Carboxylic Acid Derivatives
Nucleophilic Acyl Substitution (With Negatively Charged Nucleophiles)
Last updated: May 28th, 2026 |
Nucleophilic Acyl Substitution (With Negatively Charged Nucleophiles)
Yes, there are a lot of reactions of carboxylic acid derivatives to learn! In this article we’ll explore what is hands-down the most important pathway: nucleophilic acyl substitution.
There is good news, though:
- with negatively charged nucleophiles nucleophilic acyl substitution tends to follow a simple two-step mechanism (addition-elimination)
- furthermore, for all intents and purposes, NAS reactions behave a lot like acid-base reactions. If you recall that “stronger acid plus stronger base gives weaker acid plus weaker base”, (otherwise known as The Principle of Acid-Base Mediocrity) you’ll be well on your way to being able to predict whether or not a given nucleophilic acyl substitution reaction will happen.
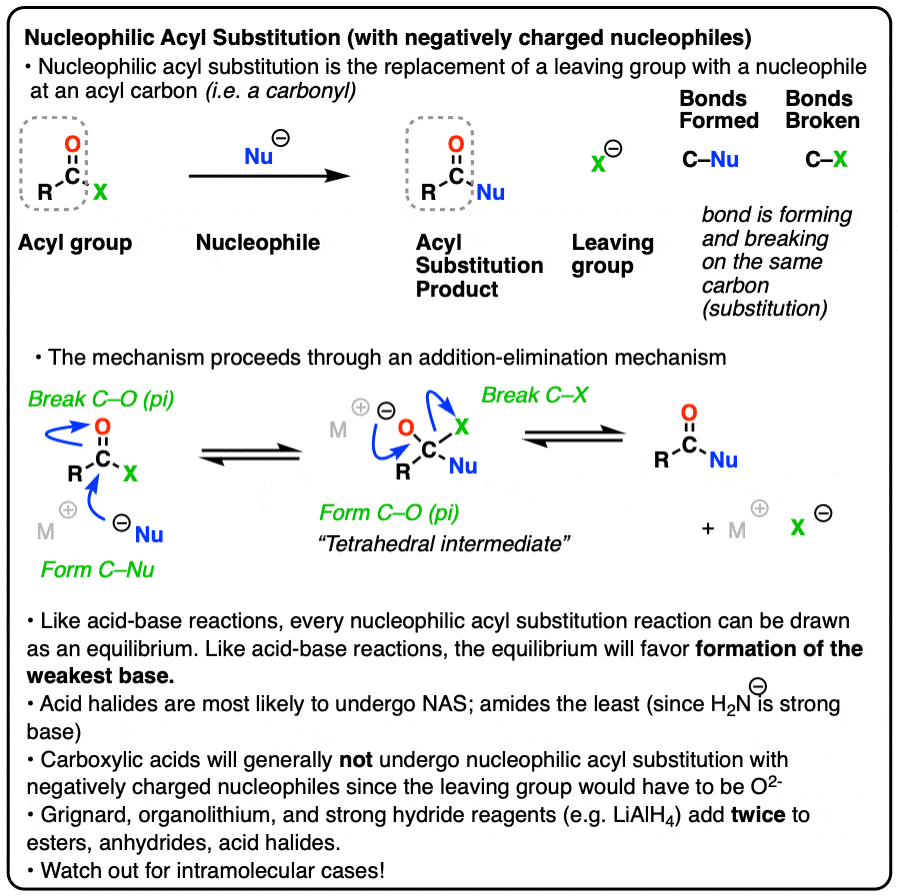
Table of Contents
- Nucleophilic Acyl Substitution (NAS)
- Three Nucleophilic Acyl Substitution Reactions (That Work Pretty Well)
- The Addition-Elimination Mechanism for Nucleophilic Acyl Substitution With a Negatively Charged Nucleophile
- Some Nucleophilic Acyl Substitutions That DON’T Work
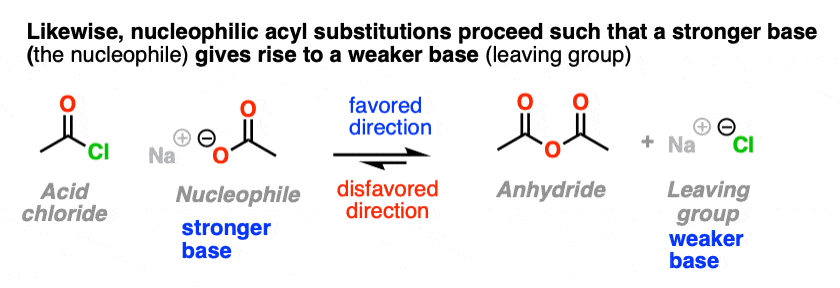
- NAS Reactions Are Favored When The Leaving Group Is A Weaker Base Than The Nucleophile
- Some Quick Exercises
- Carboxylic Acids… Are Acids!
- Saponification
- Intramolecular Nucleophilic Acyl Substitution
- Grignard Reagents and LiAlH4 Add Twice
- Neutral Nucleophiles and Acid Catalysis
- Summary
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Nucleophilic Acyl Substitution
Nucleophilic acyl substitution is a reaction where a nucleophile forms a new bond with the carbonyl carbon of an acyl group with accompanying breakage of a bond between the carbonyl carbon and a leaving group.
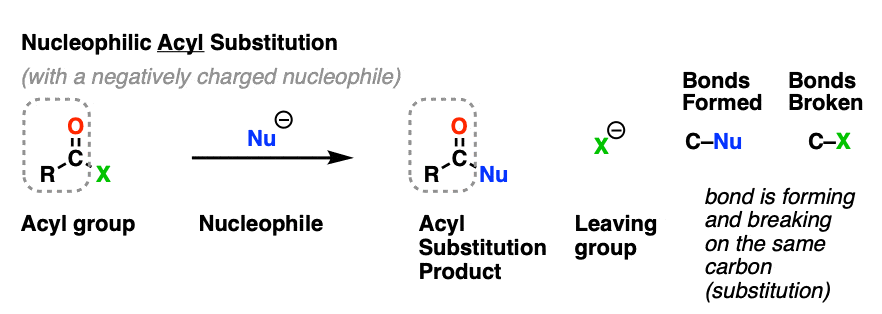
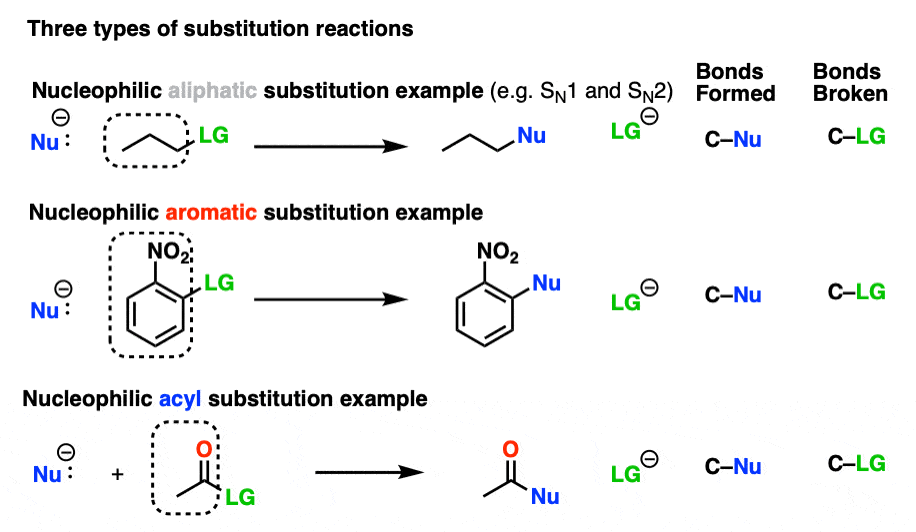
This is classified as a substitution reaction because we are forming and breaking a bond on the same carbon. A carbon-nucleophile bond forms, and a carbon-leaving group bond breaks.
Although the mechanism is different, nucleophilic acyl substitution superficially resembles substitution reactions we’ve seen before such as nucleophilic aliphatic substitution and nucleophilic aromatic substitution.
Hover to see other families of nucleophilic substitution reactions or click on the link.
{kind=link}
In this article we will specifically cover examples of nucleophilic acyl substitution reactions with negatively charged nucleophiles. In a subsequent article we’ll cover cases where an acid catalyst is employed for nucleophilic acyl substitution, such as the Fischer esterification, acidic hydrolysis of esters, and many other examples.
(For neutral nucleophiles, as well as nucleophilic substitution under acidic conditions, see Addition-Elimination of Neutral Nucleophiles)
2. Three Nucleophilic Acyl Substitution Reactions (That Work Pretty Well)
Here are three classic examples of nucleophilic acyl substitution reactions that work well.
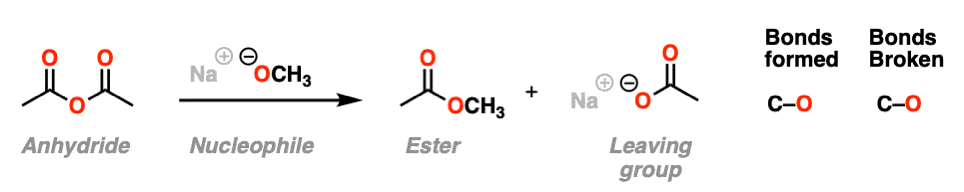
In the first, acid chlorides react with carboxylates (the conjugate base of carboxylic acids, acting as a nucleophile here) to give an anhydride. A new C-O bond is formed and a C-Cl bond is broken.
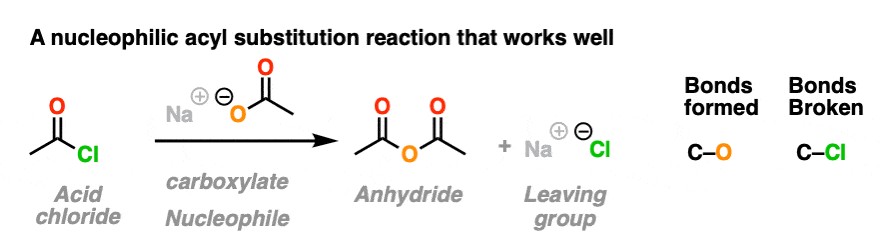
In the second example, we treat an anhydride with an alkoxide and form a new ester. A C-O bond is formed and a C-O bond is broken. In this case the carboxylate is our leaving group.
