A Primer On Organic Reactions
What Makes A Good Nucleophile?
Last updated: May 28th, 2026 |
Factors That Determine Whether A Species Is A Good Nucleophile
If you read the last post, you’ll recall that a nucleophile is a species that donates a pair of electrons to form a new covalent bond. Nucleophilicity is measured by comparing reaction rates; the faster the reaction, the better (or, “stronger”) the nucleophile.

Table of Contents
- Reminder: Nucleophilicity Is Measured By Reaction Rate
- The Role Of Charge: Nucleophilicity Increases As An Atom’s Electron Density Increases
- Electronegativity: Across The Periodic Table, Nucleophilicity Increases With Decreasing Electronegativity
- The Choice Of Solvent (Polar Protic vs. Polar Aprotic) Can Drastically Affect Nucleophilicity Trends
- Nucleophilicity Decreases With Increasing Steric Hindrance (“Bulkiness”)
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Reminder: Nucleophilicity Is Measured By Reaction Rate
When discussing nucleophilicity we’re specifically talking about donating a pair of electrons to an atom other than hydrogen (usually carbon). When a species is donating a pair of electrons to a hydrogen (more specifically, a proton, H+) we call it a base.
This post attempts to address one of the most vexing question to students of organic chemistry. What are the factors that make a good nucleophile?
For our purposes, there are at least four key factors contributing to nucleophilicity.
- Charge
- Electronegativity
- Solvent
- Steric hindrance
The first two should hopefully be familiar from the discussion of what makes something a strong base. After all, basicity and nucleophilicity essentially describe the same phenomenon, except basicity concerns donation of lone pairs to hydrogen, and nucleophilicity concerns donations of lone pairs to all other atoms. It’s the third and fourth points where extra factors come into play.
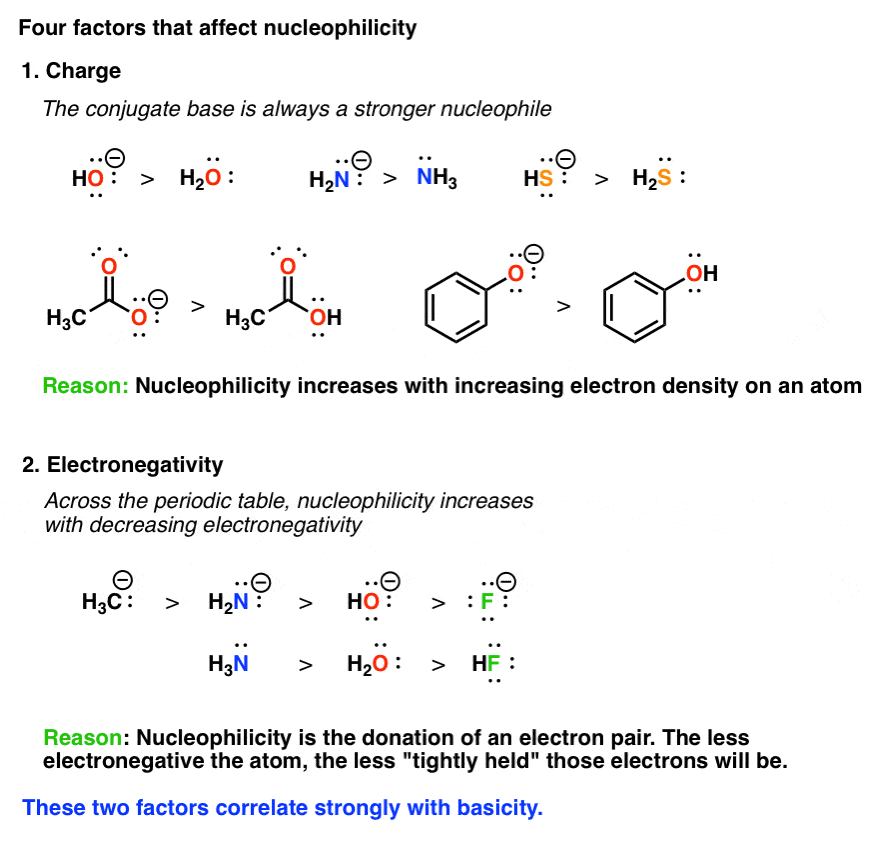
2. The Role Of Charge: Nucleophilicity Increases As An Atom’s Electron Density Increases
Since a nucleophile is a species that is donating a pair of electrons, it’s reasonable to expect that its ability to donate electrons will increase as it becomes more electron rich, and decrease as it becomes more electron poor, right? So as electron density increases, so does nucleophilicity.
A handy rule to remember for this purpose is the following: the conjugate base is always a better nucleophile.
3. Electronegativity: Across The Periodic Table, Nucleophilicity Increases With Decreasing Electronegativity
Assuming an atom has a pair of electrons to donate, the ability of a species to donate that pair should be inversely proportional to how “tightly held” it is. The key factor for determining how “tightly held” an electron pair is bound is the familiar concept of electronegativity. Bottom line: as electronegativity increases, nucleophilicity decreases. Note: It’s important to restrict application of this trend to atoms in the same row of the periodic table; for instance, C N O F, or Si P S Cl. Going down the periodic table, another factor comes into play (next)
4. The Choice Of Solvent (Polar Protic vs. Polar Aprotic) Can Drastically Affect Nucleophilicity Trends
Nucleophilicity is not a property inherent to a given species; it can be affected by the medium it’s in (otherwise known as “the solvent”). [For an introduction to the different classes of solvents, see this post]
A polar protic solvent can participate in hydrogen bonding with a nucleophile, creating a “shell” of solvent molecules around it like mobs of screaming teenage fans swarming the Beatles in 1962.
