Enols and Enolates
Decarboxylation
Last updated: May 28th, 2026 |
Decarboxylation
In this article we’ll cover decarboxylation, most commonly encountered in the context of the loss of CO2 from beta-keto acids (and malonic acids) in the malonic ester synthesis and acetoacetic acid synthesis, respectively.
We’ll go through some examples, show how not to do the arrow pushing (and how to avoid a common mistake) and then try to explain why decarboxylation happens readily with beta-keto acids but not with “normal” carboxylic acids. We’ll also have some quizzes and exercises along the way.
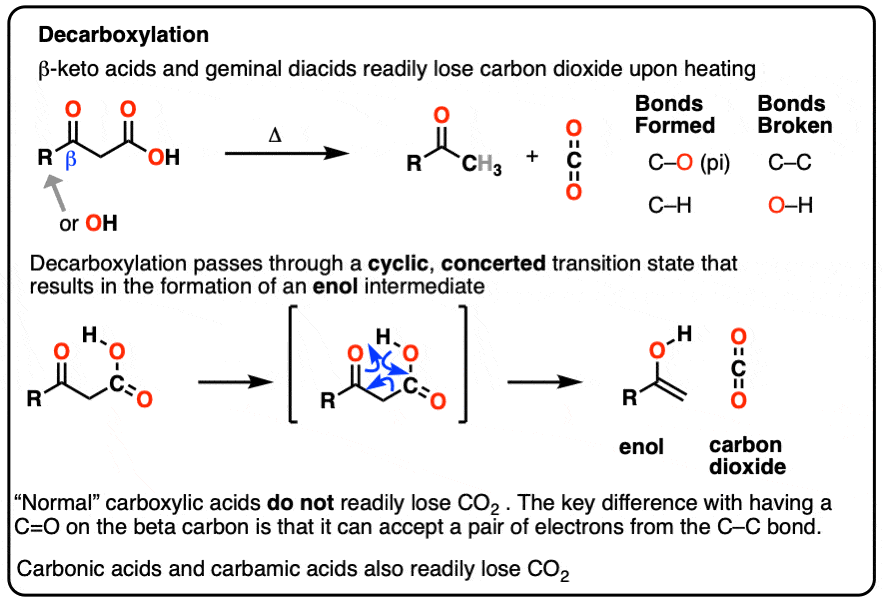
Table of Contents
- Some Decarboxylations That Work
- Decarboxylations That DON’T Work!
- How Do Decarboxylations Work?
- Decarboxylations As A Type of “1,2-Elimination”
- The Mechanism
- Decarboxylation In The Acetoacetic and Malonic Ester Syntheses
- A Beta-Keto Acid That Doesn’t Decarboxylate
- Some Other Examples of Decarboxylation
- Conclusion
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Some Decarboxylations That Work
In this article we’ll talk about some notorious CO2 emitters: beta-keto acids and gem-dicarboxylic acids.
beta-keto acids are carboxylic acids that contain a C=O group two carbons over from the carboxylic acid.
When beta-keto acids are heated, they spontaneously lose CO2 to give a ketone, like this:
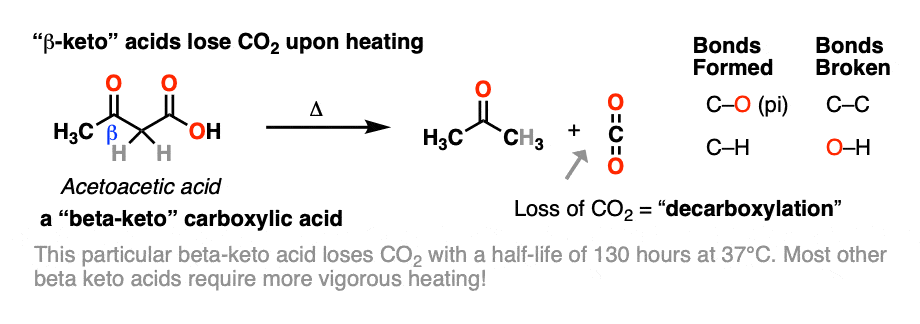
This is called “decarboxylation”.
While decarboxylation often requires heating, acetoacetic acid, the simplest example (above), spontaneously loses CO2 at room temperature! [note 1]
Another member of the greenhouse-gas-emitting club are derivatives of malonic acid (a gem-dicarboxylic acid) [note 2] which resembles a beta-keto acid. Technically I guess you could think of them as “beta-acid acids” but I’ve never heard anyone use that term.
When malonic acid is heated, it gives off CO2 to give a mono-carboxylic acid (acetic acid in this case).
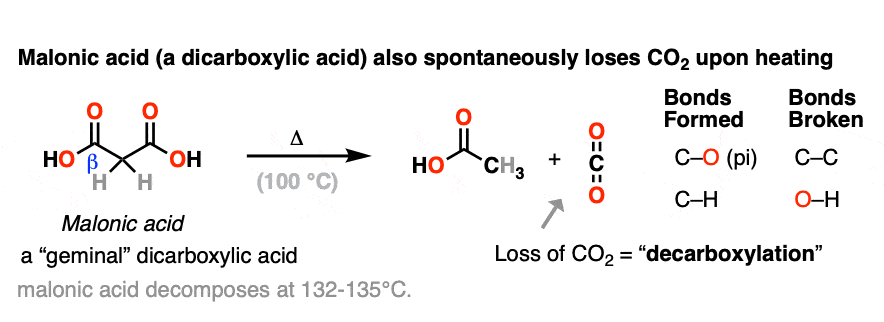
In each case of decarboxylation a C-C bond is broken and a C-O pi bond is formed in addition to a new C-H bond.
These reactions come up mostly in the context of the Acetoacetic Ester synthesis and the Malonic Ester synthesis, covered previously. [See post: The Malonic Ester Synthesis]
2. Decarboxylations That Don’t Work
So is it really that easy? All we have to do is heat up a carboxylic acid a little bit and poof! off pops CO2?
In other words, can we just heat any old carboxylic acid and lose CO2?
No. When our familiar friend acetic acid (vinegar – a sadly underappreciated condiment for french fries, BTW) is heated to 100°C, it doesn’t lose CO2. It just boils. Likewise, long-chain carboxylic acids like butanoic acid and valeric acid don’t lose CO2 readily upon heating. They just boil. I don’t personally recommend boiling them without a very well-functioning fume hood, however, since the smell of fresh puke (butanoic acid) and stinky feet (valeric acid) will not make you any friends.
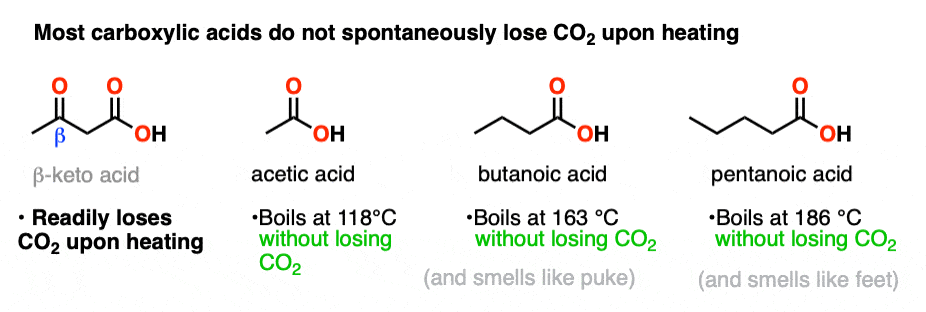
Since normal long-chain acids don’t spontaneously lose carbon dioxide upon heating, then perhaps there’s something special about the presence of the C=O ?