The third example involves the use of the bulky hydride source di-isobutyl aluminum hydride (DIBAL-H) to an ester at low temperature to form an aldehyde. A new C-H bond is formed and a C-O bond breaks.
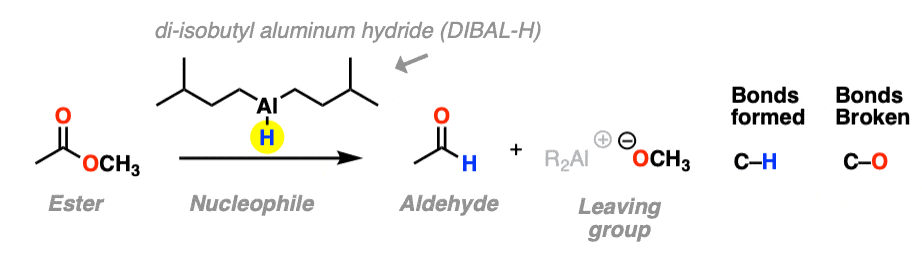
At low temperature (–80° or so) this works well; higher temperatures result in over-reduction. [Note 1]
3. The Addition-Elimination Mechanism for Nucleophilic Acyl Substitution With a Negatively Charged Nucleophile
So how might these reactions work? They’re not SN2-type reactions, since SN2 really only is effective on sp3 hybridized carbons. Nor have they been determined to be SN1-type reactions where the leaving group leaves first and then the nucleophile attacks (although this describes the Friedel-Crafts acylation pretty well!)
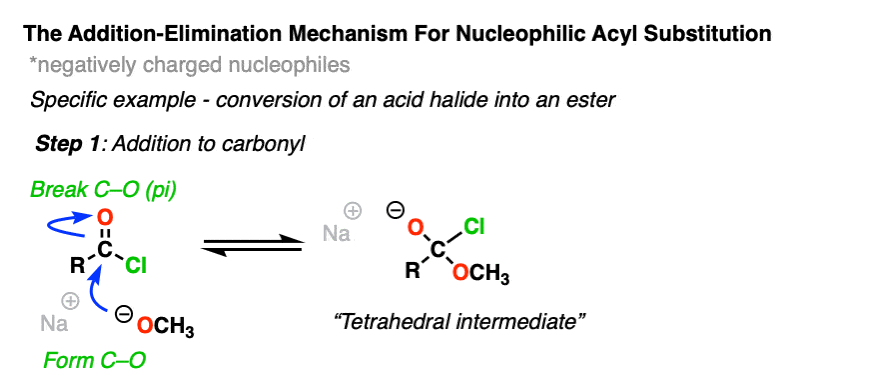
Instead, experiments provide strong evidence for a two-step addition-elimination mechanism that proceeds through a tetrahedral intermediate. [Note 2 ]
The first step should be very familiar from the reactions of aldehydes and ketones. [See – The simple two-step pattern for 7 key reactions of aldehydes and ketones]
The carbon of a C=O group is electrophilic and reacts readily with the negatively charged nucleophile through an addition mechanism [Step 1, form C-Nu, break C-O (pi) ]
Now what? Through a reversal of the addition mechanism, which goes by the name elimination, the tetrahedral intermediate could revert back to starting materials, regenerating the starting acyl group.
Or, if we have a sufficiently good leaving group, elimination could result in re-formation of the C-O pi bond with loss of a leaving group.
[Step 2, form C-O (pi), break C-LG]
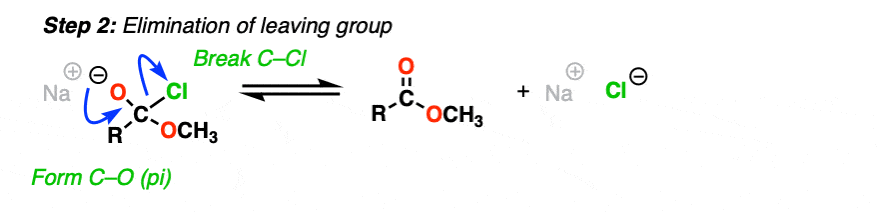
The result is that a substitution has occurred.
This two-step mechanism is known as addition-elimination and it is extremely common in the chemistry of carboxylic acid derivatives (i.e. acyl groups). In cases where a neutral nucleophile is used, or acid catalysts are present, some intervening proton transfer steps can lengthen the mechanism (e.g. PADPED) but the core process is still addition-elimination. (For more on carbonyl elimination – see Addition-Elimination)
4. Nucleophilic Acyl Substitutions That Don’t Work
So far, so good, right? We just add our nucleophile to the acyl group. The nucleophile adds, the leaving group leaves, and we get our new substitution product. Simple!
Here’s two more examples of nucleophilic acyl substitution reactions.
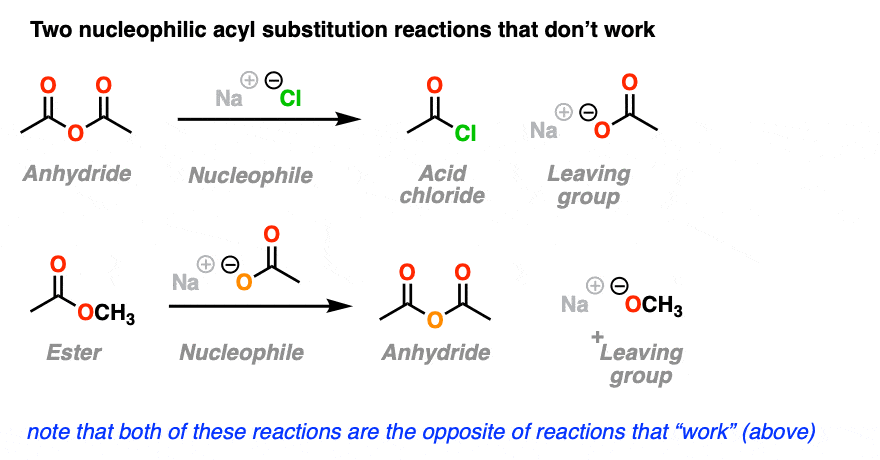
You may have noticed that the reactions shown here are the exact opposite of the nucleophilic substitution reactions that “worked” in the section above, except we’ve just switched the identities of the nucleophile and the leaving group.
(Recall that a leaving group is just a nucleophile acting in reverse, and vice-versa).
The trouble is, although you can draw these up “on paper” as a nucleophilic acyl substitution reactions, in the real world you could wait around for continental drift to make Pangaea II and they still wouldn’t happen.
So why is nucleophilic acyl substitution favored in one direction and not in the other?
5. NAS Reactions Are Favored When The Leaving Group Is A Weaker Base Than The Nucleophile
Let’s take a very brief trip back to Org 1, where we learned the Golden Rule of Acid-Base Reactions, otherwise known as the Principle of Acid-Base Mediocrity: the favored direction of an acid-base reaction is one where a stronger acid reacts with a stronger base to give a weaker acid and a weaker base. (See post: How to Use a pKa Table)
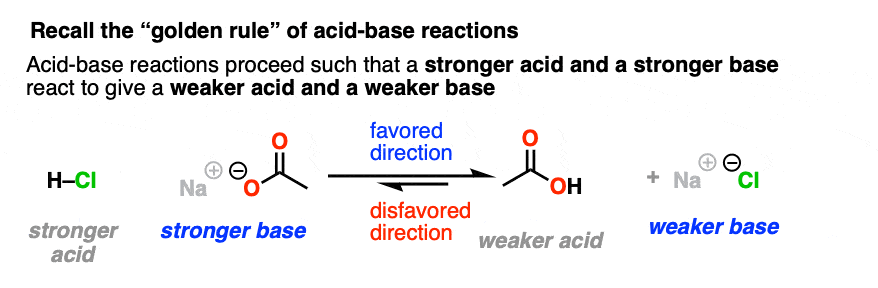
We intuitively know by this point that adding table salt to water will not result in a raging cloud of HCl gas and caustic sodium hydroxide, whereas adding concentrated HCl to pellets of NaOH is not something a sober person does without a lot of safety precautions.
So when trying to consider which direction is favored in a nucleophilic acyl substitution reaction, expect that it will favor formation of the weaker base.
Just like water flows downhill, NAS reactions will “flow” such as to give the more stable anion (weaker base).
(It might also help to think of “weaker base” as another way of saying, “a better-stabilized electron pair”. )
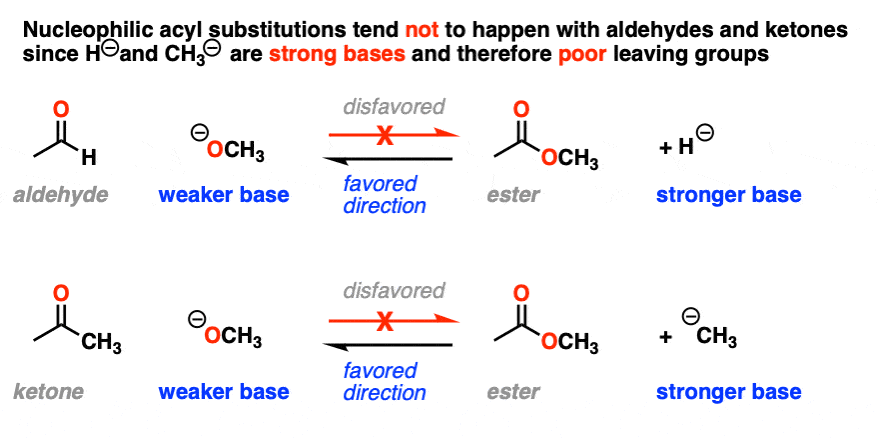
It also helps us understand why nucleophilic acyl substitution reactions don’t work with aldehydes and ketones; it would result in a hydride ion (H-) or carbanion (R -), both of which are very strong bases. [Note 3]
It might be helpful to put this into the form of a chart, if we chart pKa (of the conjugate acid) versus nucleophile/leaving group.
Reactions that involve a “downhill” flow from nucleophile to leaving group are favored, whereas those that require an “uphill” conversion of a weaker base to a stronger base will be disfavored.
