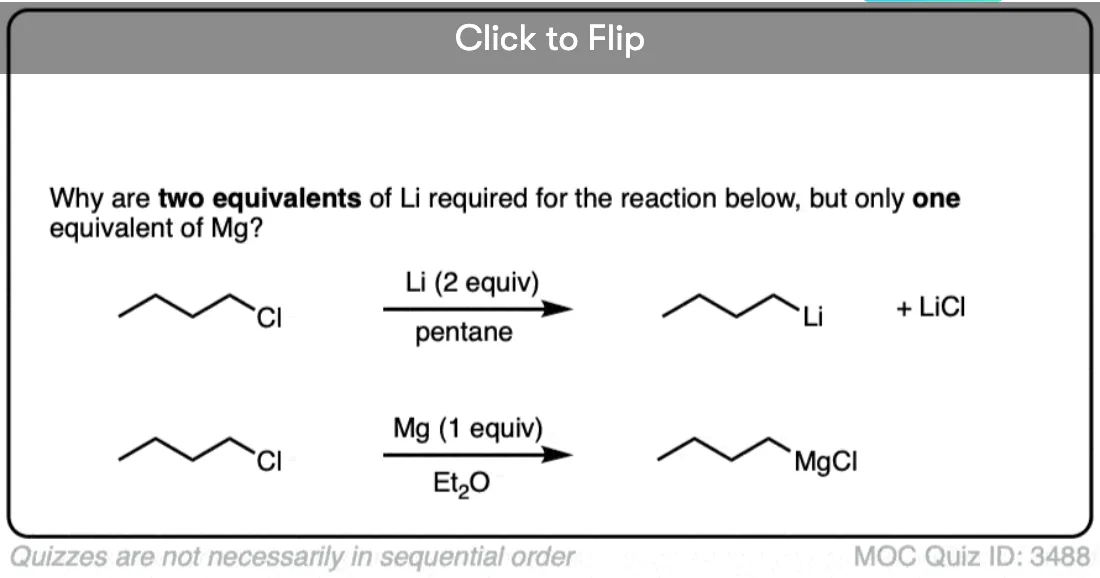
Lithium, having a very low ionization energy (i.e. it loses its electron easily) is a powerful reducing agent. Since lithium only has a single valence electron, however, we must add two equivalents if we are to complete the reduction reaction.
To make organolithium reagents, we start with alkyl halides, and add powdered lithium metal (Li or sometimes written as Li0 to distinguish it from the ion Li(+) ).
Occasionally the solvent for this reaction is written below the arrow. A common solvent is pentane. Some instructors like to include it. Some don’t. Regardless of whether it’s written there or not, it doesn’t participate in the reaction.
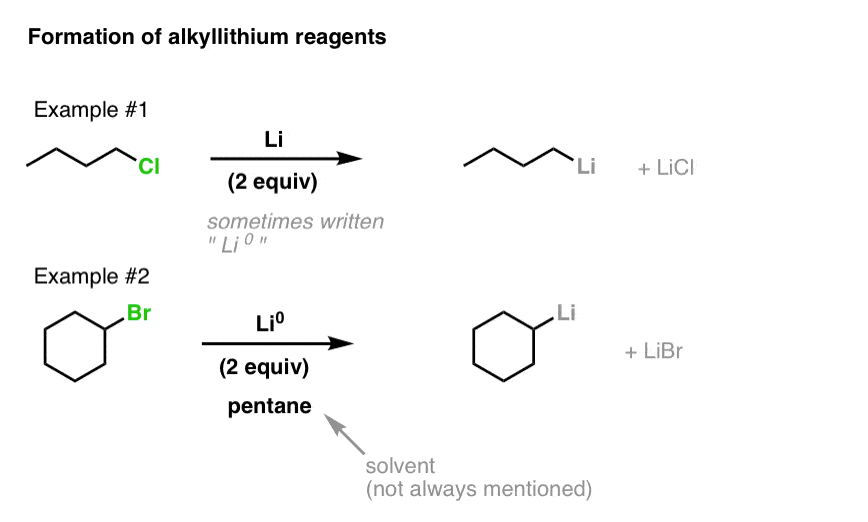
This reaction works for alkyl chlorides, bromides, and iodides, as well as alkenyl halides (fluorides excepted).