Pyruvic acid, CH3C(O)CO2H, is an alpha-keto carboxylic acid. Levulinic acid, CH3C(O)CH2CH2COOH is a gamma-keto carboxylic acid.
So how well do these molecules decarboxylate upon heating, in contrast to a beta-keto carboxylic acid?
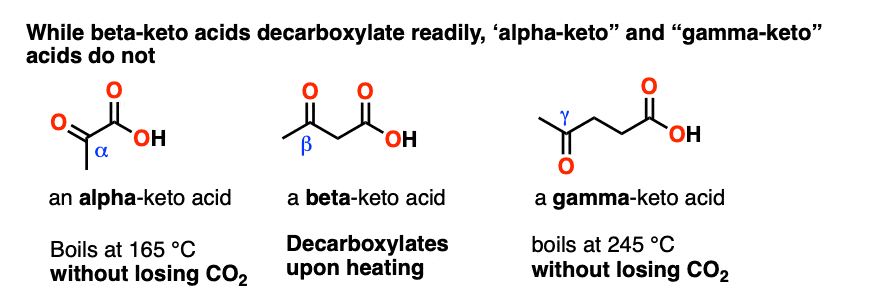
The short answer is that only beta-keto carboxylic acids decarboxylate readily. The others don’t. (The same applies to alpha- and gamma- dicarboxylic acids) [note 3]
So there is clearly something special about having a C=O two carbons away from the CO2H that allows decarboxylation to occur.
What is it?
3. How Does Decarboxylation Work?
In the decarboxylation reactions above, we’re breaking C-C and forming CO2.
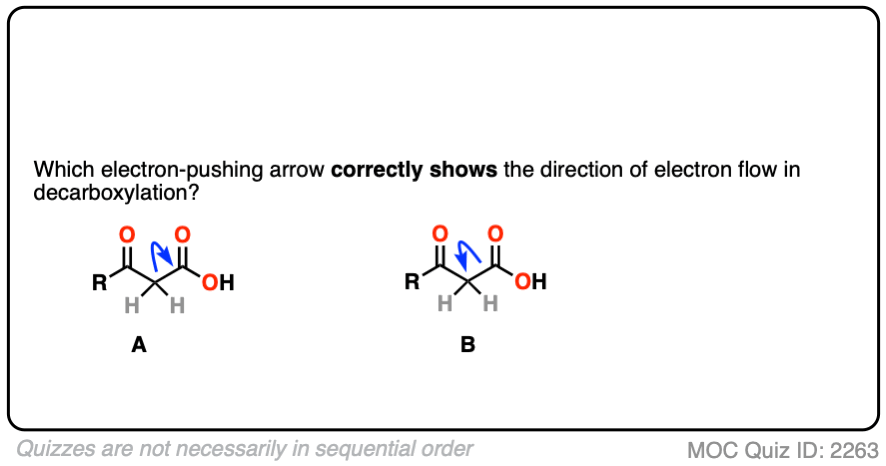
We could imagine two different ways this might happen, which we can visualize with a curved arrow.
In the first, we’d draw an arrow that shows the pair of electrons from the C–C bond moving toward CO2H and allowing CO2H to leave as a “leaving group”.
In the second, we’d draw an arrow that shows the pair of electrons from the C–C bond moving away from the CO2H and toward the carbon adjacent to the C=O.
Which one is correct?
 Click to Flip
Click to Flip
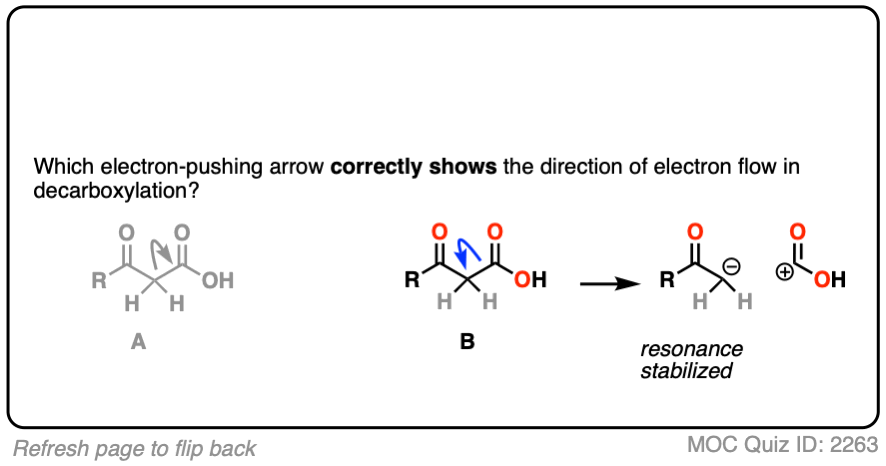
If you chose “A”, congratulations! You’ve made a common mistake that many others before you have made, and now you’ll be armed against it in the future.
Although it’s tempting to draw the arrow this way, since CO2 is “leaving”, it is incorrect.
Why? Look closely at the consequences of breaking that C–C bond in that direction. Every curved arrow makes the formal charge at the tail more positive by 1 and the formal charge at the head more negative by 1. [See: Curved Arrows For Reactions]
The result is a carbocation at carbon, and this negatively charged CO2H(-). This is not carbon dioxide! [note 4]
![]()
And no, that carbocation is not resonance stabilized. [note 5]
4. Decarboxylation As A Type of “1,2-Elimination”
The trick with decarboxylation is to look at it the same way you’d look at any other 1,2-elimination from a carbonyl group. [See post: The 1,2-elimination]
When that happens, we form C–O (pi) and break off LG(-).
![]()
That means that when a decarboxylation happens, we should look at whatever is attached to CO2H as the leaving group.
Like this:
![]()
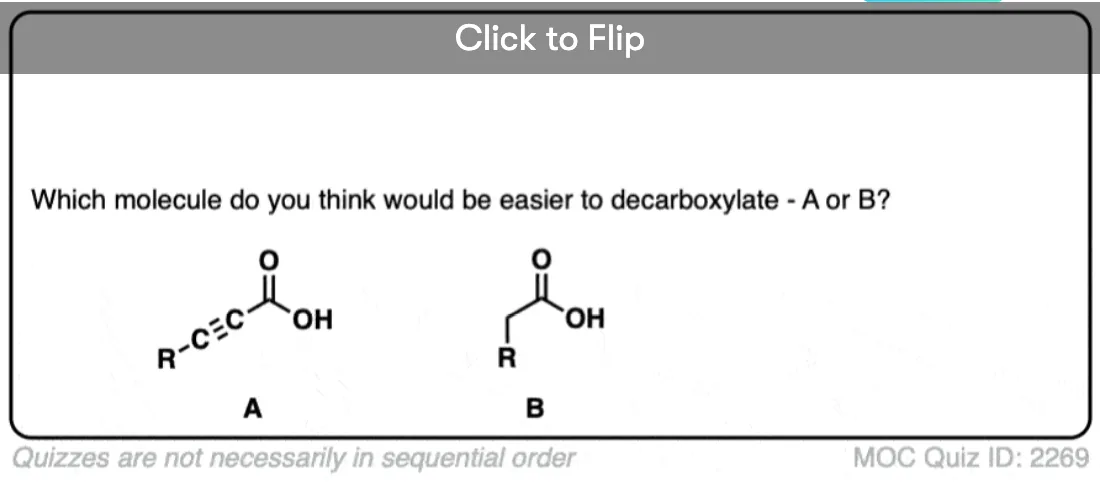
So why should decarboxylation happen in the case of beta-keto acids but not happen with the examples we showed above (straight chain acids, alpha- and gamma- keto acids, and so on).