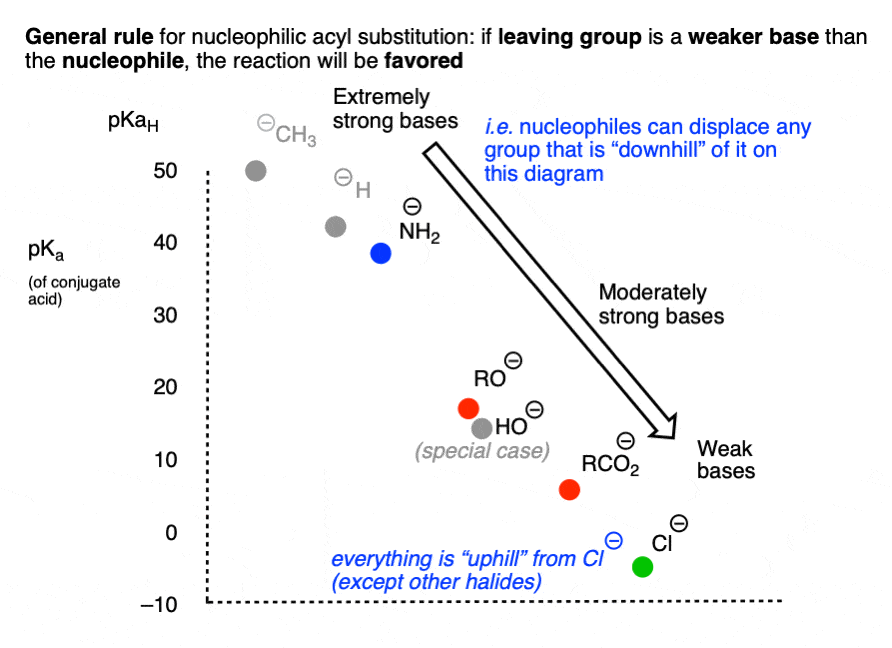
Another way of expressing the same idea is to make a chart like the following, where the weakest base (Cl-) is incapable of performing any nucleophilic acyl substitution reactions which might result in a stronger base:
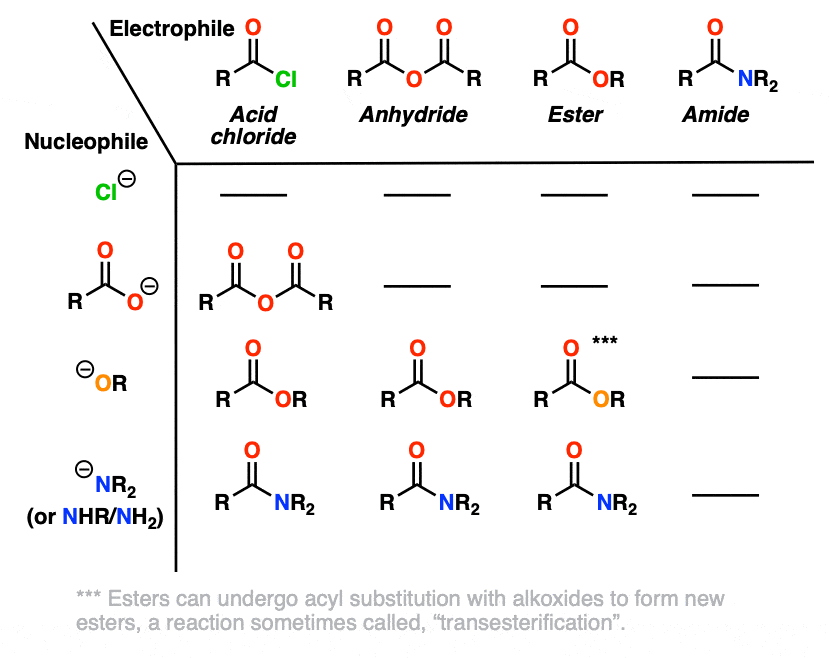
For reasons that will soon become clear, HO(-) has been omitted from this chart. [Note 4]
Hover on this link to see a simplified reaction energy diagram or click on this link.
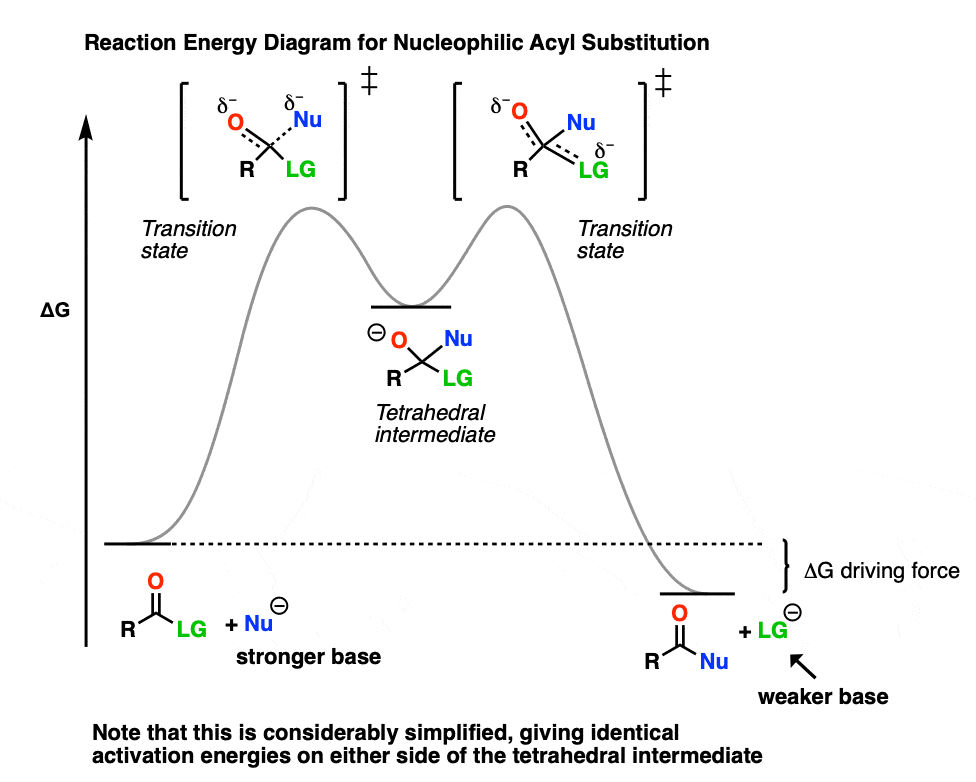{kind=link}
6. Some Quick Exercises
To let this concept settle in, why not quiz yourself with some quick exercises?
First up, thioesters.
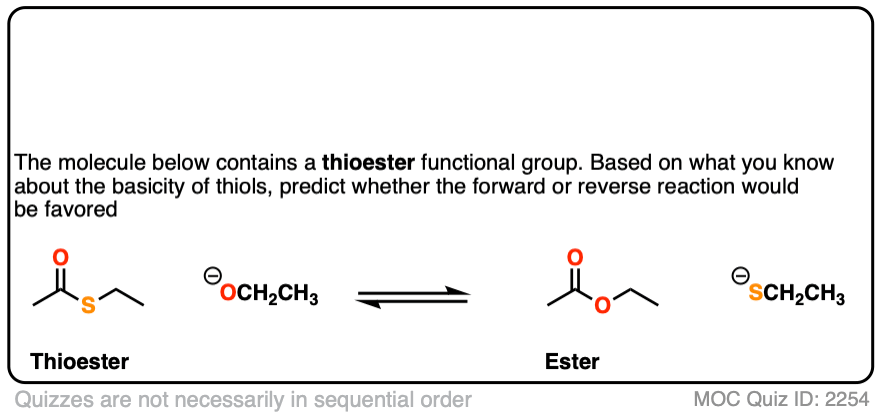 Click to Flip
Click to Flip
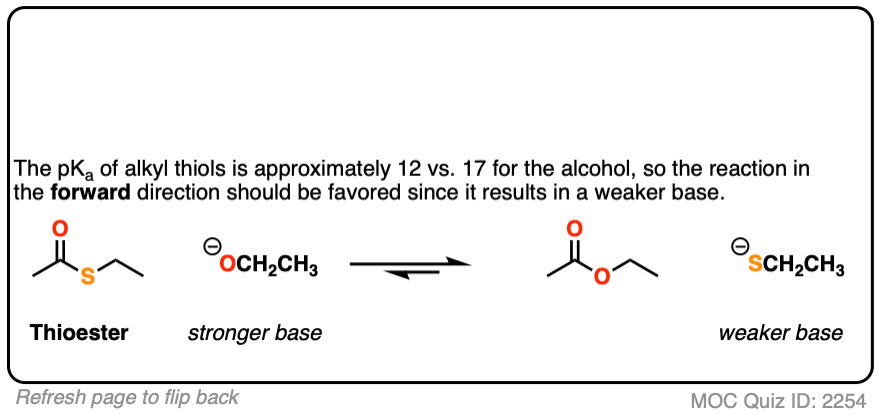
[Note 5]
Second, an example of converting an ester into another ester, a reaction called, “transesterification”.
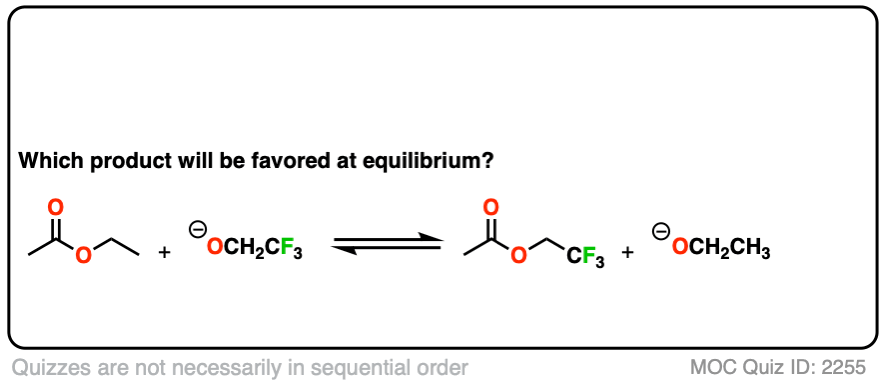 Click to Flip
Click to Flip
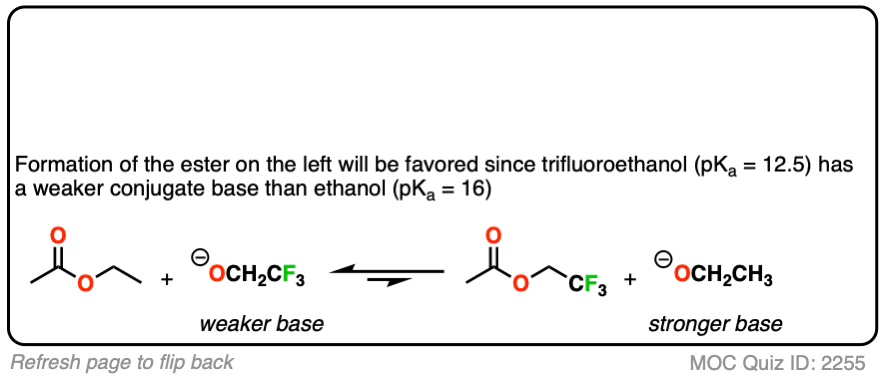
You might ask – what if there isn’t a big pKa difference between the nucleophile and leaving group? What if we are starting with the ethyl ester, for example, and want to convert it into the methyl ester?