In so doing, the nucleophile is considerably less reactive; everywhere it goes, its lone pairs of electrons are interacting with the electron-poor hydrogen atoms of the solvent.
The ability of nucleophiles to participate in hydrogen bonding decreases as we go down the periodic table. Hence fluoride is the strongest hydrogen bond acceptor, and iodide is the weakest. This means that in protic solvent, the lone pairs of iodide ion will be considerably more “free” than those of fluoride, resulting in higher rates (and greater nucleophilicity).
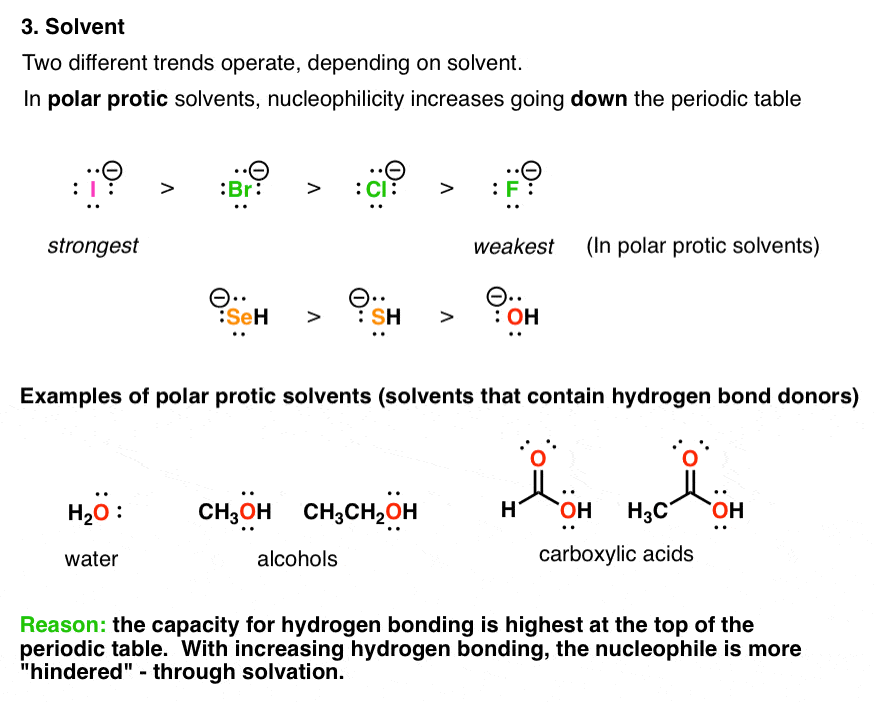
A polar aprotic solvent does not hydrogen bond to nucleophiles to a significant extent, meaning that the nucleophiles have greater freedom in solution. Under these conditions, nucleophilicity correlates well with basicity – and fluoride ion, being the most unstable of the halide ions, reacts fastest with electrophiles.

[Often asked: why don’t we care about “non polar solvents” here? Remember “like dissolves like”? If we want a reaction to take place, we need to use solvents that will actually dissolve our nucleophile. Many nucleophiles are charged species (“ions”) – they don’t dissolve in non-polar solvents.]
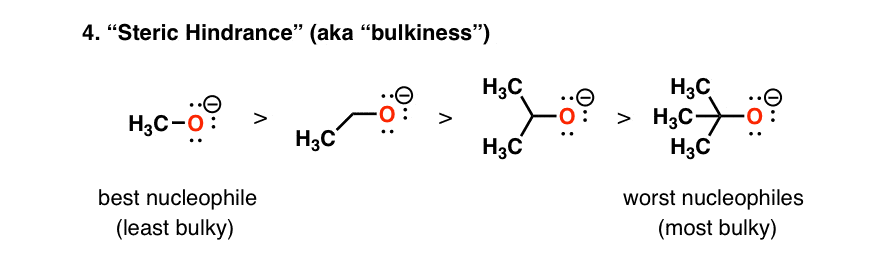
5. Nucleophilicity Decreases With Increasing Steric Hindrance (“Bulkiness”)
Since, when discussing nucleophilicity, we’re often discussing reactions at carbon, we have to take into account that orbitals at carbon that participate in reactions are generally less accessible than protons are. An effect called “steric hindrance” comes into play.
The bottom line here is that the bulkier a given nucleophile is, the slower the rate of its reactions [and therefore the lower its nucleophilicity].
So comparing several deprotonated alcohols, in the sequence methanol – ethanol – isopropanol – t-butanol, deprotonated methanol (“methoxide”) is the strongest nucleophile, and deprotonated t-butanol (“t-butoxide”) is the poorest (or “weakest”) nucleophile.
Next Post: What Makes A Good Leaving Group?
Notes
Note 1. Are there other factors? Yes. This list of four covers the basics, but several other factors are worth noting. 1) the identity of the electrophile 2) atoms with lone pairs adjacent to the nucleophile (i.e. the “alpha-effect”) 3) in the case of ions, the identity of the counter-ion [i.e. positively charged species] can be significant.