Well… it all comes down to basicity (again)
You’ve probably noticed by now that one of the features of organic chemistry is that the same old lessons come back to you in different forms, repackaged. This is what makes it fun! (Your opinion may vary)
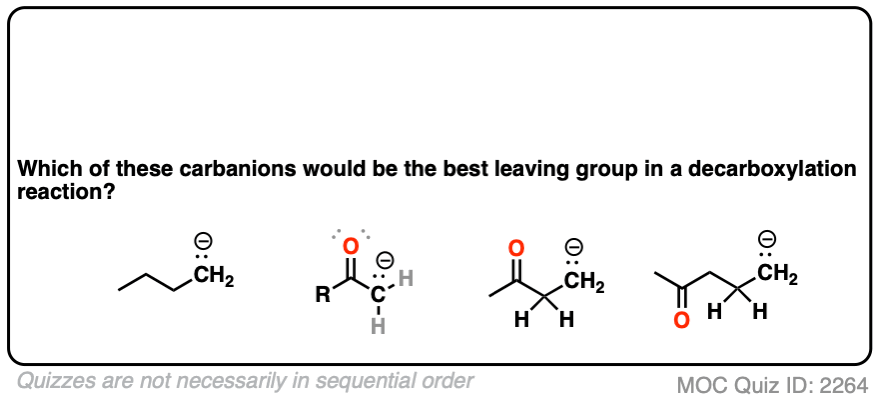
Stop me if you’ve heard this before, but, “the weaker the base, the better the leaving group“. [See post: What Makes A Good Leaving Group?]
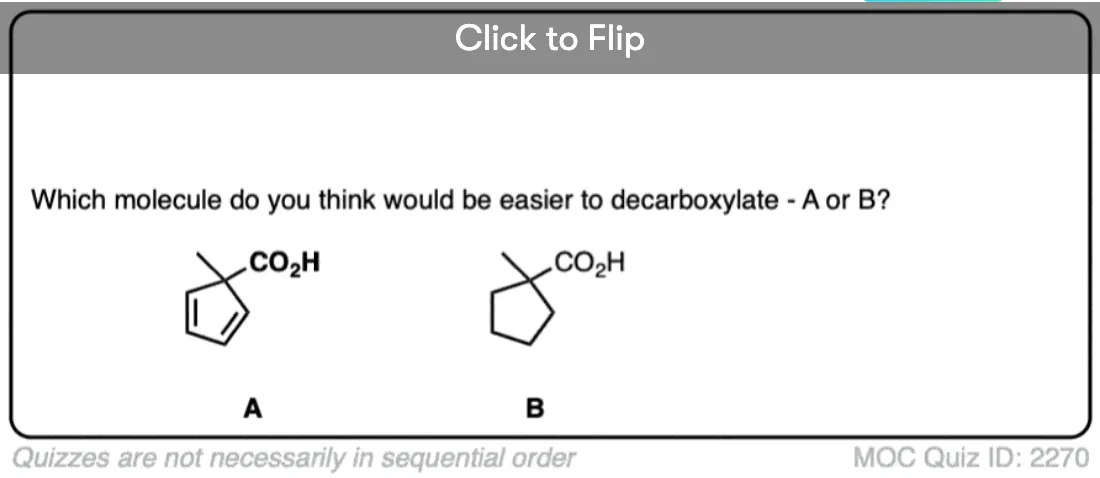
If you look at these decarboxylations as kicking off leaving groups, what do you notice?
 Click to Flip
Click to Flip
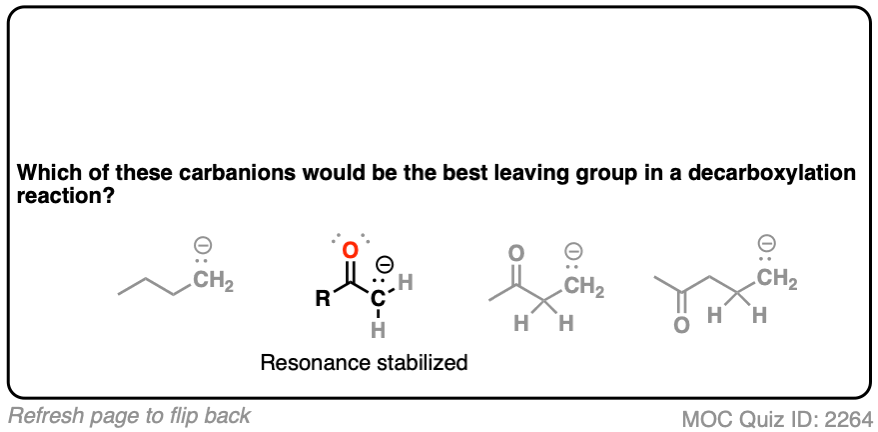
Hopefully you said something like, “aha! the enolate is different because it has a negative charge that can be delocalized to the oxygen by resonance, and is therefore a much weaker base, and therefore a better leaving group!”.
Exactly!!!
That’s why the C=O has to be two carbons away. We would form an enolate, and the negative charge can get moved to the oxygen that way. Oxygen, being more electronegative, does a much better job of stabilizing a negative charge than carbon does.
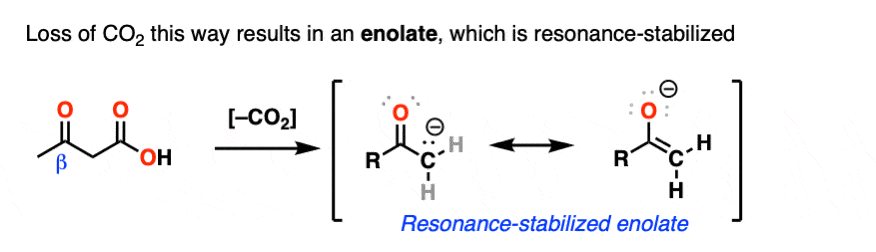
5. The Mechanism
Thinking of decarboxylations as a type of elimination from a carbonyl group is a good place to start. (In early studies of decarboxylation, a mechanism very much like that was proposed.)
Experimental evidence paints a slightly different picture. [note 6 for more on mechanism]
Our best working model is that we have a concerted mechanism with a cyclic transition state where the C–C bond breaks at the same time that the O-H and C-C pi bonds are formed. It happens all at once with no charged intermediates, similar to our old friends the sigmatropic rearrangements.