Since the pKa‘s are similar, one way to do it would be to “flood the zone” with nucleophile (i.e. use a large excess) to drive the equilibrium towards the desired product.
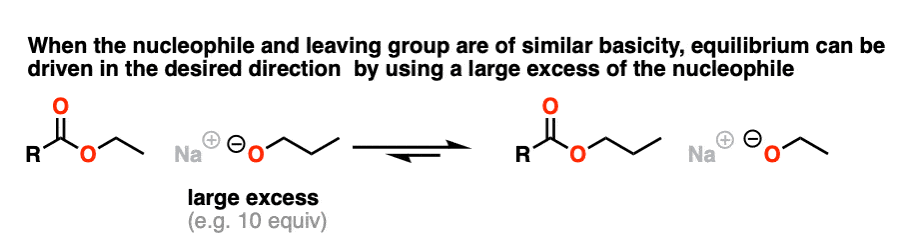
I’m not saying this is the best way (converting to an acid halide or anhydride followed by treatment with an alcohol would be superior) but it’s still workable.
7. Carboxylic Acids…. Are Acids!
Now that we have a handle on the key factor that affects nucleophilic acyl substitutions, let’s look at an example that should seem straightforward… but isn’t.
NaOH is the conjugate base of water (pKa 14) and NaOCH3 is the conjugate base of CH3OH (pKa 16).
So given everything we know, the reaction should slightly favor the product.. right?
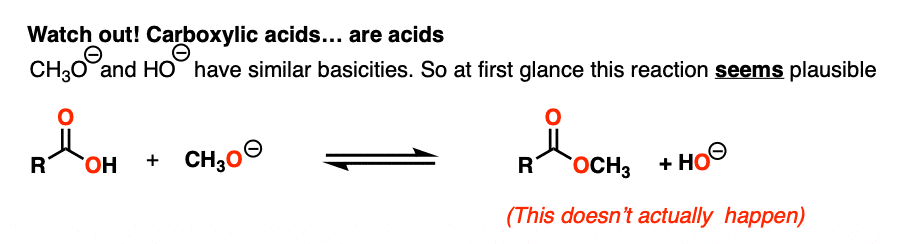
Actually… no!
That’s because there’s an extra factor we need to consider first. Carboxylic acids are acids, and acid-base reactions are fast. (See: Acid-Base Reactions Are Fast)
So the first thing that happens is not addition, but deprotonation of the carboxylic acid to give a carboxylate.
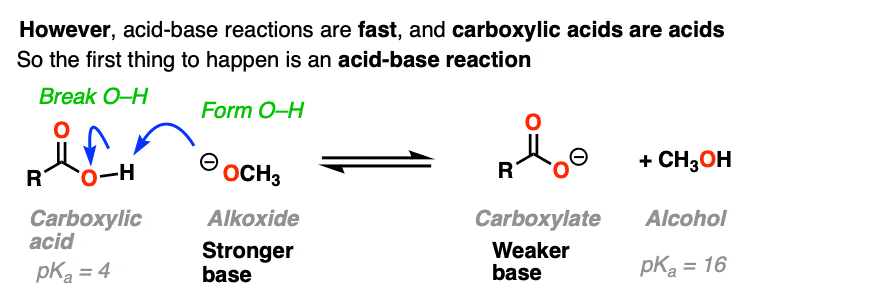
If nucleophilic acyl substitution were to happen at this point, it would have to add to this carboxylate which would make a di-anion with two negative charges on the same molecule (if that sounds unstable – it is!!).
Furthermore, the leaving group to make the ester would not be HO(-).
It would be O (2-) !
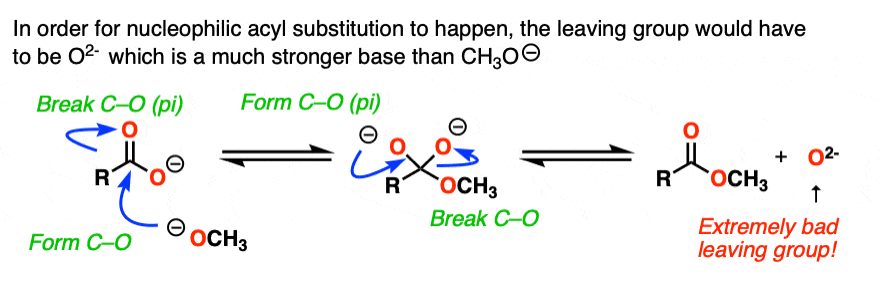
With one notable exception [Note 6] , that won’t happen.
Note that carboxylic acids can be converted to esters, but that the reaction requires acid (e.g. the Fischer esterification). In this article we’re focusing on negatively charged nucleophiles.
8. Saponification of Esters With Base
If adding RO(–) to a carboxylic acid just results in deprotonation, what about the reverse: adding a hydroxide ion to an ester?
This is a well-known process known as saponification (or just, “basic hydrolysis”) and works extremely well.
The substitution portion of the reaction proceeds through the familiar addition–elimination process. This results in a carboxylic acid. However, since carboxylic acids are acids, and the reaction occurs under basic conditions, the resulting acid will rapidly be deprotonated by hydroxide to give the carboxylate.
Therefore to obtain the neutral carboxylic acid at the end of the reaction, one has to add acid during the workup step.
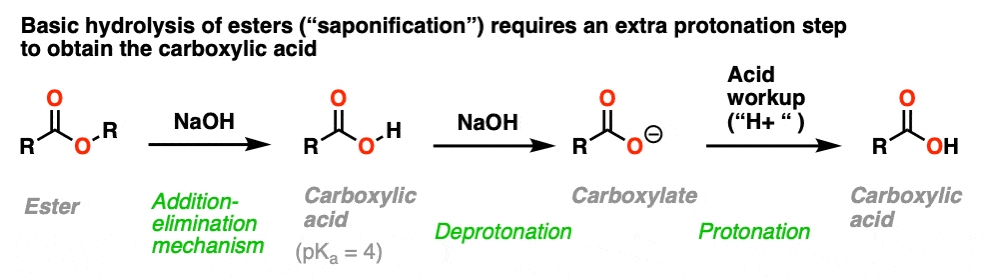
The name “saponification” comes from the classic use of this reaction in making soaps (carboxylates of long-chain fatty acids) by treating fats (which are esters of glycerol with long-chain acids) with lye (a generic term for alkali metal hydroxides).
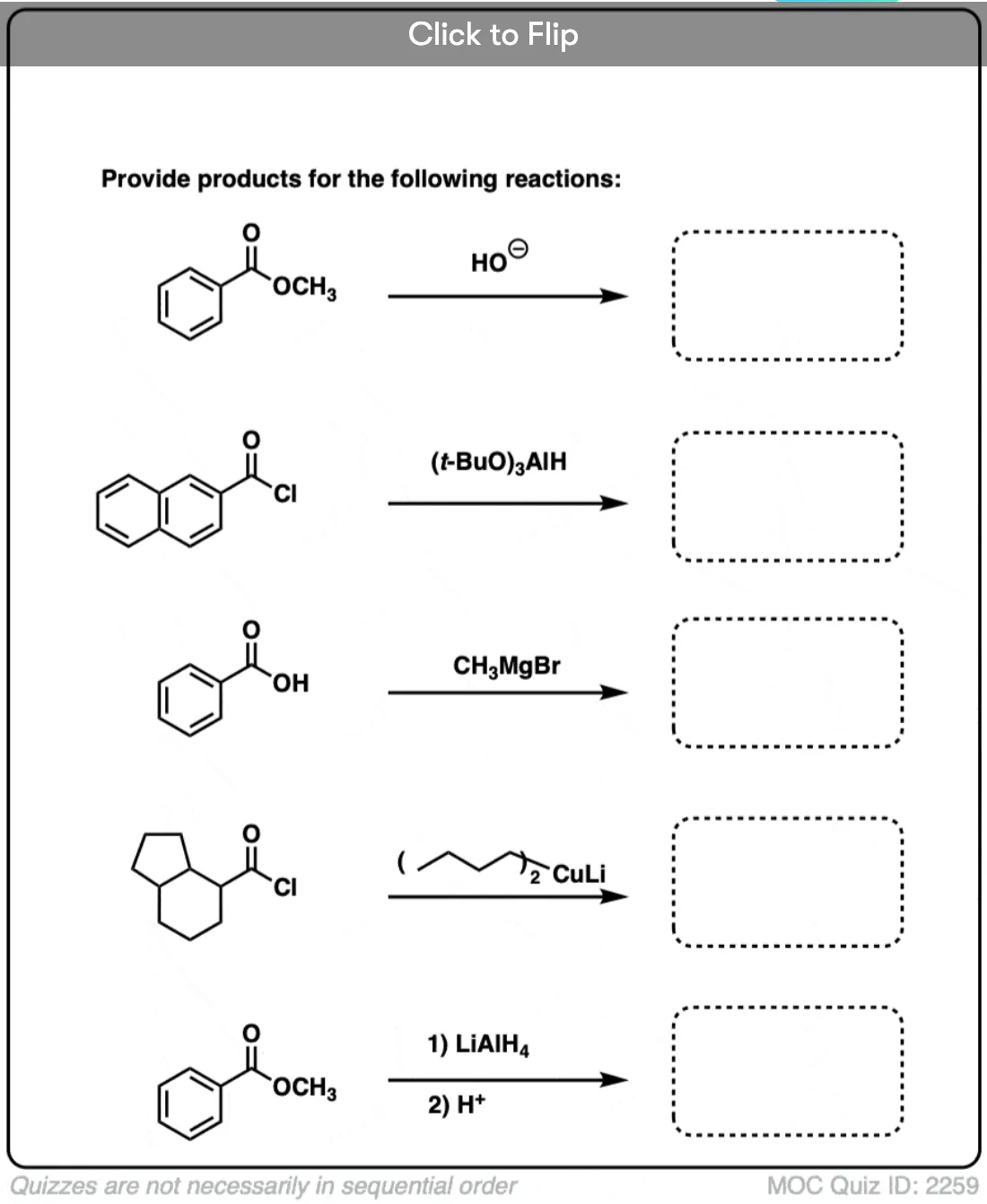
9. Intramolecular NAS
It’s always helpful to think about the intramolecular variant of any new reaction you learn. Nucleophilic acyl substitution is no exception!