If you recall that alkyl halides can be made from halogenation of alkanes, this method thus gives us a 2-step method for formation of highly basic alkyl lithium species from alkanes.
For a pop-up view of the mechanism hover here or click on the link.
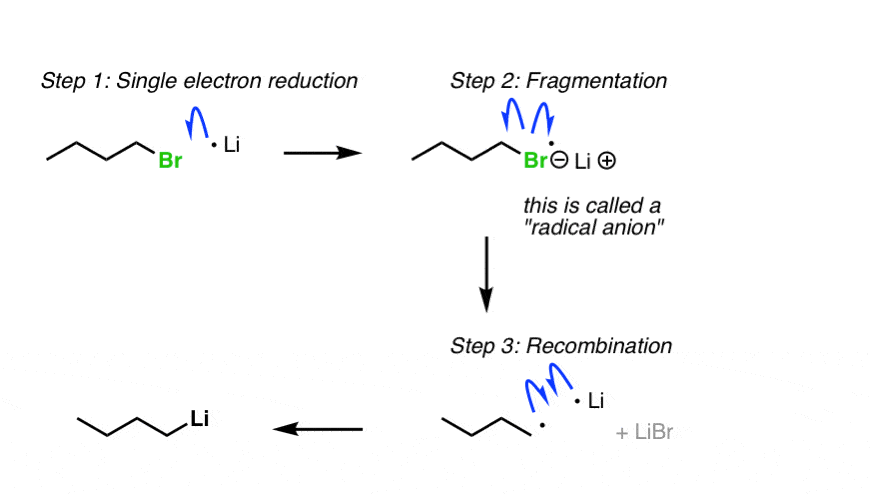{kind=link}
We’ll cover the many useful applications of organolithium reagents in a future blog post.
I’m going to skip organoberyllium reagents here (beryllium is highly toxic, and rarely sees use) and move straight across from sodium over to magnesium, which comprises a second very important family of organometallic reagents.
The process for making Grignard reagents is very similar to making organolithium reagents: start with an appropriate alkyl halide and add magnesium. Since magnesium has two valence electrons, only one equivalent of Mg is required to balance the reaction. Here’s two examples, showing formation of alkyl and alkenyl Grignard reagents.
Note that Grignards can be made from alkyl or alkenyl chlorides, bromides, and iodides – but not fluorides.
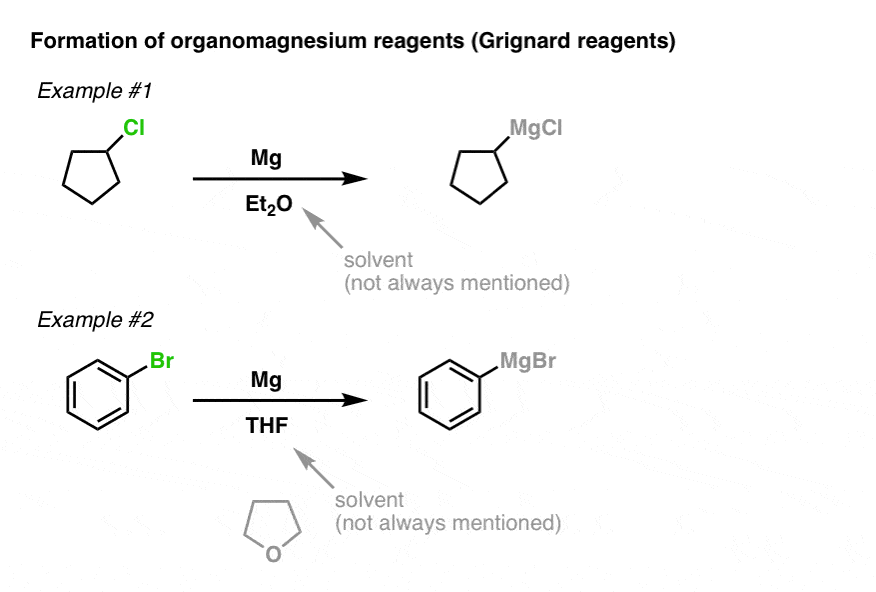
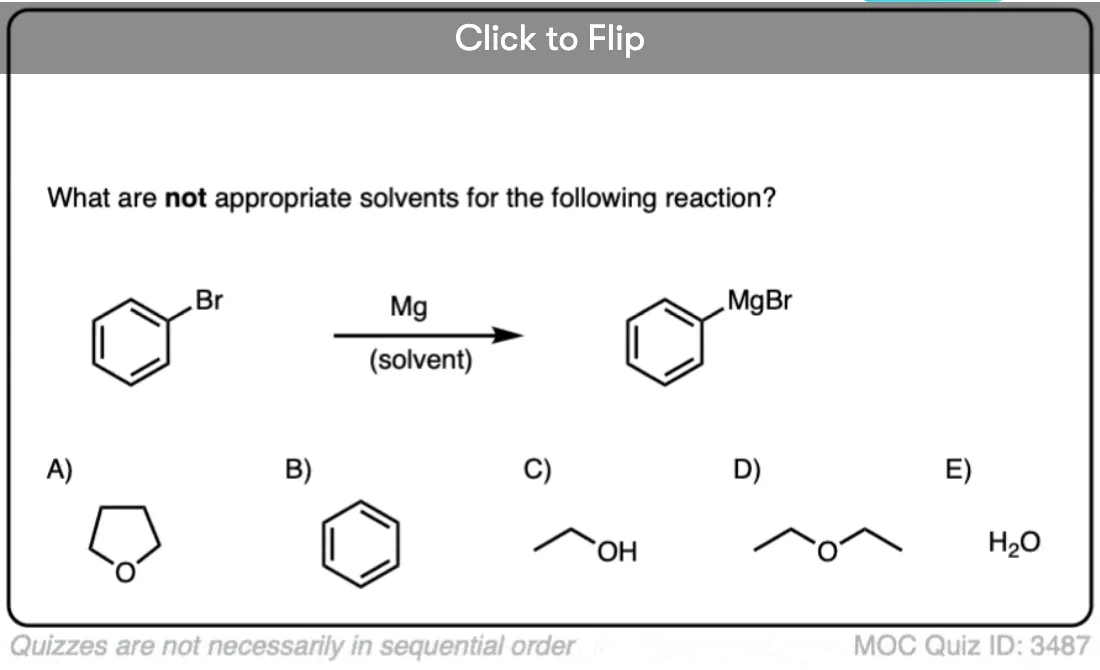