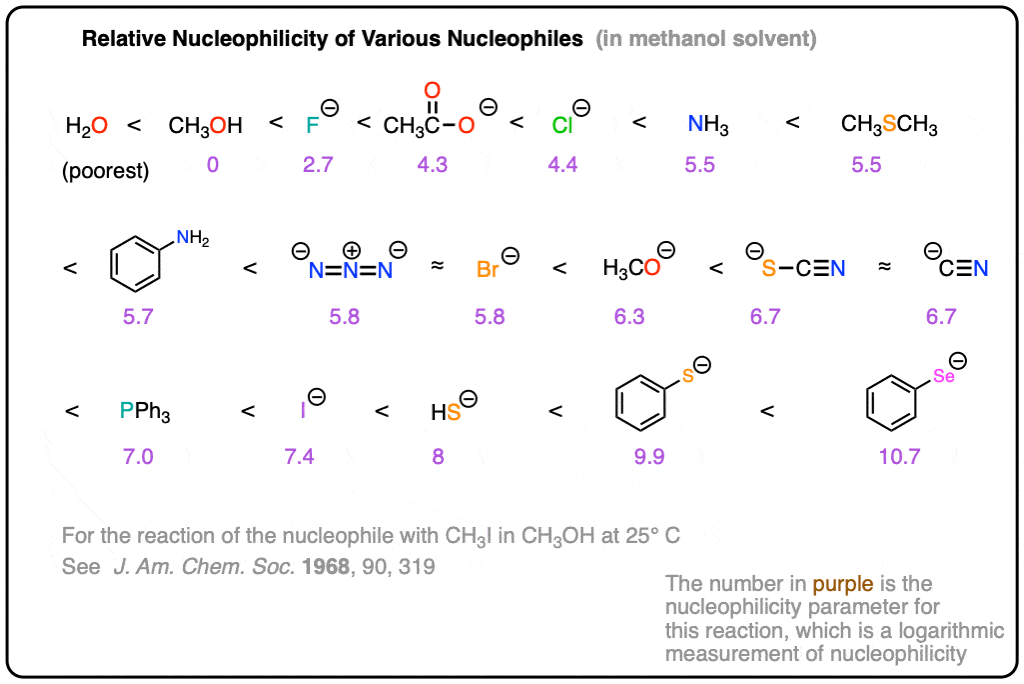
Note 2. A table comparing the relative nucleophilicities of various nucleophiles. This is specifically for the SN2 reaction of the nucleophile with CH3I in methanol solvent at 25 °C . From J. Am. Chem. Soc. 1968, 90, 319-326
Quiz Yourself!

Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- Nucleophilic reactivity constants toward methyl iodide and trans-dichlorodi(pyridine)platinum(II)
Ralph G. Pearson, Harold R. Sobel, and Jon Songstad
Journal of the American Chemical Society 1968 90 (2), 319-326
DOI: 10.1021/ja01004a021
This article contains rate constants and a relative nucleophilicity scale for the reaction of over 50 nucleophiles with CH3I in methanol at 25 °C. - Quantitative Correlation of Relative Rates. Comparison of Hydroxide Ion with Other Nucleophilic Reagents toward Alkyl Halides, Esters, Epoxides and Acyl Halides
Gardner Swain and Carleton B. Scott
Journal of the American Chemical Society 1953, 75 (1), 141-147
DOI: 10.1021/ja01097a041
This paper features one of the earliest nucleophilicity scales (Table II). - Correlation of Relative Rates and Equilibria with a Double Basicity Scale
John O. Edwards
Journal of the American Chemical Society 1954, 76 (6), 1540-1547
DOI: 10.1021/ja01635a021
This paper also attempts to develop a nucleophilicity scale, correlating nucleophilicity with other chemical properties (basicity and reduction potential). - Reactivity of Nucleophilic Reagents toward Esters
William P. Jencks and Joan Carriuolo
Journal of the American Chemical Society 1960, 82 (7), 1778-1786
DOI: 1021/ja01492a058
One of the first papers describing the “alpha effect”, the increased nucleophilicity of an atom due to the presence of an adjacent (alpha) atom with lone pair electrons. - The rate of displacement of toluene-p-sulphonate relative to bromide ion. A new mechanistic criterion
H. M. R. Hoffmann
J. Chem. Soc. 1965, 6753-6761
DOI: 10.1039/JR9650006753 - The Factors Determining Nucleophilic Reactivities
John O. Edwards and Ralph G. Pearson
Journal of the American Chemical Society 1962, 84 (1), 16-24
DOI: 1021/ja00860a005
This paper features a rudimentary nucleophilicity scale, the order being HS– > I– > CN– > Br– > Cl– > HO– > F– in polar protic solvents for attack on R-O-O-R. This paper also discusses the difference between nucleophilicity and basicity, a common source of confusion for students. - Chemical reactivity and the concept of charge- and frontier-controlled reactions
Gilles Klopman
Journal of the American Chemical Society 1968, 90 (2), 223-234
DOI: 1021/ja01004a002
Tables IX and X also support the same nucleophilicity order on the basis of MO calculations. This paper is the origin of the familiar “Klopman equation”, which treats bonding interactions as consisting of a charge component and an orbital overlap component. - Do general nucleophilicity scales exist?
Herbert Mayr and Armin R. Ofial
Phys. Org. Chem. 2008, 21 (7-8), 584-595
DOI: 10.1002/poc.1325
Prof. Herbert Mayr (LMU) has done tremendous work in quantifying and developing scales for nucleophilicity and electrophilicity. As Prof. Mayr explains in this paper, the challenge is developing suitable reference electrophiles when trying to compare similar nucleophiles, and vice-versa. Several challenging experimental techniques had to be employed for this work, including stopped-flow techniques and laser flash photolytic generation of reactive intermediates.