If a five- or six-membered ring can be formed through nucleophilic acyl substitution, it generally will. In this example, treatment with the strong base sodium hydride (NaH) results in an alkoxide that forms a new ring, displacing methoxide [CH3O (–) ]
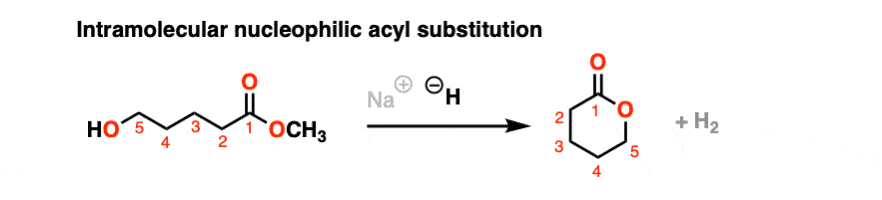
hover on this link to see the mechanism or click on this link.
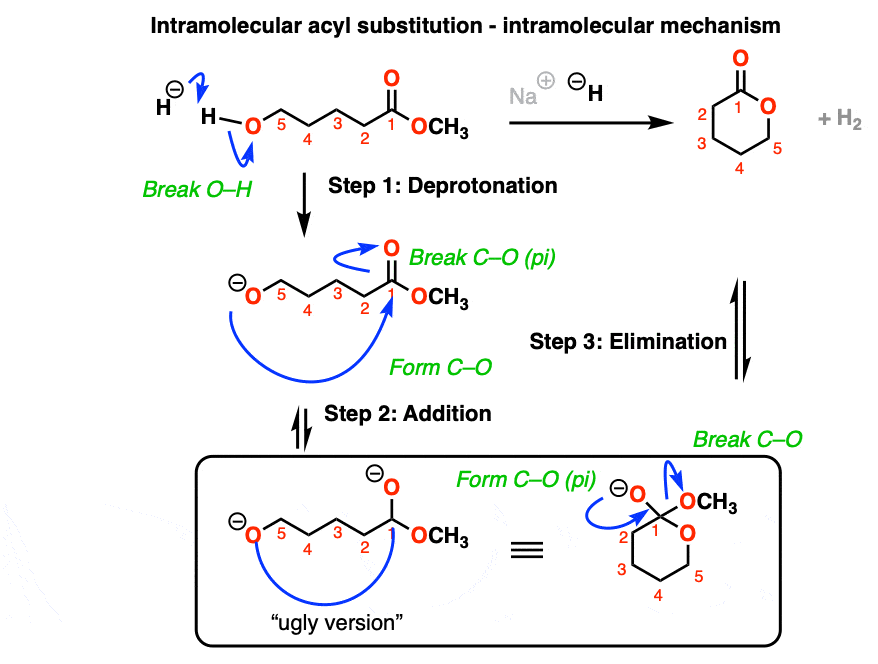{kind=link}
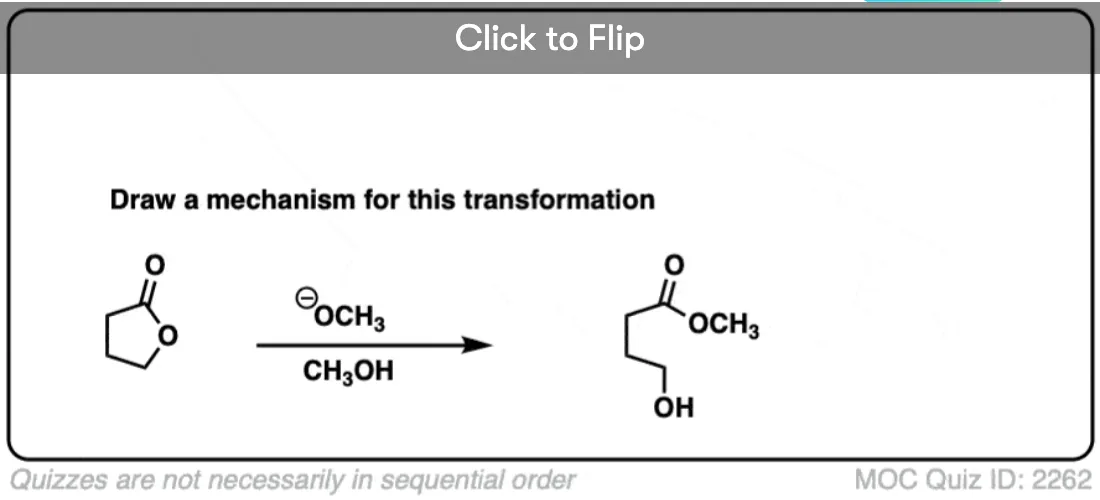
Here’s a fun example. What is happening here?
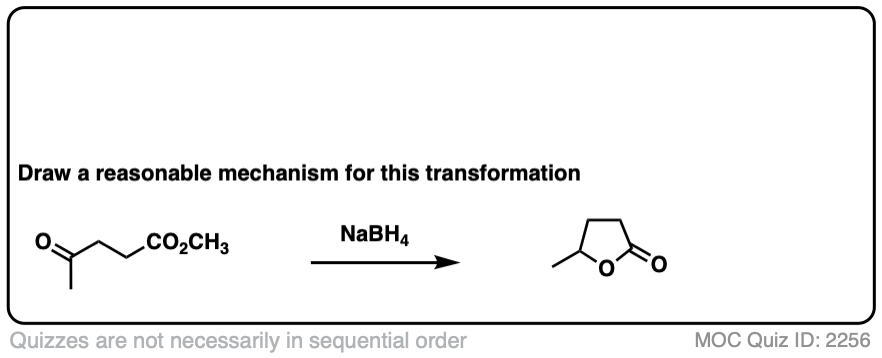 Click to Flip
Click to Flip
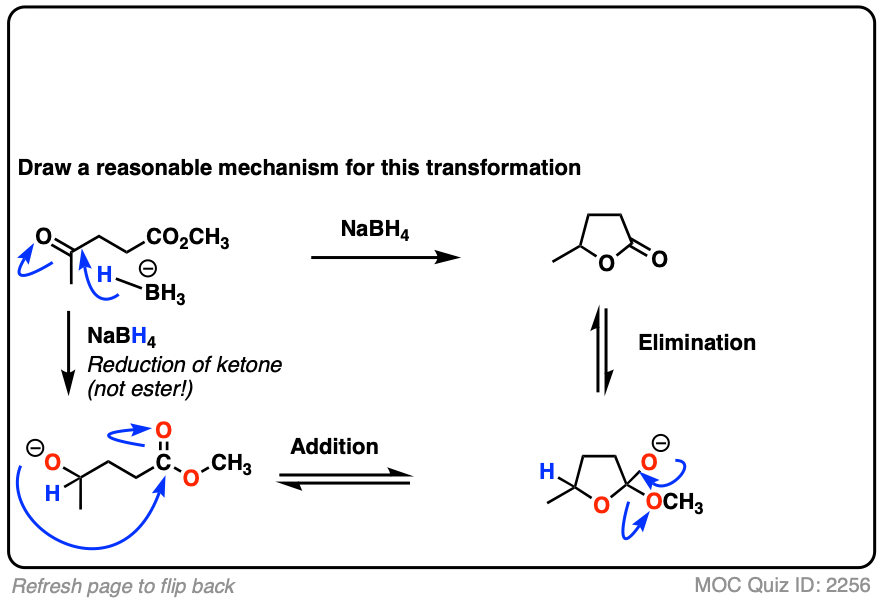
(Hint: deprotonation isn’t the only way of making an alkoxide)
10.When The Nucleophile Adds Twice – Grignards and LiAlH4
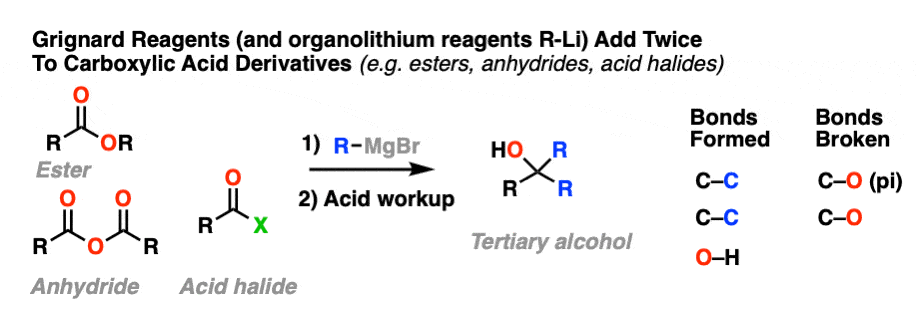
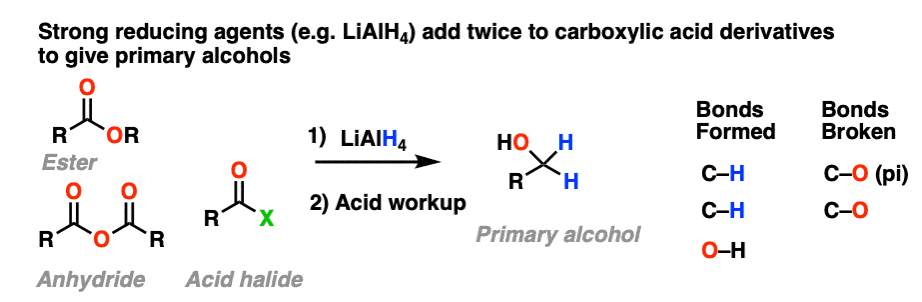
Grignard reagents and strong reducing agents like LiAlH4 tend to add to carboxylic acid derivatives twice.