What, you might ask, are Et2O and THF? These are solvents, which are often written in the reaction scheme, but don’t actually participate in the reaction itself. Et2O (diethyl ether, or, sometimes, “ether”) and THF (tetrahydrofuran) are popular choices. If you’re an introductory student, you probably don’t want to know the deeper reasons why, nor do you need to, so don’t click this link to find out.
In contrast with most of the reagents and reactions we talk about at MOC, you’ll likely have personal experience doing this reaction!
Sitting around for a few minutes staring at your flask containing Mg, ether, and organohalide do absolutely nothing is a rite of passage for every student of organic chemistry.
One key contributor to the “finicky” nature of forming Grignard reagents is that the reaction occurs on the surface of the magnesium metal. For this reason the reaction is highly surface area dependent. Breaking the Mg up into very small chunks will accelerate the reaction. Furthermore, Mg that has been sitting out in the open for awhile often has a surface coating of magnesium oxide (MgO) which is unreactive with alkyl halides. Breaking up the surface helps to expose fresh, unoxidized Mg to the reactants. A pinch of iodine (I2) or 1,2-dibromoethane can also help to kick-start things.
4. Formation of Grignard Reagents: The Mechanism
What’s the mechanism of Grignard formation? Usually not covered – it involves free radicals – but if you’re curious, hover here for a pop-up view or click on this link.