Hi! I thought phosphorus would be a better nucleophile than sulfur due to the fact that it’s less electronegative. They have similar polarized ability because they are in the same row on the periodic table, but phosphorus is less electronegative. Also, would this fact change if the atoms are carrying a negative charge?
PPh3 vs (-)SR is not directly comparable since sulfur bears a negative charge. A better comparison is R-S-R vs PPh3 where P is superior.
The conjugate base is always a better nucleophile than the parent compound.
Which one is stronger nucleophile among methoxide ion and hydroxide ion?
They are essentially the same. Perhaps methoxide is slightly more nucleophilic because it is slightly more basic.
Dear Sir.
You are the best.
I love the way in which you explain these terms in correct order and sequence….starting from the very basic terms.
You have cleared, all my confusion.
Everyone can easily understand,as you have explained in very proper manner.
This was life saving for me!
But, tell me which one has more nucleophilicity CN(-) or OH(-)…and which electron pair is actually transferred in CN(-)..that of carbon or nitrogen?
Cyanide ion is more nucleophilic, and the lone pair that is transferred is the carbon. Otherwise the result would have a positively charged nitrogen and a negatively charged carbon.
Please tell which is stronger nucleophile
Hydroxyl
Or methoxide anion?
According to the reactivity scale of Ernst Mayr, methoxide is approximately 1000 times more nucleophilic than hydroxide ion, although it does depend on solvent. For a mix of 90/10 methanol acetonitrile the Mayr parameter for methoxide is 14.5, and for 50/50 water acetonitrile the parameter for hydroxide is 10.4 .
https://www.cup.lmu.de/oc/mayr/reaktionsdatenbank/fe/showclass/51
Can. J. Chem. 2005, 83, 1554-1560
DOI: 10.1139%2Fv05-170
J. Am. Chem. Soc. 2003, 125, 286-295
DOI: 10.1021/ja021010y
I’ve heard that ammonia is stronger nucleophile than Primary, Secondary and Tertiary amines. Is it true?
If not then is it secondary or tertiary amine that is the strongest nucleophile?
No. See this post on nucleophilicity of amines. https://www.masterorganicchemistry.com/2018/05/07/nucleophilicity-of-amines/
Is hydroxide or fluoride a stronger nucleophile in a polar aprotic solvent?
I think it is hydroxide because oxygen is less electronegative than fluoride.
According to these principles, you would be correct in assuming HO- would be a better nucleophile than F- in polar aprotic solvent.
OH is a greater nucleophile or a CN ion
Cyanide ion.
What is the relation between leaving group and nucleophile in halogenoalkane reactions?
E.g I studied that iodine is a good nucleophile as well as a good leaving group.
But that doesn’t make sense because if its a good nucleophile why would it leave the halogenalkane ( with electrophilic carbon) easily?
Which is more nucleophilic among (CH3CH2)3N and (CH3CH2)3P??
Ans given is the first one. But in case of F and Cl;Cl is more nucleophilic(in Polar aprotic solvent).
Why?? Please explain.
Why are we considering the bulkiness of nucleophile? Because its job is to donate an electron pair, but how easily the acceptor gets those lone pairs should be the matter of concern. For that shouldn’t we be considering the bulkiness of the acceptor?
Long story short: I don’t understand how does bulkiness of the nucleophile effects its ability to donate a pair of electron.
Yes, of course the bulkiness of the electrophile is most important but the bulkiness of the nucleophile matters as well. For example, take the two primary amines, n-propylamine and t-butylamine. t-butylamine is about 100 times less nucleophilic due to the bulky t-butyl group adjacent to the NH2.
RO(-). And OH(-) …….Which have greater nucleophilicity??
They are roughly the same, at least within a single order of magnitude. Alkoxides will be slightly more nucleophilic because they can donate electron-density to the oxygen through inductive effects.
My question is as you said as bulky group increase, nucleophilicity decreases but as more methyl or ethyl groups are when present in nucleophile it should increase the nucleophilicity as per +I or electron donating effect ?
Yes, it is a fine balance. More alkyl groups increase nucleophilicity, but at a certain point steric effects come into play. In particular one sees this with amines.
So, I’m seriously confused.
My professor gave us the correct answers to our homeworks after we handed them in. And I don’t understand this order:
“Sort the following nucleophiles by their veolcity in Sn2 reactions.”
The correct answer is:
F- < OH- < Cl- < CH3COO- < S-
This makes no sense to me.
That F- is slower than Cl- imakes sense, and that F- is slower than OH-, and that Cl- is slower than S-.
But the other relationships don't make sense.
CH3COO- has a massive surface area, giving it great for polarizability, why isn't this first rather than S? And why is OH- slower than Cl-?
All of the constituents of CH3COO- are either in the same group as F or to the left of it as well, so I can't see any reason why this is slower.
No information about solvent in your exam question? Absent that information, it’s hard to make absolute judgements.
Nope, none, unfortunately. But I’d assume that – since according to him that’s what is most currently used for SN2 reactions – we are talking about a reaction in a polar aprotic solvent.
Also, someone has suggested to me that the professor might have made an error, but he doesn’t usually answer emails…
Why RO- is more Nucleophilic than OH- I want to know reason
Alkyl groups are electron-releasing, they will increase the electron density on the oxygen and make it a better nucleophile.