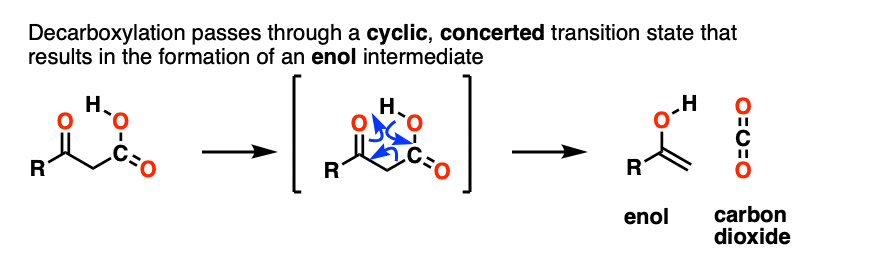
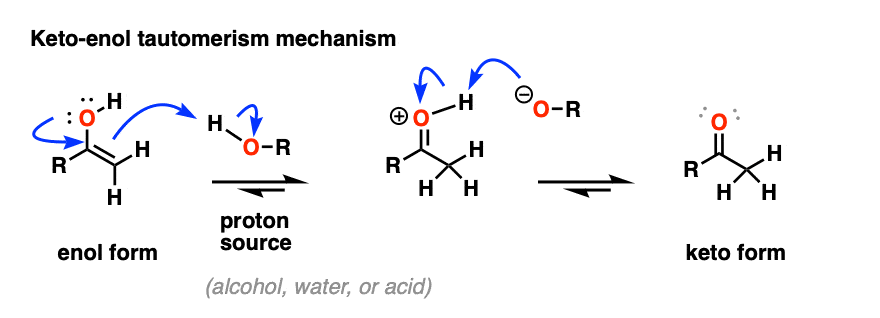
An enol is formed as an intermediate. The enol then undergoes keto-enol tautomerism to give us the ketone
To see the keto-enol tautomerization mechanism, hover here and an image will pop up or click the link.
{kind=link}
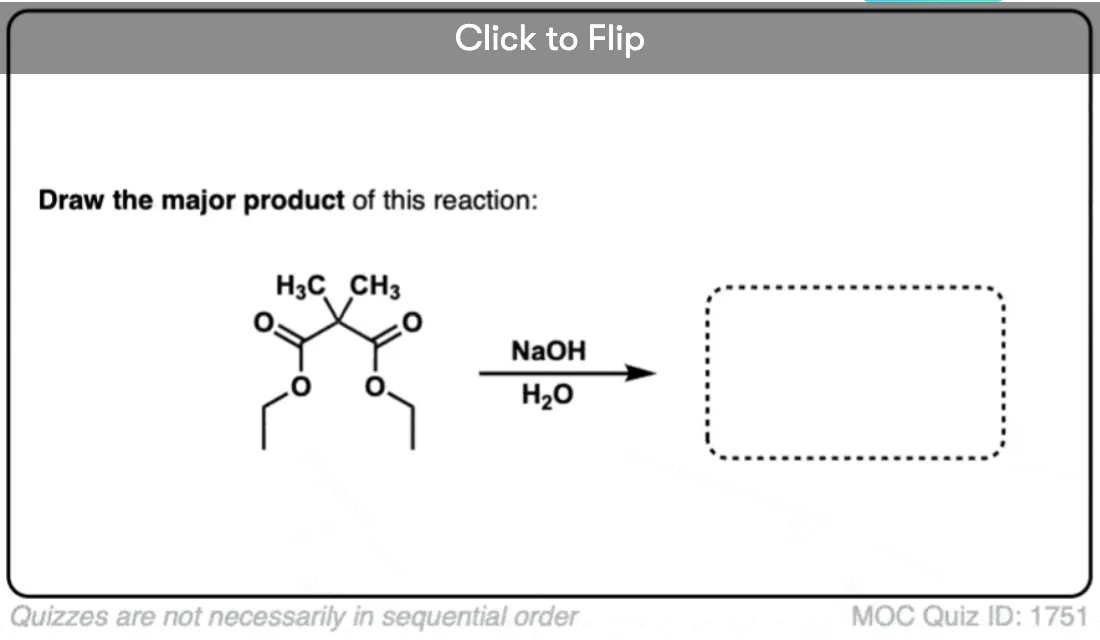
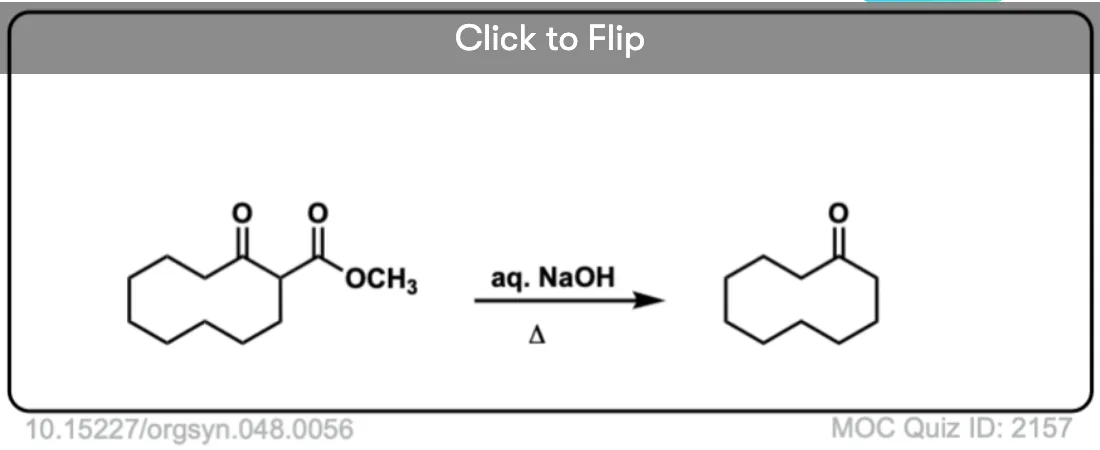
6. Decarboxylation in the acetoacetic ester and malonic acid syntheses
In the malonic ester and acetoacetic ester syntheses, an enolate is formed (e.g. with NaOCH3) which is then alkylated with an alkyl halide (e.g. CH3I). A second alkylation can be performed if desired. The ester is then hydrolyzed, either under basic (NaOH) or acidic (H3O+) conditions.
Decarboxylation is the final step in the malonic ester and acetoacetic ester syntheses.
Here are some examples of decarboxylation from these syntheses as found in the wild. Can you draw the final product?