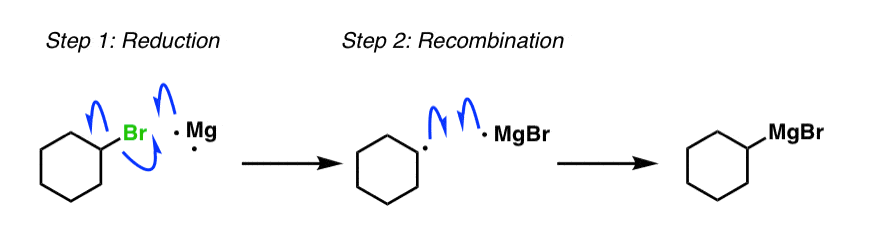{kind=link}
From a practical perspective, one key thing to make sure of when preparing organolithium or Grignard reagents is that the solvent and glassware are completely dry. Water (pKa 14) is death to Grignard and organolithium reagents, which as we said above, act as the equivalent of highly basic alkyl and alkenyl anions.
In the next post, we’ll talk about this and also some other complications of making Grignard reagents.
Next post: Organometallics Are Strong Bases
Notes
What about Organosodium Reagents?
So if lithium works, why not go further down the column of the periodic table? Why not use sodium?
This is an excellent idea – sodium is a great reducing agent, after all. The trouble is, when we try to make organosodium reagents from alkyl halides, what tends to happen is that the carbanions that form then go on to react with our starting alkyl halide (in an SN2 process). The result is a pretty useless reaction you likely don’t need to care about that we call Wurtz coupling.
That’s OK, however. Organolithium reagents are plenty reactive enough for almost every purpose that we’d otherwise want organosodium reagents to do. I mean, who needs Chuck Norris when you’ve got Jackie Chan?
Likewise for organopotassium reagents. The one application of organopotassium reagents that sees common use is a reagent called Schlösser’s Base , which is strong enough to deprotonate allylic C-H bonds, something not easily done by organolithium reagents.
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
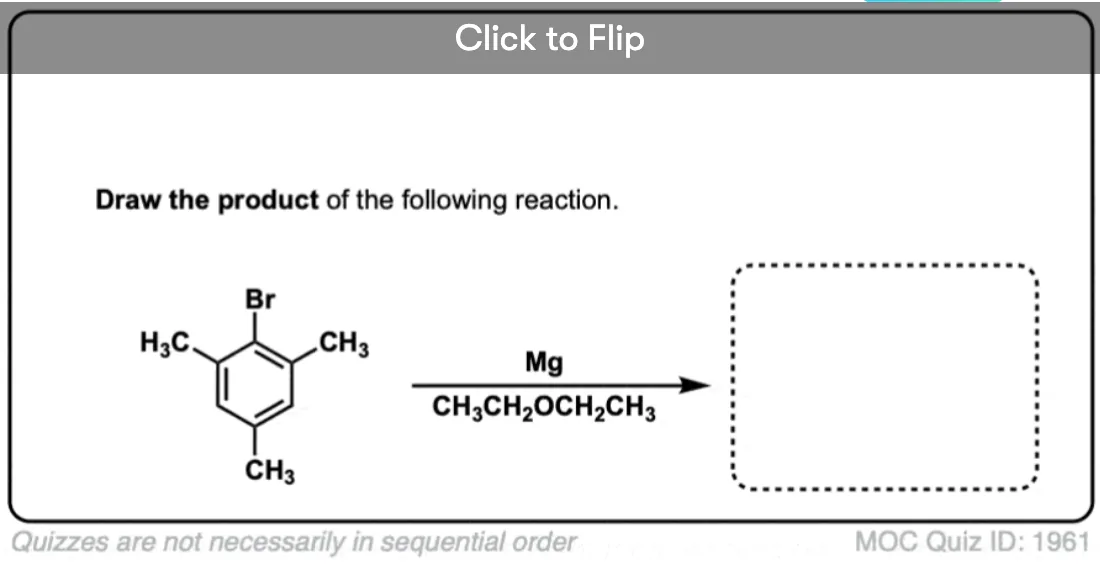
Become a MOC member to see the clickable quiz with answers on the back.