Grignard reagents (and organolithium reagents) give tertiary alcohols from esters, acid halides, and anhydrides.
Strong reducing agents (e.g. LiAlH4) give primary alcohols from esters, acid halides, and anhydrides.
So how do these “double addition” reactions work?
Addition of the nucleophile R(-) or H(-) followed by elimination of the leaving group initially give a ketone (with Grignards) or an aldehyde (with LiAlH4).
If the elimination process is fast relative to addition, then the aldehyde or ketone will be formed in the presence of the strong nucleophile before all the starting ester (or other carboxylic acid derivative) is consumed.
Since ketones (and aldehydes) are better electrophiles than esters (i.e. they react faster with nucleophiles), a situation results that I sometimes affectionately call a Cookie Monster Reaction: the reaction forms a product that is more reactive towards the nucleophile than the starting material.
(because the Cookie Monster can’t stop after just one cookie).
This results in a second addition reaction occurring, which becomes an alcohol after addition of acid. (Even if one equivalent of Grignard is used, the product is still a tertiary alcohol and unreacted ester.)
So the final sequence ends up being addition–elimination–addition followed by a final workup (protonation).
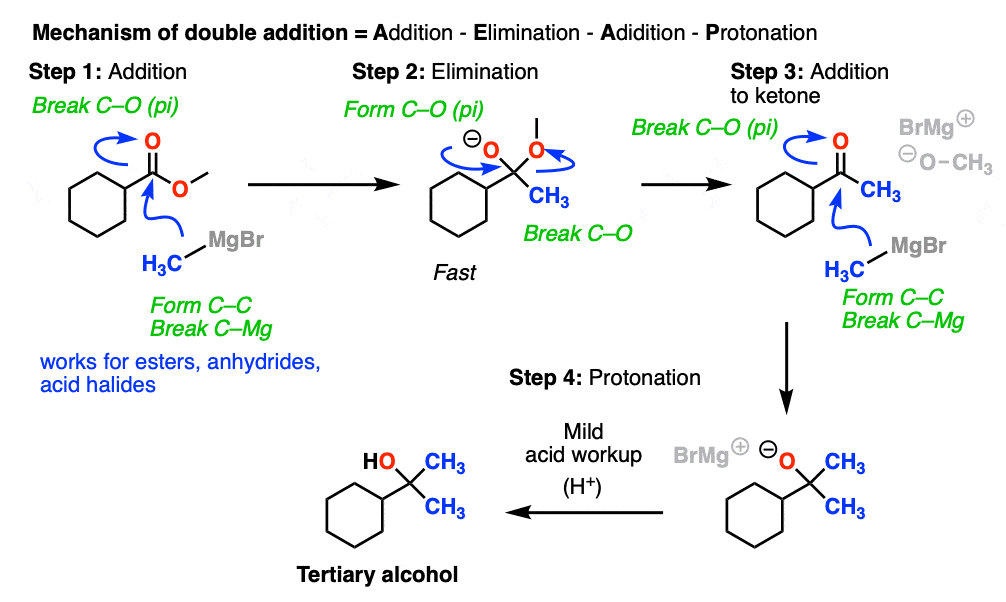
Here is a specific example:
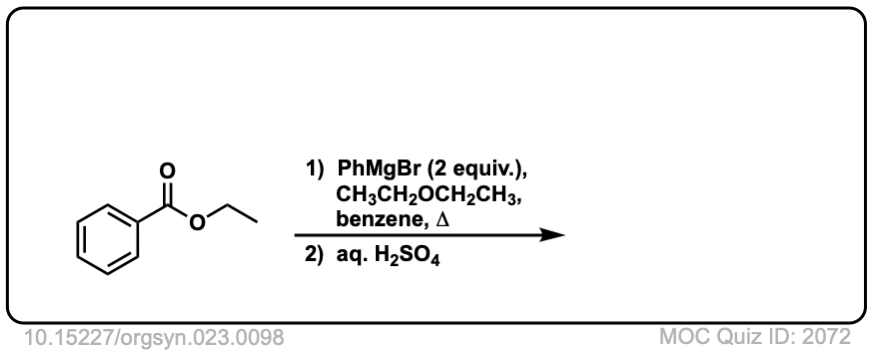 Click to Flip
Click to Flip
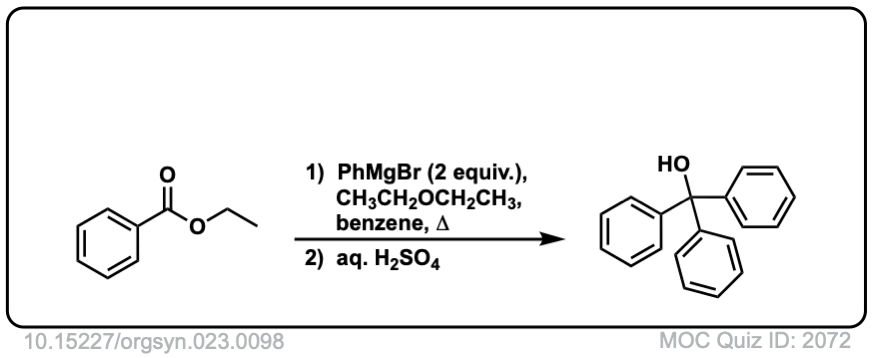
It is possible to get addition to acid halides to stop at the ketone stage by using organocuprates (Gilman reagents) which are not nearly as ravenous as Grignard or organolithium reagents.
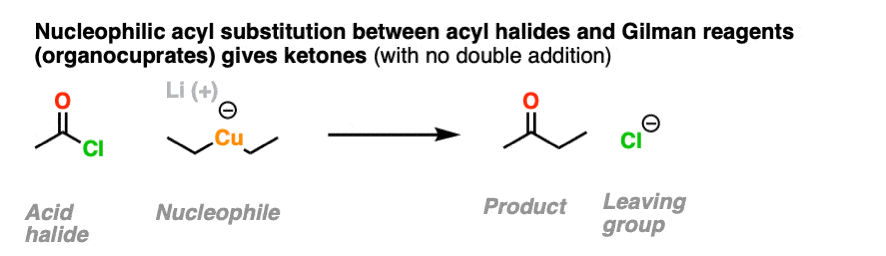
11. Neutral Nucleophiles and Acid Catalysis
This whole article has been confined to the reactions of negatively charged nucleophiles, such as Grignards, hydrides, amide bases, alkoxides, carboxylates, and halides.
We’ve seen that under these conditions, the direction of the equilibrium is almost entirely dictated by the stability of the leaving group. This makes it extremely difficult to perform the substitution reactions of amides, for example, since R2N(-) is such a strong base.
Is there another way?
Yes. We should expect that adding acid would help with performing nucleophilic acyl substitutions, since the conjugate acid is always a better leaving group.
And that is indeed the case. See Addition-Elimination Reactions With Neutral Nucleophiles (Including Acid Catalysis)
The tradeoff here is that acidic conditions are not compatible with extremely basic nucleophiles such as Grignards and hydride reagents.
We’ll explore nucleophilic acyl substitution under neutral and acidic conditions in the next article.
12. Summary
Nucleophilic acyl substitution reactions with negatively charged nucleophiles proceeds through an addition-elimination mechanism.
- In the first step (addition) the nucleophile attacks the carbonyl carbon, leading to a tetrahedral intermediate.
- In the second step (elimination) the carbon-oxygen pi bond is re-formed and a leaving group is displaced.
- Since we are forming and breaking a bond on the same carbon this is classified as a substitution reaction.
- The forward and backward reactions can be treated as an equilibrium. Like an acid-base reaction, the equilibrium will favor the direction which produces the weakest base.
- Carboxylic acids do not generally undergo nucleophilic substitution under basic conditions since they are so easily deprotonated and the resulting leaving group (O2-) is highly basic.
- However, esters do undergo basic hydrolysis with hydroxide ions (“saponification”) to give carboxylic acids (after protonation).
- Intramolecular nucleophilic acyl substitution reactions can occur, especially when five- and six-membered rings can be formed.
Notes
[Note: This article significantly updates a previous article from 2011 entitled, “Simplifying the Reactions Of Carboxylic Acid Reactions, Part 1].
Note 1 – Technically the first step of DIBAL-H reduction is coordination of the carbonyl oxygen to the Lewis-acidic aluminum, followed by delivery of hydride. But for our purposes we can just look at it as addition of H(-) to the carbonyl carbon.