Out of OH- and ch3o- which is a strong base and which is a strong nucleophile and why?
Which will be a stronger base? Can you find a pKa table to give you the values for H2O and CH3OH? That will tell you which will be a stronger base, since the weaker the acid, the stronger the conjugate base.
Hi James , I want to know why a HYDROXIDE ion i.e ( OH- ) is better nucleophile than an ethoxide ion ( C2H5O- ) despite the fact that an ethoxide ion is bulkier and offers greater steric hindrance.
Thanks in advance and job well done. !!!
It’s hard to answer this question without more information, since the relative nucleophilicity of hydroxide and ethoxide is roughly within the same order of magnitude and can be sensitive to factors such as solubility (ethoxide will be more soluble in organic solvents). Hydroxide is less sterically hindered but since ethoxide is a primary hydroxide steric hindrance will not be a large factor.
More significant is the difference in nucleophilicty between ethoxide and t-butoxide. The latter is significantly streakily hindered and will react at a slower rate than ethoxide with most electrophiles.
What does steric effect actually mean?
“Steric effect” is the effect of the (non-bonding) electron clouds of atoms colliding with each other. It is a repulsive effect. In another post, I say “steric hindrance is like a fat goalie”. https://www.masterorganicchemistry.com/2011/07/18/steric-hindrance-is-like-a-fat-goalie/
Alkyl grups are electron donating ..do they not increase the nucleophilic character of of the grup attached i?
They do, to some extent. For example the acidity of alcohols goes from about pka 16 (primary alcohol, least most acidic) to pKa 18 (tertiary alcohol, least acidic) because donation of electron density from the alkyl groups will make the conjugate base more electron-rich (and slightly less stable). However when discussing nucleophilicity we are usually talking about the O- attacking a hindered sigma* orbital, which is much more sensitive to steric effects than acid-base reactions. So the increased electron density is generally offset by the retarding effect of steric bulk. See https://www.masterorganicchemistry.com/2011/07/18/steric-hindrance-is-like-a-fat-goalie/
which is stronger nucleophile, hydroxide ion or benzoate ion ????
Hydroxide ion – it is a stronger base.
Hi James, thank you for this awesome post! I’m a chemistry beginner and following your posts to learn more. One question is that, in your another post regarding basicity, higher basicity is better stability of the anion, i.e., F(-) > OH(-) > NH2(-) > CH3(-), where F(-) is the most “stable”. Was this only applicable for polar protic solvents?
In the current post, in polar aprotic solvents, why is F(-) is most “unstable”? High electronegativity means tight binding of electron pair. So as you wrote above, shouldn’t F(-) be hard to donate electron pair?
I really enjoy your postings. Thanks!
Just wanted to thank you for your passion in this field and channeling it into this fantastic website. As a 5th semester Bio student taking organic chemistry, I have done very well with concepts and applications essentially up until SN1 SN2 E1 E2 – a common complaint, so I am told.
Regardless, you have an excellent way of explaining the “why’s” and the “how’s” without getting too complex but not keeping it “too simple.”
Love your site, so happy I stumbled upon it.
Thanks for sharing your wealth of knowledge and passion for organic chemistry with the rest of us confused people
Best, RT
PSU
Thank you very much RT. I greatly appreciate you stopping by and your kind words.
which is a better nucleophile a peroxide ion or a hydroxide ion and why??
Peroxide, due to the alpha effect, which I don’t discuss here. Briefly, an adjacent atom with a lone pair will raise the energy of the HOMO (due to repulsion) resulting in greater nucleophilicity. https://en.wikipedia.org/wiki/Alpha_effect
order of nuclephilicity?
1:CH3CO`,OH`, CH3COO`, H2O
2;CH3COO`, CH3O`, OH`, H2O
3:OH`, CH3O`, CH3COO`, H2O
(here ` represents minus charge)
1. Charged species generally more nucleophilic than neutral species (“the conjugate base is always a stronger nucleophile”).
2. The more basic a species is, generally, the more nucleophilic it is. Lots of exceptions, but not within the series you mention here.
What a BS, nucleophilicity in polar aprotic solvents is still incrasing going down in a group (when we talk about Sn on saturated carbon) because of higher homo energy and bigger size. Compare Sn2 done in polar aprotic solvents reaction rates and youll see that RSH is still better than ROH, and so does I- compared with F-.
Is there any good publication comparing nucleophilicity of halides (including F-)? I’m thinking of comparing rates of SN2 reactions depending on the nucleophile. I find many statements comparing these halides in protic and aprotic solvents but there is no reference given.
Yes, I would suggest visiting the website of Herbert Mayr. I’m including the part of the site where halides are covered. https://www.cup.lmu.de/oc/mayr/reaktionsdatenbank/fe/showclass/39
your explanations are just awesome and totaly logical ¬!!
Once again, very well explained. Though I find the reason for nucleophilicity in polar aprotic solvents a bit confusing:
“here, nucleophilicity is essentially correlating with basicity”
The sentence implies somewhat, that there is a positive correlation as seen in the role of charge and the role of electronegativity. But I- is clearly more basic than F-. Maybe this aspect cannot be simplified in this manner? Or am I missing something?
Nevermind, I got it:
The basicity of F- is higher than the basicity of I- because it’s less solvatized if the solvent is polar aprotic (i.e. not water). So cannot correlate THIS basicity with pKa-values obtained from water. Right?