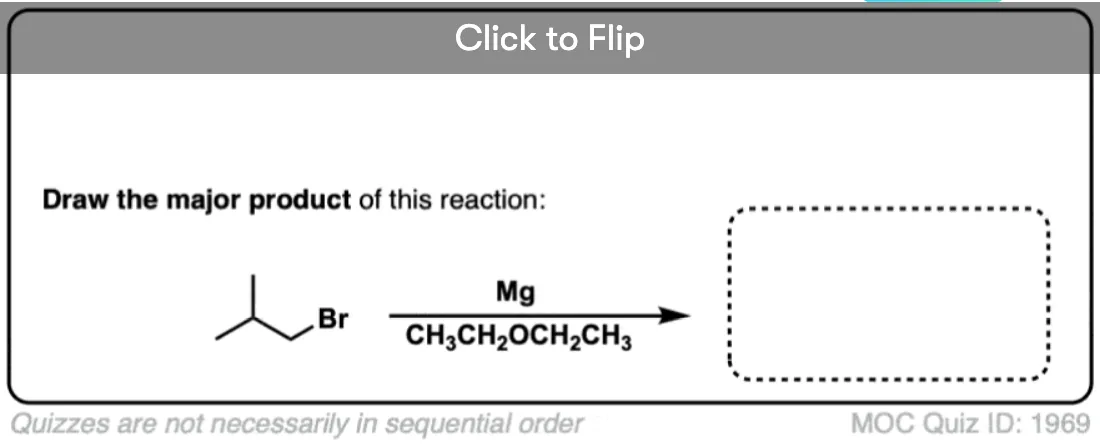
Become a MOC member to see the clickable quiz with answers on the back.
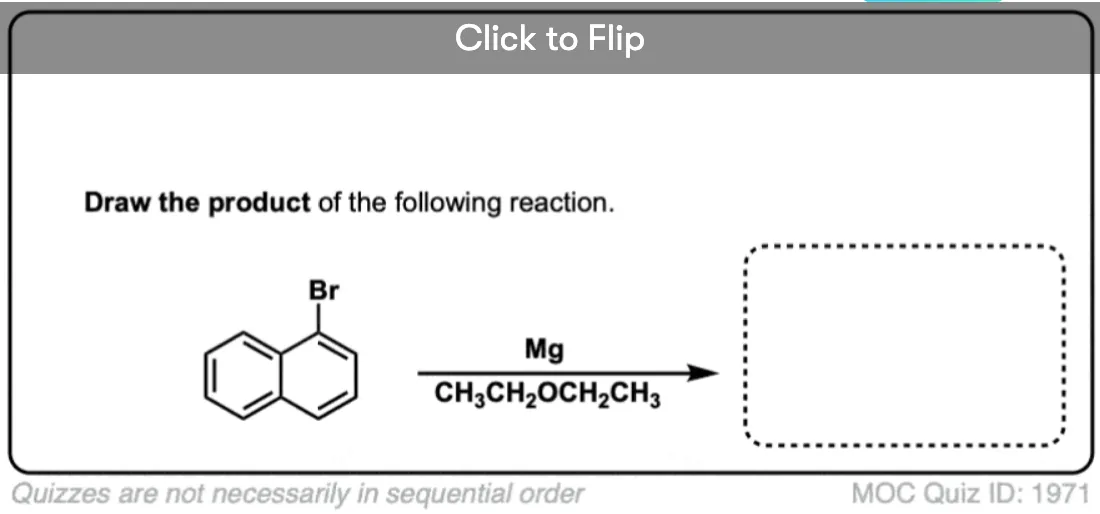
Become a MOC member to see the clickable quiz with answers on the back.