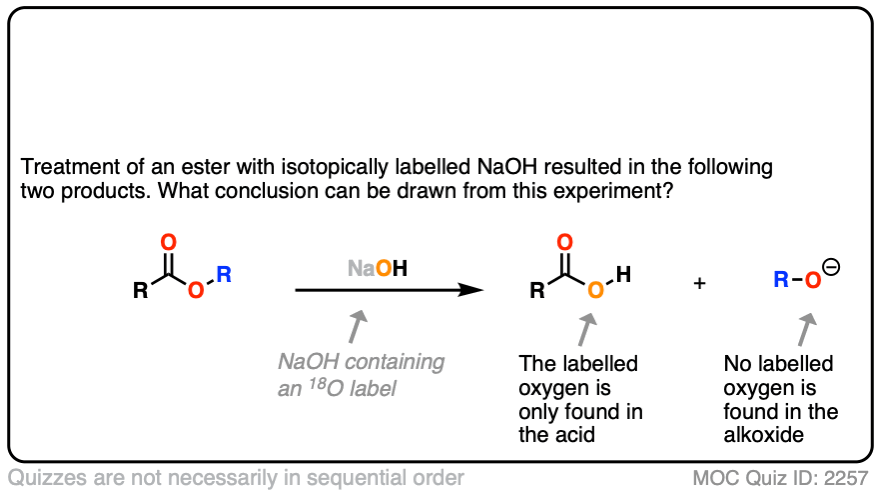
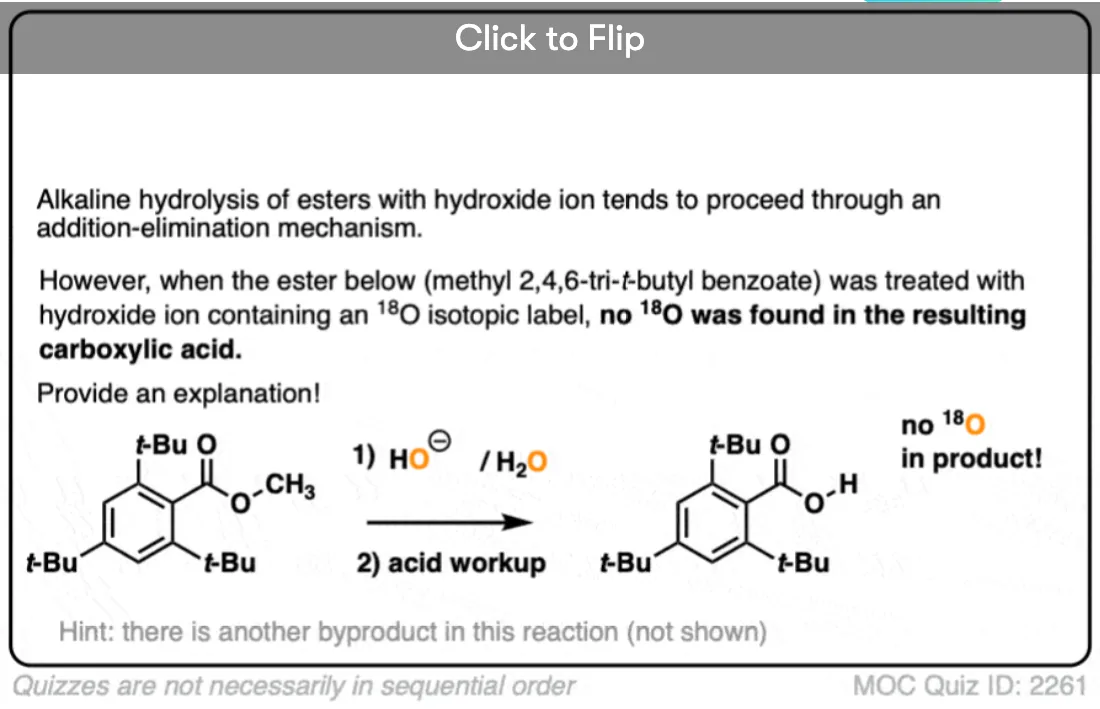
Note 2. Let’s put the evidence for the mechanism in the form of two quizzes.
In the first experiment [Ref], an ester was treated with hydroxide containing isotopically labelled oxygen (18O versus “normal” 16O ). None of the recovered alcohol contained the isotopic label.
 Click to Flip
Click to Flip
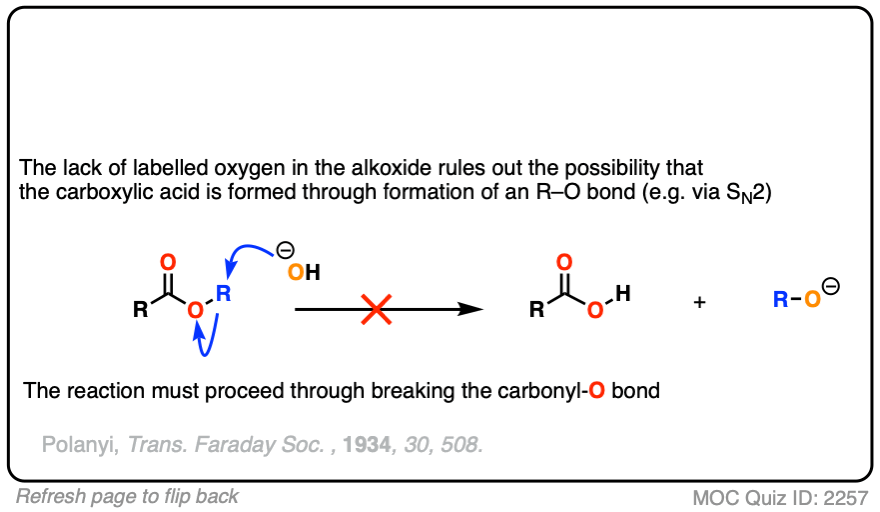
The conclusion to draw here is that the carbonyl-oxygen bond is broken, not the alkyl-oxygen bond (e.g. in some kind of SN2 process). Similar experiments have been done with esters attached to chiral alcohols and the hydrolyzed alcohols were found to maintain their optical purity.
In another classic experiment [Ref] an ester was treated with 18O labelled hydroxide and the reaction was stopped before completion.
The recovered starting ester showed some incorporation of 18O on the carbonyl oxygen. What conclusion can be drawn from this?
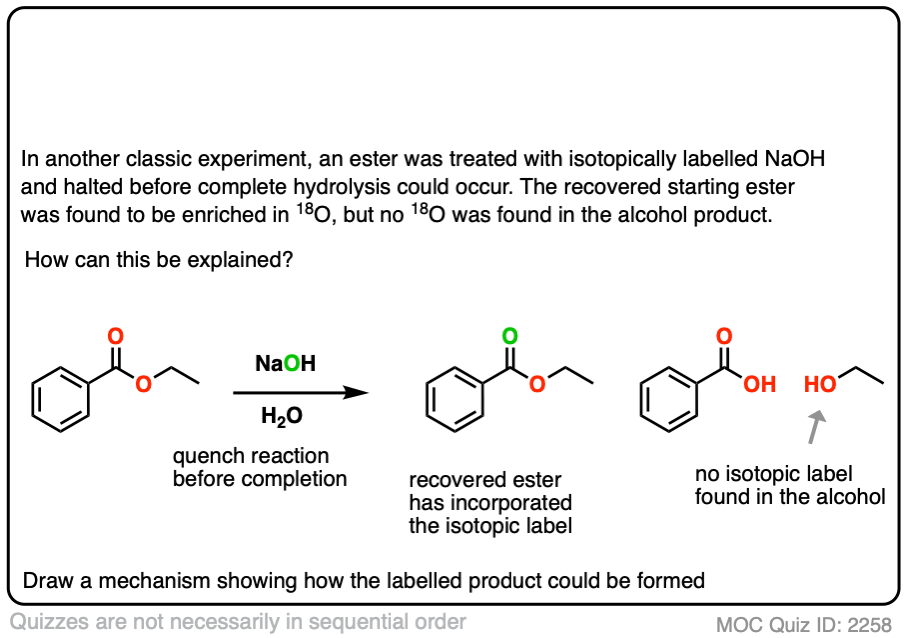 Click to Flip
Click to Flip
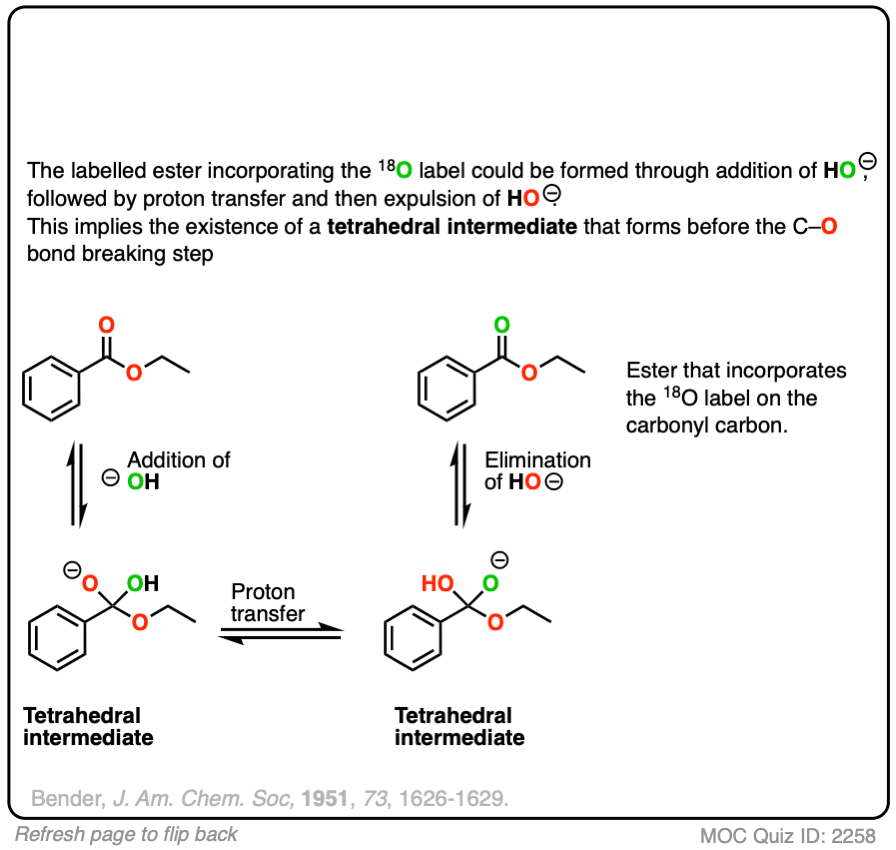
Reversible addition of 18O labelled HO(-) leads to a tetrahedral intermediate. Assuming that the 18O and 16O labelled oxygen have essentially identical reactivity (they do, within a few percent), proton transfer followed by elimination of 16OH would lead to a starting ester enriched in 18O. No exchange with the -OR group should occur.
This was a key experiment that established the existence of the tetrahedral intermediate.
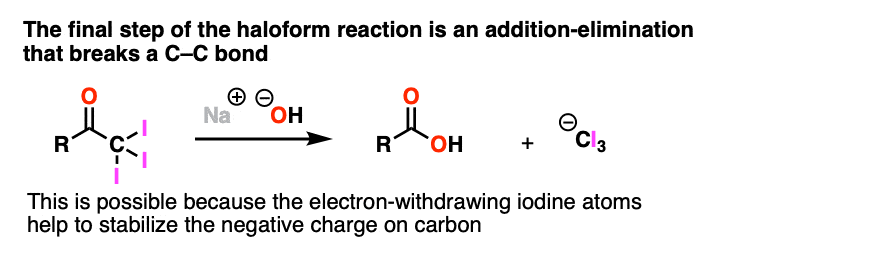
Note 3. One notable exception to this is the final step of the Haloform reaction (see: The Haloform Reaction) where addition-elimination with NaOH as nucleophile results in expulsion of (-)CI3 as a leaving group. This only works because the electron-withdrawing halogens help to stabilize the resulting carbanion.