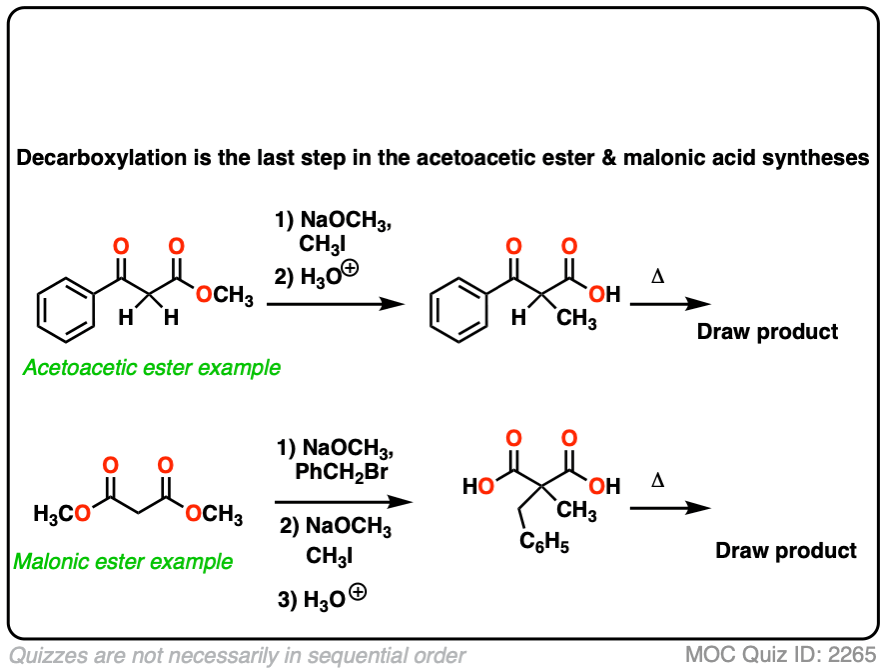 Click to Flip
Click to Flip
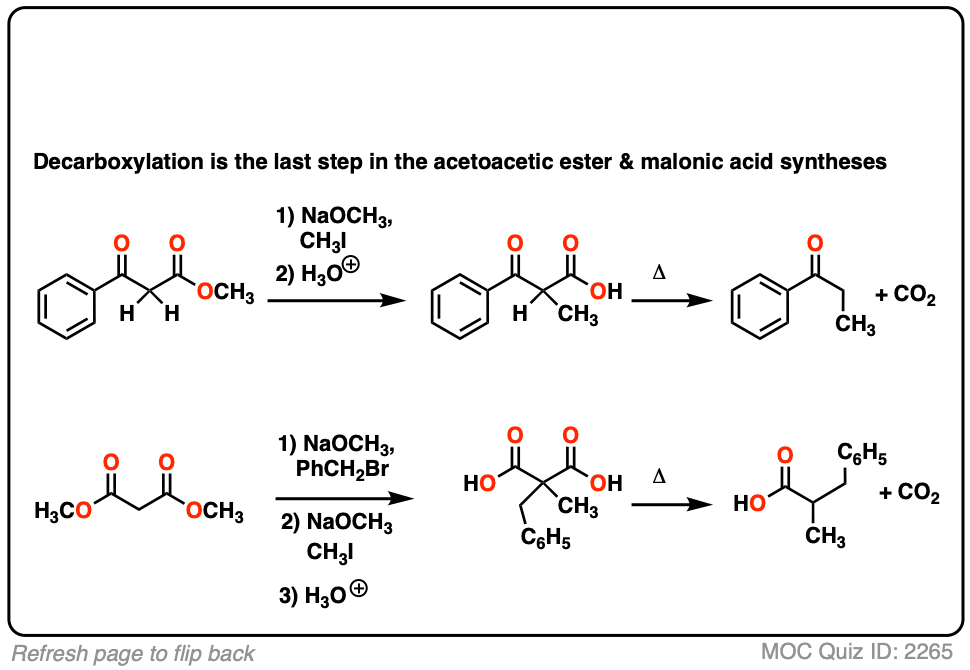
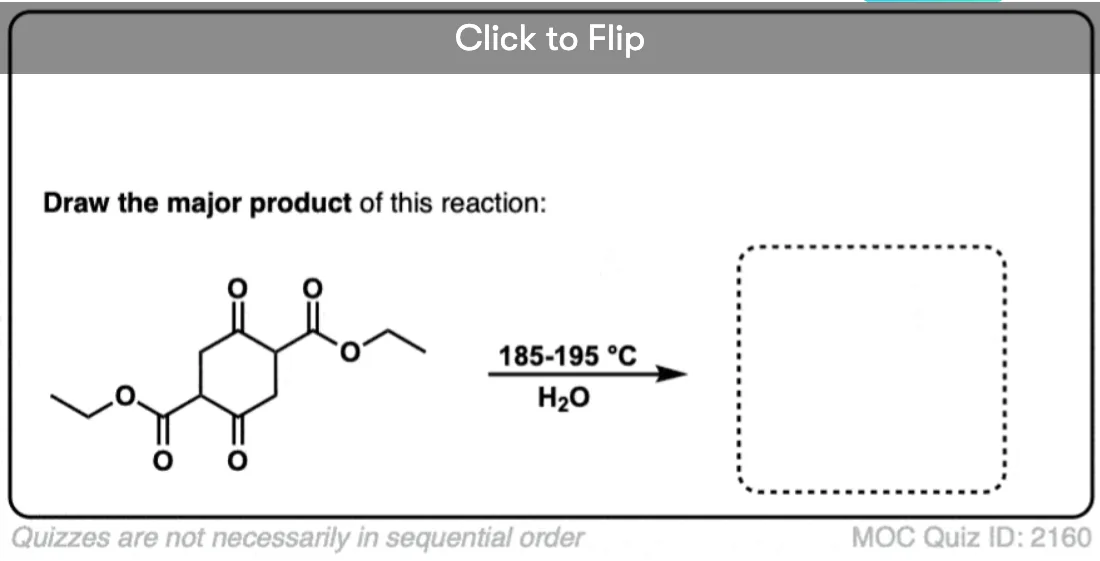
7. A beta-keto acid that doesn’t decarboxylate
Alright. We’ve seen that having a C=O two carbons over from a carboxylic acid is the key to decarboxylation.
Let’s look at a fun exception. Here’s an example of a beta-keto carboxylic acid that doesn’t readily decarboxylate upon heating.
Why not?
 Click to Flip
Click to Flip
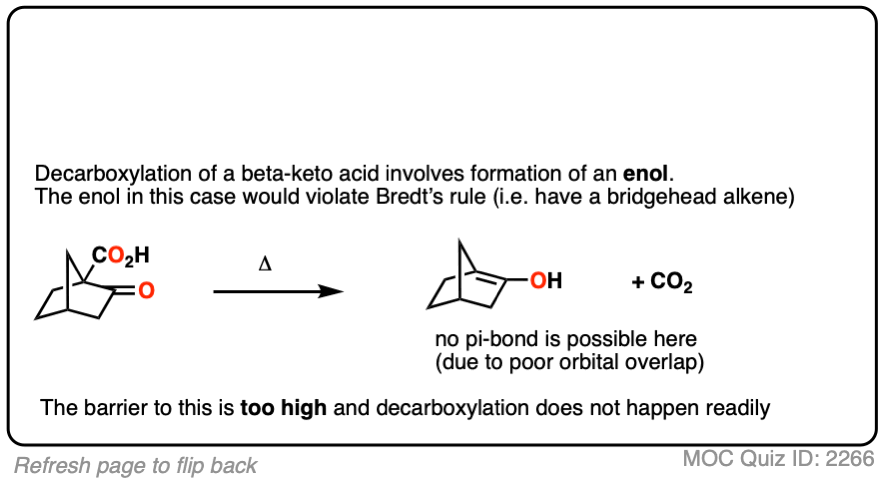
This is one of those little examples that helps to prove the rule.
Since the reaction proceeds through an enol intermediate, and the enol has a C-C bond, it turns out that enols don’t form very well if it means that a bridgehead double bond has to form.
8. Some Other Examples of Decarboxylation
I’ve gotten so far without covering the most familiar example of decarboxylation.