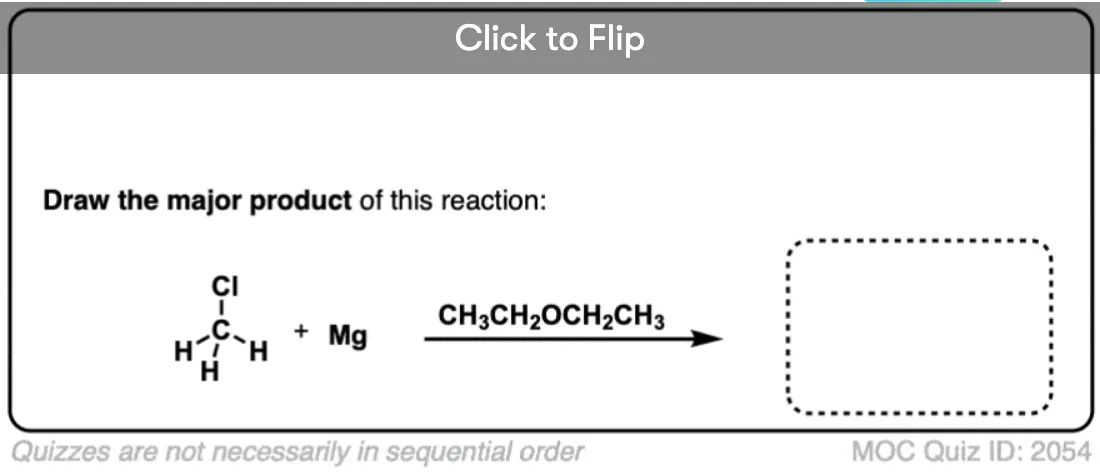
Become a MOC member to see the clickable quiz with answers on the back.
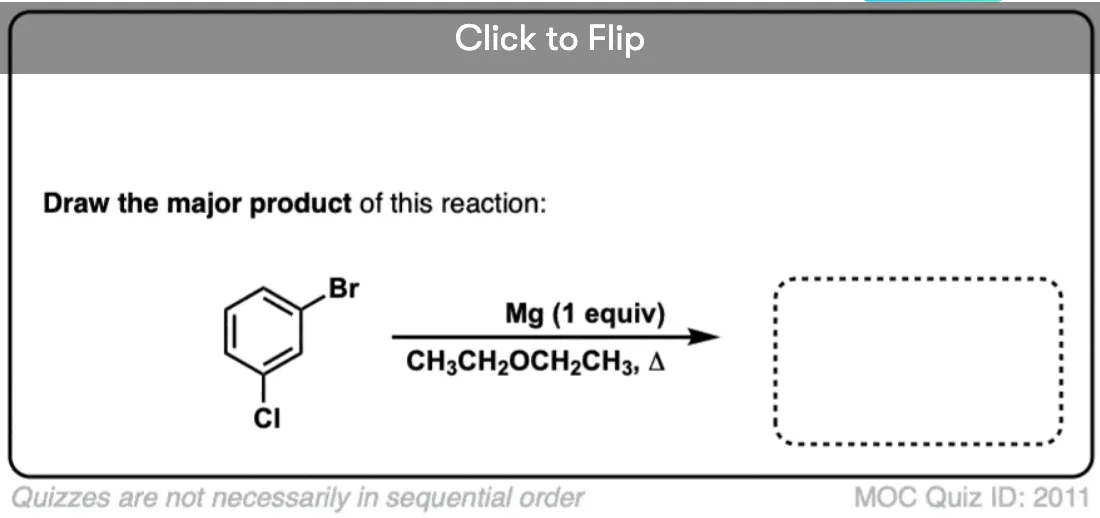
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading:
- Secondary and Tertiary Alkyllithium Compounds and Some Interconversion Reactions with Them
Henry Gilman, Fred W. Moore, and Ogden Baine
Journal of the American Chemical Society 1941, 63 (9), 2479-2482
DOI: 1021/ja01854a046
Prof. Henry Gilman (Iowa State) was a pioneer in organometallic chemistry in the first half of the 20th century. In this paper he describes the synthesis and reactivity of various alkyllithiums (n-butyllithium, s-butyllithium, isopropyllithium, and t-butyllithium). The synthesis is from the alkyl halide and lithium metal, as can be seen in the experimental section. - t-Butyllithium
Paul D. Bartlett, C. Gardner Swain, and Robert B. Woodward
Journal of the American Chemical Society 1941, 63 (11), 3229-3230
DOI: 1021/ja01856a501
This communication is from some legendary figures in organic chemistry and describes the preparation of t-butyllithium. - 2-PHENYLPYRIDINE
C. W. Evans and C. F. H. Allen
Org. Synth. 1938, 18, 70
DOI: 10.15227/orgsyn.018.0070
The first step in this procedure is a preparation of phenyllithium from bromobenzene and lithium metal. Organic Syntheses is a reputable source of reproducible and independently tested synthetic organic procedures. - The mechanism of the lithium – halogen Interchange reaction : a review of the literature
Bailey, W. F.; Patricia, J. J.