But pKa-tables obtained from DMSO also tell me that I- is more basic than F-… now I am completely confused….
Please forget everything and delete my comments. I have no idea why I made the initial misconception of I- being more basic than F-. I definitely need a vacation… ;) Thanks again for your excellent walkthrough!
Fantastic explanation! Thank you so much!!
Really helpful :) thanks :)
sir, I have a problem that a nucleophile have a negative charge yeah it means that it is rich in electrons but there are reactions in which a molecule after attacking by a nucleophile ,their leaving group also form negative charge . Is it mean that both the leaving group and attacking nucleophile are richer in electrons?If it is yes then it means that a leaving group is also a nucleophile?
Leaving groups are nucleophiles acting in “reverse”. https://www.masterorganicchemistry.com/2012/06/25/leaving-groups-are-nucleophiles-in-reverse/
I have a doubt regarding RS- and RO- . How do we compare their nucleophilicity and their basicity?
Which will be more Nucleophilic in DMF :-
OH- or NH2- ?
NH2- would be. But if this is a practical question, I’m not sure DMF would be stable to NH2- .
Well first of all I want to thank you for this awesome post. I have a suggestion. We can use thi. Mnemonic. CASH to remember all the four points. C for Charge, A for role of atom or electronegativity (u used this in CARDIO), S for solvent and H hindrance or steric hindrance).
THANK U
Why thank you, Shadow of Death, for that useful mnemonic.
Is a molecule that is more basic less nucleophilic, or do the trends go along the same way? For example, between CN-, OH-, CH3OH, and NH3, the strongest nucleophile is CN-,which is also the strongest base, or is it NH3, the least basic?
Also, going down a group, if atoms become more polarizable since they can hold on to their electrons less (shielding), then they become better lewis bases, but they also stabilize the charge better because they are spread over larger area, so that makes them worse lewis basis, is this not contradictory? PLEASE HELP.
Please please help me someone I am getting extremely confused in order of nucleophilicity in case of aprotic solvent
In polar protic solvents, the order of nucleophilicity will be I- > Br- > Cl- > F- . This reflects the extent of hydrogen bonding around each ion (I- least, F- most). In polar aprotic solvents, the order reverses: F- > Cl > Br- > I- , since hydrogen bonding is not a factor, and F- tends to be more basic, as well as forms stronger bonds.
Thank u very much that’s exactly what I wanted :)
B/w (CH3)3N and (CH3)3P which one is a stronger nucleophile in polar aprotic solvent and polar protic solvent?
If one follows the same logic that the higher one goes on the periodic table, the more it will hydrogen-bond, then (CH3)3P will be more nucleophilic than (CH3)3N in polar protic solvents, and (CH3)3N will be more nucleophilic in polar aprotic solvents.
Is NH2- more nucleophilic than OR-???
How do we know??
Less electronegative means that it holds on to its electrons less tightly, and is – in theory – a better nucleophile. In practice, NH2- is quite a strong base and generally acid-base reactions occur first.
So is an alcohol group a weaker nucleophile than chloride?
I’d say so, yes.
In a protic solvent, is chloride a better leaving group than iodide?
I have a question I cant seem to find an answer to. I dont understand why a weak nucleophile would attack a more highly substituted carbon. I would think that it would be easier for it to access one that is less hindered.
why iso-propanol (maximum) used as a proton source in transfer hydrogenation reaction? any advantage when compared to other alcohols like methanol and ethanol?
I think you mean hydride source? The isopropyl C-H bond is weaker.
If all the factors come into play how do we prioritize them? For instance F- is bad nucleophile when we consider solvation , and a good one when we consider charge density stability and steric factor..So who how do we choose whether I- or F- is better..?Also in gaseous phase size doesn’t matter., so is F- good one as it is unstable than I-?
Comparisons using these factors are best done if one keeps all variables constant except for one. For example keep the solvent the same, and vary the nucleophile from F- through I- . Or keep the nucleophile the same and vary the solvent. If multiple variables are changing, then it’s hard to predict the effect of changing any individual factor without measuring the importance of each through experiment.
How can we differentiate between a nucleophile and a base? e.g KOH in aquous and in alcoholic medium has two opposite effect one as a nucleuphile and other as a base why? I have heard that alcoholic medium increases its basic strength but how ???
This saves my life! Very clear and helpful, thanks a lot!
Hi every one
I know -OH has more electronegativity than -NH2 , also increasing electronegativity decreases the lone pair availability; I think the lone pair on the nitrogen atom is more available to resonance with phenyl ring (because the above reason), so if lone pair of the -NH2 is shared in the resonance, the lone pair of -OH is free to play a nucleophilic role.
But in many reactions -NH2 is the nucleophile (sometimes -OH is nucleophile and sometimes both of them play this role). (http://www.chemsink.com/list_reactions/7/403/1/1/)
Is there any convincing reason? please explain with details.
Great article! But I still have a question. I understand now how one nucleophile can be stronger or weaker than another, but what would you consider to be (absolutely) strong? Just because one nucleophile is stronger than another does not mean they both cannot be weak nucleophiles, I think. What are the cuttoff points for strong nucleophiles that would be able to partake in SN2 reactions?
We can come up with a good general scale for acidity, which can be measured by equilibria. Nucleophilicity is a bit trickier because the reactions are irreversible and we’re measuring rates. The rate of the reaction is a combination of orbital overlap and charge density and by changing conditions (like solvent, reagent, etc) we can also affect nucleophilicity.