Treating a carbonate ion with weak acid leads to formation of carbonic acid, which undergoes spontaneous decarboxylation to give water and CO2.
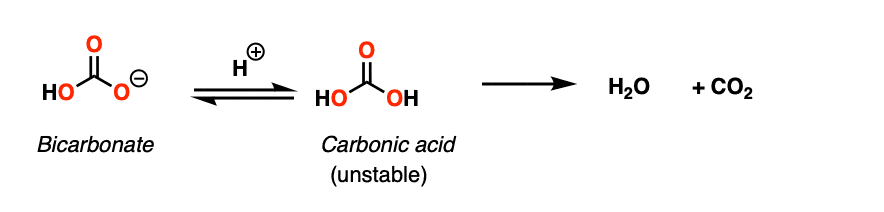
Here is a more vivid example of what it looks like in practice:
In this vein, a few similar examples of decarboxylations make their appearance in the latter part of Org 2.
The examples that spring to mind are cousins of each other. [See post: The Hofmann and Curtius Rearrangements]
In the Curtius rearrangement, an acyl azide is heated and rearranges to an isocyanate, losing N2 in the process. If an alcohol is added we get a carbamate, which is the more usual application of this reaction. However if water is added, this gives us a carbamic acid, which (like carbonic acid) spontaneously decarboxylates.
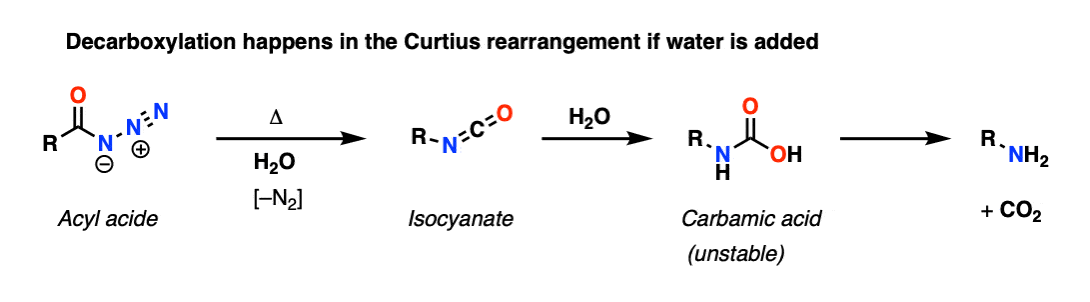
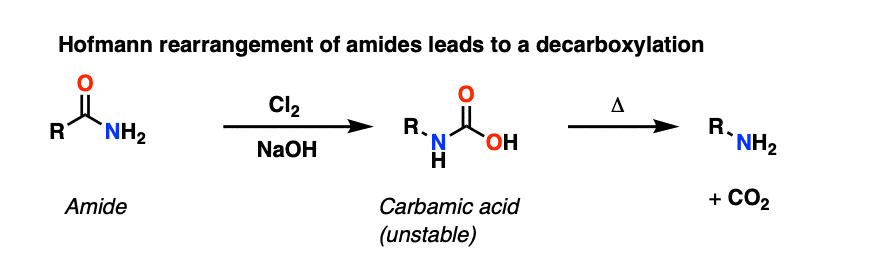
The Hofmann rearrangement is a similar rearrangement of an amide with Br2 and NaOH to give an intermediate carbamic acid, which likewise loses CO2 to give an amine.
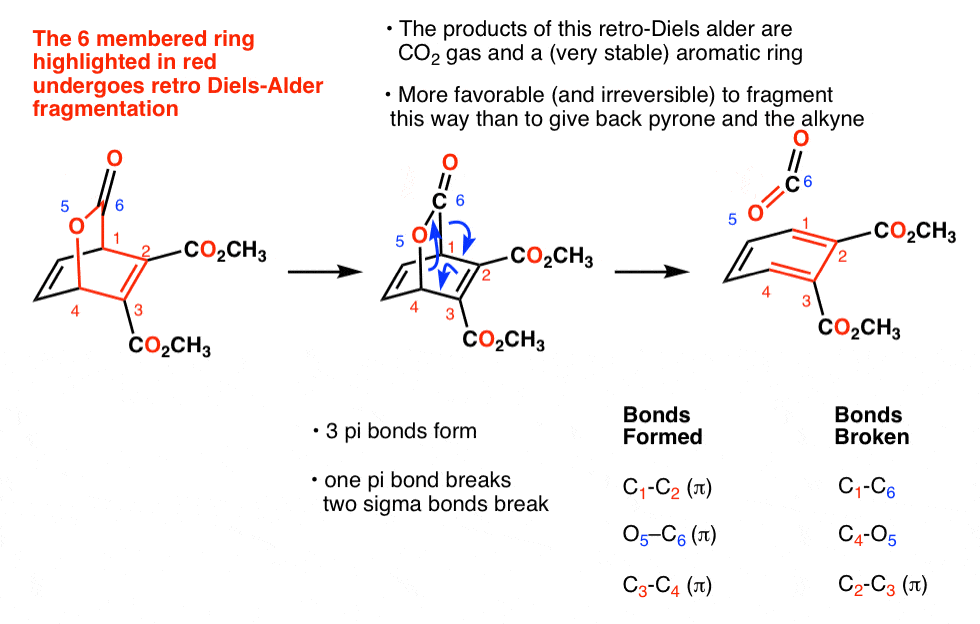
In the footnotes is a brief discussion of two more decarboxylations that are sometimes encountered in introductory organic chemistry: the Kolbe electrolysis and decarboxylation in some Diels-Alder reactions. [note 7]
9. Conclusion
Let’s recap:
- Decarboxylation occurs readily in beta-keto acids and in malonic acid derivatives. A C-C bond is broken, and a C-O (pi) bond is formed.
- The reaction proceeds through a cyclic, concerted transition state and results in an enol intermediate, which then undergoes keto-enol tautomerism
- Watch out for the common mistake of treating CO2 like a leaving group! It’s better to think of decarboxylation as a type of 1,2-elimination reaction, where forming the new C-C pi bond “kicks off” the C-C bond.
- The C-C bond can be broken because the pair of electrons can delocalize to oxygen via resonance. This path isn’t available to “normal” long-chain carboxylic acids.
- Decarboxylation also happens spontaneously in carbonic acid and carbamic acid.
Notes
Note 1 – Acetoacetic acid (pKa = 3.5) has a half-life of 140 minutes at 37°C in water. [Ref] Its conjugate base, acetoacetate, is present in the body under physiologic conditions. In nature, the decomposition of acetoacetate to acetone and carbon dioxide is catalyzed by an enzyme, acetoacetate decarboxylase.
Interesting historical note on this enzyme, from Wikipedia:
Acetoacetate decarboxylase is an enzyme with major historical implications, specifically in World War I and in establishing the state of Israel.[3] During the war the Allies needed pure acetone as a solvent for nitro-cellulose, a highly flammable compound that is the main component in gunpowder.[4] In 1916, biochemist and future first president of Israel Chaim Weizmann was the first to isolate Clostridium acetobutylicum, a Gram-positive, anaerobic bacteria in which acetoacetate decarboxylase is found. Weizmann was able to harness the organism’s ability to yield acetone from starch in order to mass-produce explosives during the war.[3] This led the American and British governments to install the process devised by Chaim Weizmann in several large plants in England, France, Canada, and the United States. Through Weizmann’s scientific contributions in World War I, he became close with influential British leaders educating them of his Zionist beliefs.[5] One of them was Arthur Balfour, the man after whom the Balfour Declaration—the first document pronouncing British support in the establishment of a Jewish homeland—was named.