Organomet. Chem. 1988, 352 (1-2), 1-46
DOI: 10.1016/0022-328X(88)83017-1
In modern organic chemistry, organolithium reagents are rarely prepared from scratch (i.e. using Li metal), due to the ready availability of alkyllithium reagents from vendors (e.g. MeLi, the BuLi reagents, PhLi, etc.). Instead, these reagents can be used to form other organolithium species through a process known as lithium-halogen exchange. - What’s Going on with These Lithium Reagents?
Hans J. Reich
The Journal of Organic Chemistry 2012, 77 (13), 5471-5491
DOI: 1021/jo3005155
Prof. Hans Reich (U. Wisconsin-Madison) has spent his career studying the behavior of organolithium species, and this is an account of his research and the surprising findings he made. This is classic Physical Organic chemistry. - Grignard, V. C. Acad. Sci. 1900, 130, 1322-1324
The original paper by Victor Grignard describing a new method for alcohol synthesis from hydrocarbons. - Victor Grignard and Paul Sabatier: Two Showcase Laureates of the Nobel Prize for Chemistry
Henri B. Kagan
Angew. Chem. Int. Ed. 2012, 51, 2-9
DOI: 10.1002/anie.201201849
For those interested in the history of science, this is a historical perspective on the lives of Victor Grignard and Paul Sabatier, and gives insight into their lives, how they made their seminal discoveries, and the impact of their work, among other things. - Mechanical activation of magnesium turnings for the preparation of reactive Grignard reagents
Karen V. Baker, John M. Brown, Nigel Hughes, A. Jerome Skarnulis, and Ann Sexton
The Journal of Organic Chemistry 1991 56 (2), 698-703
DOI: 10.1021/jo00002a039
Sometimes the formation of a Grignard reagent using Mg metal can be challenging, and various methods for activating the metal surface have been developed, including mechanical activation by dry-stirring Mg turnings under an inert atmosphere for several hours.
The following 3 papers are mechanistic studies on the formation of Grignard reagents: - The Mechanism of Formation of Grignard Reagents: Trapping of Free Alkyl Radical Intermediates by Reaction with Tetramethylpiperidine-N-oxyl
Karen S. Root, Craig L. Hill, Lynette M. Lawrence, and George M. Whitesides
Journal of the American Chemical Society 1989 111 (14), 5405-5412
DOI: 10.1021/ja00196a053 - Mechanism of Grignard Reagent Formation. The Surface Nature of the Reaction
M. Walborsky and Janusz Rachon
Journal of the American Chemical Society 1989 111 (5), 1896-1897
DOI: 10.1021/ja00187a063 - Mechanism of Grignard Reagent Formation. Comparisons of D-Model Calculations with Experimental Product Yields
John F. Garst and Brian L. Swift
Journal of the American Chemical Society 1989 111 (1), 241-250
DOI: 10.1021/ja00183a037 - 4-METHOXY-4′-NITROBIPHENYL
K. Stille, Antonio M. Echavarren, Robert M. Williams, and James A. Hendrix
Org. Synth. 1993, 71, 97
DOI: 10.15227/orgsyn.071.0097
The second step in this procedure includes the synthesis of p-anisylmagnesium bromide, which can be a tricky Grignard reagent to prepare and requires special activation of Mg with methyl iodide. - The Grignard Reaction – Unraveling a Chemical Puzzle
Raphael Mathias Peltzer, Jürgen Gauss, Odile Eisenstein, and Michele CascellaJournal of the American Chemical Society 2020 142 (6), 2984-2994
DOI: 10.1021/jacs.9b11829
Recent (and open-access) article that suggests that the reaction “does not occur via a single process but by an ensemble of parallel reactions.”