The classic example is the reaction of enolates. Under different conditions either the O or the C of the enolate can be the best nucleophile.
Later on in organic chemistry the topic of hard soft acid base theory is covered which helps to explain some of these mysteries.
Being challenged, need reasons why H2O instead of Br- became nucleophiles. Textbooks with huge price tags just presents drawings of kind of skeletons, H2O and arrows.
Here I get it.
Thank you James.
Hi! It says in my textbook that in methanol, RS- is a better nucleophile than iodide. This is something I didn’t get it. I mean, sulfur is smaller and the R group is probably making it more basic by electron-donating effect, thus making it a stronger base. This means that RS- should be the most solvated one and therefore less nucleophilic. But according to Ms. Paula Bruice, no, RS- is the better one.
By the pka table, HI is a stronger acid than RSH. So, RS- would still have the stronger base, that would still be more solvated. I really don’t get it. When I think I got it, I didn’t. I used to like Ochem a lot, but now I think I will never undestand it and it bums me out.
Another thing, in a SN2 reaction, ammonia is the nucleophile and it is asked which solvent will make the reaction faster: ether or ethanol. I would say ethanol, because it would stabitize the transition state by solvation, right? But, no, according to my professor, ether would be the best one because it would not make hydrogen bonds to ammonia. Now I don’t know if I must consider transition states, and solvents stabilizing carbocations or just the nucleophile and hydrogen bonding.
Please, help.
Hi Symara – there are many factors involved in nucleophilicity. It’s not a simple, one-variable equation.
Hydrogen bonding is most important for atoms O, N, and F (because of the large difference in electronegativity between these atoms and H). When you compare that to S (electronegativity 2.6) there just isn’t much hydrogen bonding going on so it shouldn’t affect nucleophilicity. So the above answer (RS- beats I-) and ether being a better solvent than ethanol probably makes the most sense.
As an aside, when talking about different variables like basicity or polarizability it is much clearer if you are going across a row or up and down a column of the periodic table. For example, compare polarizability going down (F- Cl- Br- I- ) or basicity going across (H3C- H2N- HO- F- ). Comparing both at once (e.g. Cl- and H2N- ) really requires looking at experimental evidence.
Hello!
My question is why flouride ion behaves as a strong nucleophile in aprotic polar solvent when nucleophilisity is related to polarizability. any specie that is more polarizable tends to be a good nucleophile. Then why not iodide is a strong nucleophile in aprotic polar solvent and also iodide is less electronegative than fluoride so it should easily donate its lone pair of elctron
Because Iodine has a VERY big electron cloud. It does not prefer to accommodate any other atom with it. Therefore fluoride is a better nucleophile than iodide.
You mean only hindrance effect make basicity and nucleophilicity different?
Not quite. Acidity/basicity is measured by equilibrium constant, whereas nucleophilicity is measured by reaction rate (since the vast majority of substitution reactions are not reversible). But steric hindrance (due to the fact that a sigma star orbital is being attacked on carbon, versus an S orbital on hydrogen) is the key difference.
Can u explain finkelstein’s reaction on the basis of the effect of polar Aprotic solvents on Nu ?
No, Finkelstein’s reaction goes forward because NaI is soluble in acetone, whereas NaCl precipitates out. That drives the equilibrium forward.
in sn2 reaction ı know why we use polar aprotic solvent, but under which conditon the reaction may retard? I mean which polar aprotic solvent may retard reaction ?
Hi, I came across a question asking ‘Which is the stronger base? Pyridine or Morpholine?’ Is it the same as asking which is the stronger nucleophile? Anyway, I think the answer is morpholine but I do not know how to explain it. Could anyone please help me on this?
I expect that, in pyridine nitrogen atom surrounded by three bons all of them with carbon atoms while in morpholine there are three bonds two of them with crbon and the thired one with hydrogen which is lower electronegative than carbon so the availability of unshered electron pair in morpholine more than that in pyridine.
Does good nucleophilies have anything to do with hard/soft base?
Hi,
This was very helpful, but I’m still confused about 1 thing, how is fluorine a better base than iodine but a worse nucleophile?
In polar protic solvents (and only in polar ptotic solvents) fluorine’s strong Lewis basicity helps it form very strong hydrogen bonds with the solvent. The resulting “shell” of solvent molecules around fluorine acts to “hinder” fluorine and therefore makes it a poorer nucleophile. Iodine does not form strong hydrogen bonds and therefore is not accompanied by a large solvent shell, so it is less “hindered” in polar protic solvents and thus a better nucleophile.
is boron quadfluoride a nucleophile
What do you think? What atom(s) on BF4(-) could donate a pair of electrons?
“Bottom line: as electronegativity increases, nucleophilicity decreases”
“Under these conditions, nucleophilicity correlates well with basicity – and fluoride ion, being the most unstable of the halide ions, reacts fastest with electrophiles.”
Which one is it? It seems to me that you are contradicting yourself and making me more confused than I was previous to visiting this page…
Maybe I should have made this clearer. It’s consistent if you consider that nucleophilicity increases with basicity EXCEPT in the case of polar protic solvent, in which nucleophilicity increases with polarizability.
Basicity: CH3(-) > NH2(-) > HO(-) > F(-) and F(-) > Cl(-) > Br (-) > I(-) [in polar aprotic solvents].
I don’t understand why F(-)>Cl(-)>Br(-)>I(-)
Electronegativity decreases down a group, so shouldn’t nucleophilicity increase down the group?