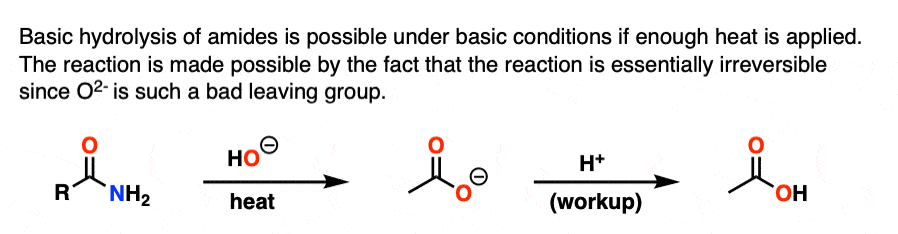
Note 4. With enough heat, it is possible to perform the basic hydrolysis of amides, which has the appearance of being “uphill” since NH2(-) is a stronger base than HO(-). However, as soon as the carboxylic acid is formed, deprotonation results in the carboxylate, which renders the reaction essentially irreversible since displacement by NH2(-) would have to give O(2-)
Note 5. Nature employs nucleophilic acyl substitution reactions extensively, and thioesters are important “acyl transfer” reagents in biochemistry. The sulfur group of Coenzyme A serves as the leaving group with a variety of nucleophilic acyl substitution reactions.
Of particular importance is the mevalonic acid pathway which leads to the biosynthesis of terpenes and steroids.
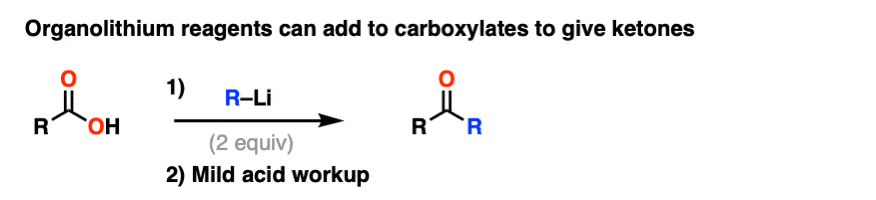
Note 6. Although Grignard reagents will merely deprotonate carboxylic acids to give carboxylates, organolithium reagents will add to carboxylates to give a stable tetrahedral intermediate that breaks down to a ketone after acidic workup.
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
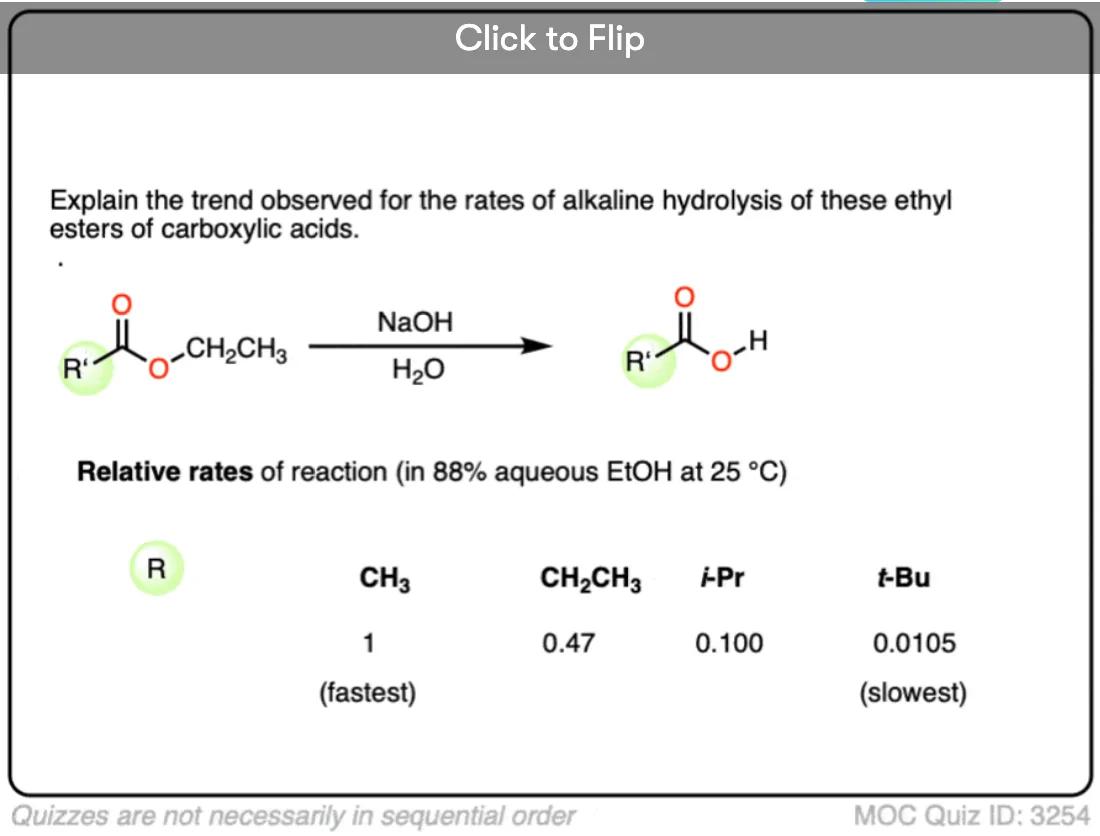
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
March’s Advanced Organic Chemistry, Carey & Sundberg Part A (Chapter 3) and B (Chapter 7) are good advanced sources on this subject. Structure and Mechanism in Organic Chemistry by C. K. Ingold is valuable for historical context.
1. On the mechanism of hydrolysis. The alkaline saponifications of amyl acetate
M. Polanyi and A. L. Szabo.
Trans. Faraday Soc., 1934, 30, 508-512
DOI 10.1039/TF9343000508
Early study on the mechanism of acyl substitution employed isotopically labelled oxygen to identify acyl-O bond fissure as the dominant mechanism in ester hydrolysis.
2. Oxygen Exchange as Evidence for the Existence of an Intermediate in Ester Hydrolysis
Myron L. Bender
Journal of the American Chemical Society 1951 73 (4), 1626-1629
DOI: 10.1021/ja01148a063
Classic experiment for establishing the existence of a tetrahedral intermediate in nucleophilic acyl substitution. Bender performed the partial acidic hydrolysis of ethyl benzoate in 18O enriched water and recovered 18O-enriched ethyl benzoate.
3. Chemical effects of steric strains—XIII: Kinetics of the reaction of sodium borohydride with carbonyl groups—a convenient tool for investigating the reactivities of aldehydes and ketones
H.C. Brown, O.H. Wheeler, K. Ichikawa
Tetrahedron, 1 (3), 1957, 214-220.
DOI: 10.1016/0040-4020(57)88041-7
Useful study on the rate constants for the addition of NaBH4 to various aldehydes and ketones.
4. Mechanisms of Catalysis of Nucleophilic Reactions of Carboxylic Acid Derivatives.
Myron L. Bender
Chemical Reviews 1960 60 (1), 53-113
DOI: 10.1021/cr60203a005
Review on the mechanisms of nucleophilic acyl substitution.
Which is the order of reactivity to NAS between the acid halides, i mean acyl fluoride, acyl chloride, acyl bromide, acyl iodide ? I believe that the increasing reactivity is acyl fluoride<acyl chloride<acyl bromide<acyl iodide, because as the leaving group gets better (decreasing basicity) the reactivity increases. This trend is affected by the size of groups which is F<Cl<Br<I. The I is the weaker base due to the large size of I, thus the large bond length of C-I. The atoms F, Cl, Br, I are on the same column of the periodic table and the difference that exists between them is the size of atom, so this differnce affects more than the electronegativity which is opposite. Is this thought correct or i am so far ? Thank you very much
You are correct that acyl fluorides are the least reactive and acyl iodides (very unstable!!) are the most reactive.
Acyl fluorides are quite stable. They can be handled without special precautions – in fact they can even be subjected to column chromatography.
One of the key factors is the ability of the fluorine atom to donate a pair of electrons to the carbon, resulting in a resonance form with a partial C-F double bond. This is similar to the reason why F is an ortho-para director. This is more effective for F relative to Cl, Br, and I since the size of the orbitals on C and F are similar (both 2p). Orbital overlap is more difficult when the orbitals are of a different size (e.g. 2p overlap with 3p)
The fact that all of this is free… you’re doing the service of god my friend.
Glad to hear you find it useful – if the username is accurate, hope you’re getting help! James
thank you james very much. It was helpful indeed.
I wish more progress for you in particular in your researc
Hi James, thanks for your web. I noticed that DIBAL-H has one more C-atom in your drawing.
Regards,
Rubén
Why does the acidity of the corresponding carboxylic acid increase due to attachment of phenyl or vinyl group , contrary to the decrease expected due to resonance effect ?
Good question. It’s likely due to the inductive effect of the sp2-hybridized carbon atoms of the aromatic ring attached to the carbonyl. You can think of them as having a greater effective electronegativity, since they have 33% s-character (vs 25% for sp3) and the electrons are held more tightly to the nucleus.
For the nucleophilic acyl substitution of an ester with OH-, why is the OR- group the leaving group?
In the graph showing the pKa of leaving groups, it appears that OH- is a slightly better leaving group than OR-. If so, why does the reaction still occur? The graph states that this is a special case, but I’m not really sure why.
Great question. The product of nucleophilic acyl substitution is a carboxylic acid.
Now you have a carboxylic acid (pKa = 4) in the presence of an alkoxide (conjugate base of an alcohol, pKa about 16).
It is then deprotonated to give a carboxylate.
Now what happens?
If you add RO(-) to the carboxylate, in order for nucleophilic acyl substitution to occur, your leaving group would have to be O(2-). That’s impossible, basically.
Beautifully crafted! Such a gem for the avid learners :)
thank you
“4. Nucleophilic Acyl Substitutions That Don’t Work”
Labelling of ester and anhydride is mixed up.
I really like your website, thank you! :)
Thank you for the heads up! Fixed.
God bless you!! Phenomenal work for such a difficult course!!
Your website is so precious. It helped me to fall in love with organic chemistry and ace my exam. Thank you so much.
Hi Mary – thank you, I’m so glad you’ve found the website helpful. Best wishes, James
Really great representation. Really useful for advanced competitive exams.
Dear Sir,
Really helped me a lot in my chemistry preparation that I am doing for IIT JEE.
~Abhay