Note 2 – The term “geminal” (from the Latin geminus, “twinned”) refers to a molecule containing two identical substituents (e.g. gem-diols are another name for hydrates, gem-dihalides are made through double addition of HX to alkynes). Maleic acid is a gem-diacid since there are two CO2H substitutents on the carbon.
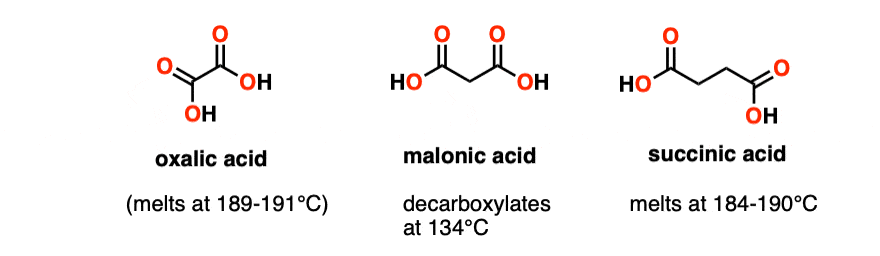
Note 3 – The equivalent diacids would be oxalic acid (melts at 190°C without decomposition) and succinic acid (melts at 184-190°C). Both of these molecules happily melt without decarboxylation.
Note 4 – The species (-)CO2H is technically a conjugate base of formic acid (HCO2H) obtained through deprotonation of the aldehydic hydrogen. It is (by far) more unstable than the much more commonly encountered formate ion HCO2(-).
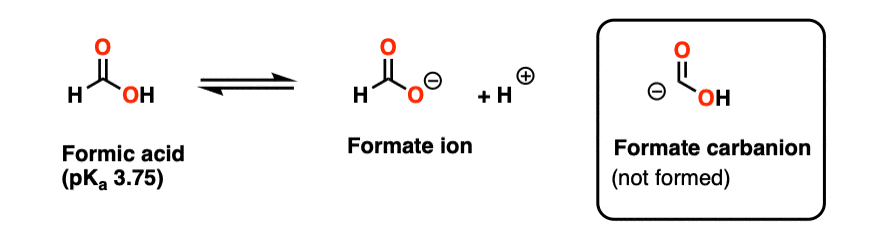
The carbon in formic acid is at an oxidation state of +2, in contrast the +4 oxidation state of carbon in CO2. So this species would need to lose two electrons in order to form CO2. (It turns out that formic acid is a useful source of H(-) in some transition-metal catalyzed reactions).
Note 5 – Why isn’t this carbon “resonance-stabilized”? This seems like a good opportunity for a quiz.
 Click to Flip
Click to Flip
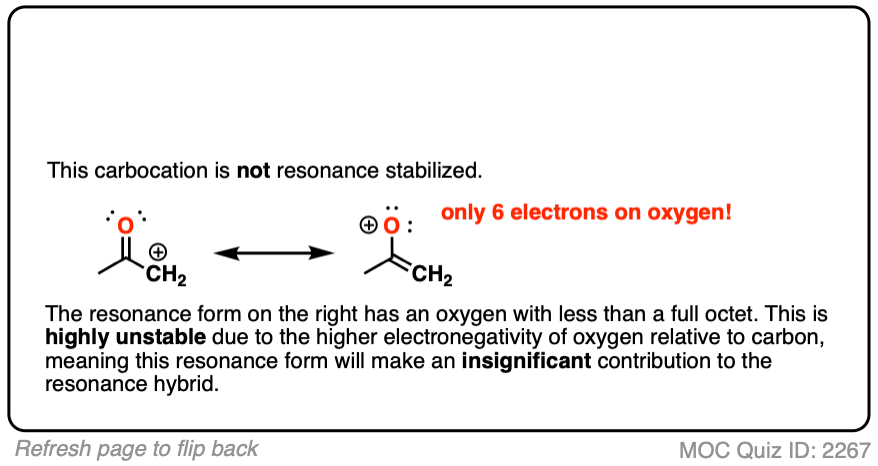
Moving a pair of electrons to form a new C-C pi bond would result in an oxygen atom with less than a full octet.
The stability of atoms with less than a full octet greatly decreases as one goes from left to right along the periodic table from boron (most stable) to carbon, nitrogen, oxygen, and fluorine (least stable).
As the electronegativity of the nucleus increases, so does its electron affinity. While carbocations are well-known and can even be isolated in some cases, there is no way that an oxygen (electronegativity 3.4) will give up its pair of electrons to a wimp like carbon (electronegativity 2.5).
For an entertaining taste of what kind of chemistry an oxygen atom with less than a full octet might perform, I highly recommend this article by Derek Lowe: “Things I Won’t Work With: Dioxygen Difluoride“, which covers the chemistry of FOOF.
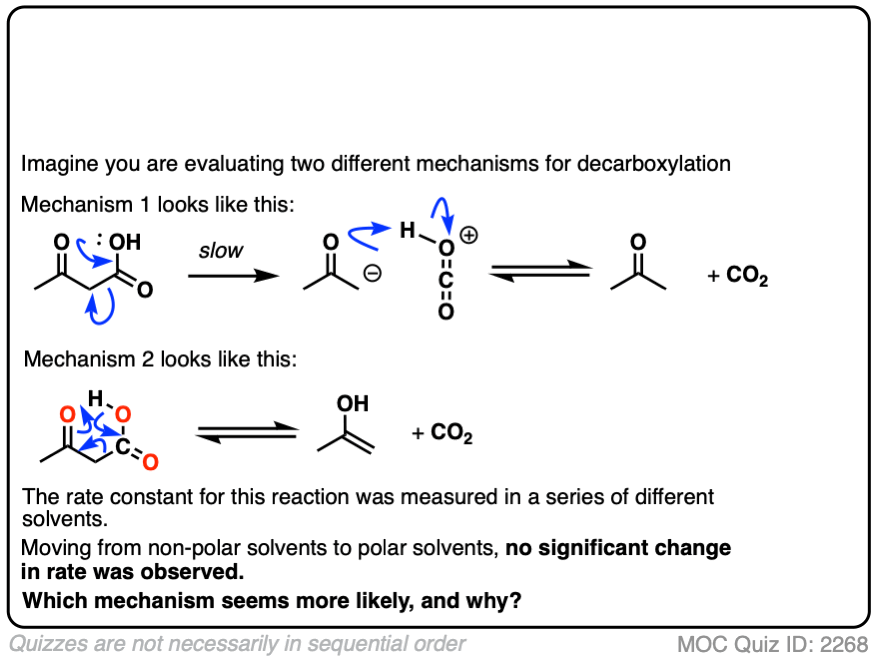
Note 6 This seems like a good opportunity for an exam-level quiz.
Let’s say it’s 1941, and the mechanism of decarboxylation is not known with certainty, but you have two hypotheses as to how it might happen.
So you conduct a series of experiments where you measure the rate constants in a variety of solvents of differing polarity.