We are all aware that doctoral students have to remember a lot of things when studying. It has been observed in various instances that if memorization is practiced on a regular basis and in certain instances, the student may forget something, however, when we apply the method of scientific memorization, it’s very simple.
To help you, here is a fantastic mnemonic method site I discovered while researching.
https://sciencemnemonic.com
Should be a carbanion. Is there a typo somewhere I’m not seeing?
The grignard reagent (say Ch3mgbr) can act as a base here thereby abstracting the acidic hydrogen from terminal triple bond carbon. But still we get a carbocation!!
Why can’t 3-bromoprop-1-yne form grignard reagent?
First, draw out (or at least imagine) the Grignard that would form from 3-bromoprop-1-yne.
Next, think about how basic this species is. Remember the pKa’s of alkanes are around 50 (this one would be slightly less, but still).
Are there any acidic protons around that could react with this very basic Grignard reagent?
Thanks for sharing!
So because of Schlenk equilibrium, Et2O or THF is necessary for the synthesis of Grignard reagents, right? My research project now requires Grignard reagents, but THF or Et2O is not good for the whole reaction system, I’m trying to synthesize Grignard reagents in some non-polar solvents such as toluene, I’m wondering if it’s possible?
Do they need to be Grignards? Or can they be organolithium reagents?
Yeah, we need Grignard reagents because lithium introduces impurity to our system
What happen when lithium bromide reacts with water ? Do it dissociate into ions or not?
Also can you tell me the reaction when lithium bromide added to silk fibrion protein?
Yes, LiBr dissociates, although you’d have to look up the solubility in water to get a precise number.
how activate the lithium metal for synthesis of organolithium on industrial and laboratory scale?
From my friend Jeff, who regularly used Li metal to make lithium di-tertbutyl diphenylide (LDBB).
Lithium prep:
tweezers
1 pair of needle-nose pliers
Lithium wire (usually with a small % of Na in it) in oil
3 beakers: hexane, THF, and MeOH, and then a 4th with reaction solvent (or just the reaction itself, with an ARGON atmosphere, preferably)
Cut wire into pieces with scissors or pliers and put into hexanes
Use tweezers to fish out a piece of Li and squish with pliers and return to hexanes
Once all the pieces have been pressed, use tweezers and take a piece out of hexanes and dip in THF then MeOH then THF again and then into the reaction solvent/flask.
Repeat with remaining pieces of Li
is there a reason that orgo students learn about both grignard and organolithium reagents if they do the same thing which acts as a strong nucleophile/base? is there a certain advantage one has over the other
Good question. For most purposes it’s like choosing between Chuck Norris and Jackie Chan. They both get the job done. Organolithium reagents are are generally less compatible with a range of functional groups than Grignards are.
One thing that organolithium reagents can do that Grignards cannot is add to carboxylic acids. For your purposes that’s likely the most important distinction. [Advanced purposes: organolithium reagents will also do a reaction called “lithium halogen exchange” which Grignards generally don’t do].
Is this covered in your revision guide?
It’s in the reagent guide, yes!
Thank you for sharing. Hope to hear more from you.
May also want to mention that these days, chemists usually buy their Grignards from Aldrich rather than make them.
True enough! Although it’s handy to know how they’re made if you need a fairly obscure one.
Great post. Two observations, though:
– “Note that Grignards can be made from alkyl or alkenyl chlorides, bromides, and iodides…”; you could add aryl halides, especially since Example #2;
– the last link (for the mechanism of Grignard formation) actually leads to the last figure in this post.
thank you, as always.