SO CONFUSED!
The “electronegativity decreases, nucleophilicity increases” rule applies only to atoms within the SAME ROW in the periodic table. Also, that rule only applies for polar protic solvents. F(-) tends to H-bond with the solvent more, making it less reactive as a nucleophile, as compared to a nucleophile containing carbon.
The reverse of the rule is what actually applies in polar aprotic solvents. Since the solvent does not H-bond to the halide nucleophiles, fluorine basically becomes the most reactive among the halides.
It took me way too long before I finally understood this whole nucleophile thing, but I hope my answer helped.
Hello,
Is it accurate to say that primary amines are more strongly nucleophilic than carboxylic acids? I presume this is so given the charge delocalization in COO- and the steric hindrance in COOH. Do you happen to know of a reference in the literature that compares these two species’ nucleophilicity?
Many thanks for your help and time.
Jim
How do we decide that from anisole, nitrobenzene and benzene, what will be the correct order of rate of electrophillic substitution? And can you possibly link me to an article related to it?
PS AMAZING site! You are soon gonna get a lot of Indian visitors. :D
It should be anisole > benzene > nitrobenzene since the rate of ESR depends on the charge density on benzene. In OCH3 oxygen has got a lone pair due to which can be shared with the benzene ring through resonance hence increasing its charge density whereas nitrogen in NO2 has not got lone pair( due to formation of dative bond with O) and has higher electronegativity than C so it withdraws charge from benzene ring. Hope it helps!
Hi, I was wondering why (CH3)2N- is better nucleophile than CH3NH- which is better than H2N-, when (CH3)2N- and CH3NH- has more steric hindrance? I understand that the presence of electron donating groups (eg. methyl groups) would increase the nucleophilicity, but how do I know which factor is more important?
Thanks!
Great question. These types of tradeoffs are what can make organic chemistry difficult. In advance, it’s hard to know exactly which factor is most important until you actually see the results from experiment.
Good discussion here:
http://wavefunction.fieldofscience.com/2010/11/why-are-secondary-amines-most-basic.html
Also, I think “Polarizability” was a factor that may have been missed, or perhaps stated in a more subtle manner…
Excellent post, but…
If this list does not take into account all the factors that make a good nucleophile, where is a more detailed treatment of the ones that are remaining?
Also, in the case of polar aprotic solvents, one may mention the idea of the cation being solvated, while the anion (nucleophile) less so, and so it is more reactive.
Once again, excellent post.
What’s missing is hard-soft acid base (HSAB) theory which involves discussion of molecular orbitals. I’ve chosen to defer that discussion for now. One example is the differing selectivity of enolates for C vs. O alkylation; depending on the nature of the solvent, counter-ion, and electrophile, either dominant O vs. C alkylation can be achieved. For a discussion I’d refer to Carey and Sundberg but there are many other online sources which discuss HSAB.
How is polarizability related to nucleophilic strength? Would it fit into any of these categories?
Polarizability plays a role when you take the solvent into account. In polar protic solvents, hydrogen bonding occurs between the partial positive hydrogen (H attached to N or O usually) and the nucleophile. Smaller nucleophiles become more solvated than larger nucleophiles, which means that smaller nucleophiles in polar protic solvents will not be able to react as well and thus are poorer nucleophiles.
For example, Florine anions become so heavily solvated in polar protic solvents that they wont even react, but Iodine, being much larger, is much less solvated and can still react.
In aprotic solvents, hydrogen bonding does not occur to any significant extent and the stronger base is usually the stronger nucleophile.
THANK YOU!
With the “bulkiness,” does that have to do with Beta-branching?
Yes it does.
My understanding with I>F in polar protic solvents wasn’t because F has MORE hydrogen bonding but rather that it is more affected by the hydrogen bonding due to being smaller in size. Roughly, the larger I ion has the same hydrogen bonds but can still “wiggle” the electron pair through them since it is a “looser coat” of hydrogen bonds…
is this correct?
It’s correct in that fluoride, being smaller, has a larger charge density, and the interactions with the partial positive charges on the hydrogen of water will be stronger. Iodide, being larger, will have a lower charge density and interactions with hydrogen will be weaker. Does that make sense to you?
Its actually the opposite. Since flourine is smaller, its charge is confined to a smaller space and it therefore has a higher electron density. Florine is effected by hydrogen bonding more than Iodine (in polar protic solvents) but the reason is because Florine’s smaller size makes it more easily solvated (surrounded by solvent molecules) than iodine so it can’t react as well. Remember that nucleophilicuty is a measure of how well/fast something reacts, while basicity is a measure of how “willing” an atom is to give up a lone pair. They are correlated most of the time but not always.
O is more powerful nucleophile as well as strong base than F due to less electronegativity but F has less nucleophilicity than I but strong base than I because F has small size but tendency to loose electron is low and I is large in size due to which it’s charge gets dispersed and it is more stable then fluoride anion and therefore its basicity is low because we know the more the stable anion the less it’s basicity.
my question is why we don’t consider size factor when we do comparison b/w F and O , as O is large in size than F therefore charge on O will be more dispersed so oxygen also should be less basicity than fluorine like Iodine ???
basically my question is why nucleophilicty runs parallel is same row or period but don’t in same group or coloumn ???
This goes back to hydrated ionic radius, being smaller in size and highest electronegativity, it attracts more water/polar protic solvent molecules, hence it nucleophilic properties gets destroyed by hydrogen molecules.