The rates are essentially constant. What conclusions can you draw?
 Click to Flip
Click to Flip
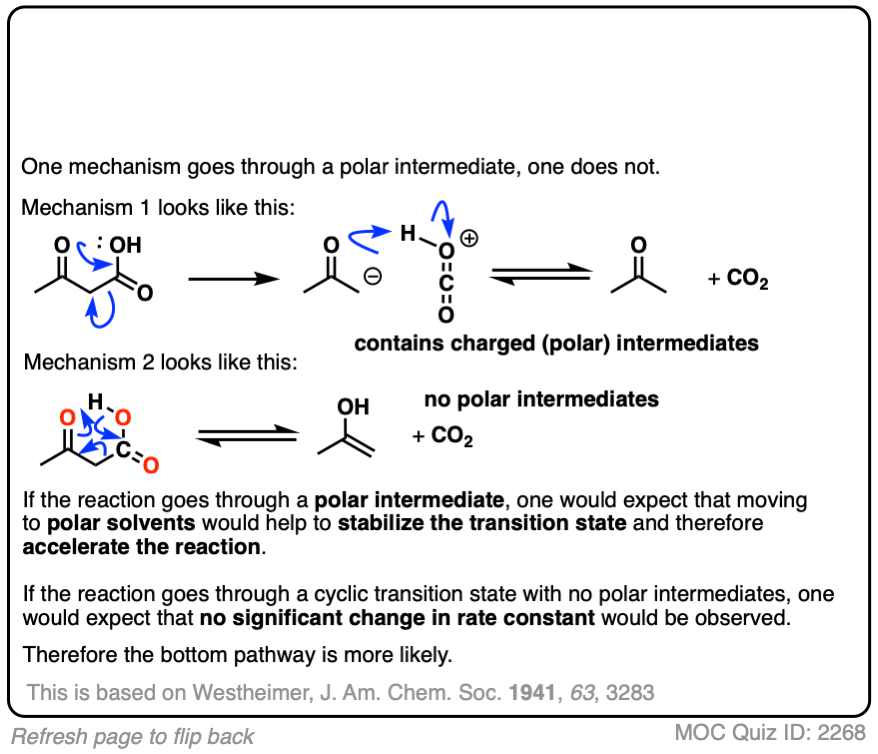
This was an important experiment for establishing the cyclic mechanism for decarboxylation.
The full story is a little more interesting. [Ref]
Note 7. Decarboxylation comes up in a few more contexts in introductory organic chemistry.
In the Kolbe synthesis, a carboxylic acid is treated with electrons, which results in loss of CO2 and a coupling of two molecules together via a radical process.
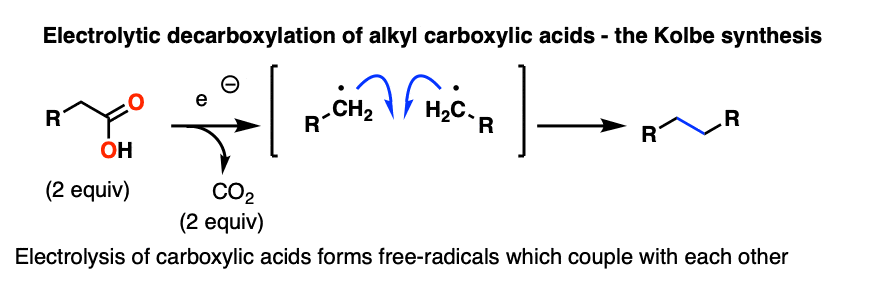
This is one area where future textbooks of organic chemistry will be rewritten. After being essentially ignored for decades, electrochemistry is undergoing a renaissance. A very interesting frontier of organic chemistry is in taking carboxylic acids and coupling them together through an electrochemical process in the presence of metal additives that ends up saving a lot of intermediate oxidation/reduction steps. Stay tuned.
Here is a very good modern example [Ref].
Another context where decarboxylation comes up is in retro Diels-Alder reactions of pyrones. Treatment of pyrones with a dienophile like electron-poor alkynes followed by a retro Diels-Alder results in a loss of CO2 plus a new aromatic ring.
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.

Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
1. Kinetics of the Decarboxylation of Acetoacetic Acid
R. W. Hay and M. A. Bond
Australian Journal of Chemistry 1967 20, 1823-1828
DOI: 10.1071%2FCH9671823
From the abstract: The decarboxylation of acetoacetic acid CH3COCH2CO2H → CH3COCH3+CO2 and its anion have been studied in aqueous solution over a range of temperature. Although the acid decomposes approximately 50 times faster than the anion at 37°, the activation energy of the acid (23.7 kcal mole-1) is comparable with that of the anion (22.9 kcal mole-1) and the large difference in reactivity is due to an entropy effect. The significance of these results is discussed and a comparison made with other β-keto acid decarboxylations.
2. Tautomeric equilibriums in acetoacetic acid
Karen D. Grande and Stuart M. Rosenfeld
Journal of Organic Chemistry 1980 45 (9), 1626-1628
DOI: 10.1021/jo01297a017
The proportion of acetoacetic acid in the enol form is highest in non-polar solvents (49% in CCl4) than polar solvents (2% in D2O). Protic solvents disrupt the internal hydrogen bond found in the enol form.
3. The Effect of Solvent on Some Reaction Rates
F. H. Westheimer and W. A. Jones
J. Am. Chem. Soc. 1941, 63, 3283
DOI: 10.1021/ja01857a017
Westheimer investigated the rates of decarboxylation in various solvents. If decarboxylation proceeds through a charged intermediate, then it would be expected that a rate acceleration would be observed in solvents with a high dielectric constant. No significant rate acceleration was found. This led Westheimer to propose the cyclic transition state model for decarboxylation.
4. Concert along the Edge: Dynamics and the Nature of the Border between General and Specific Acid–Base Catalysis
Hannah R. Aziz and Daniel A. Singleton
Journal of the American Chemical Society 2017, 139, 5965-5972
DOI: 10.1021/jacs.7b02148
A very interesting experimental and theoretical investigation of beta-keto acid decarboxylation. The study supports the existence of a cyclic mechanism where proton transfer is fast, but heavy-atom (i.e. C and O) movement is slow, leading to a dynamic “intermediate” which is not actually an energy minimum.
The study also has an interesting rejoinder to Westheimer’s 1941 study; the calculated dipole moments of the concerted (8.40 D) and “stepwise” (8.35 D) proton transfer events are not so different, meaning that one should not expect to see significant acceleration of the rate in polar solvents.
5. The Nature of The Transition State for the Decarboxylation of β-keto acids
Marshall W. Logue, Ralph M. Pollack, Victor P. Vitullo
J. Am. Chem. Soc. 1975, 97, 6868-6869
DOI: 10.1021/ja00856a047
Study of deuterium isotope effects in the decarboxylation of beta-keto acids.
6. On the transition state for decarboxylation of β-keto acids and βγ-unsaturated acids
D.B.Bigley, J.C.Thurman
Tetrahedron Letters 1967, 8, 2377-2380
DOI: 10.1016/S0040-4039(00)71610-8
7. The Decarboxylation of β-Keto Acids. II. An Investigation of the Bredt Rule in Bicyclo[3.2.1]octane Systems
James P. Ferris and Nathan C. Miller
Journal of the American Chemical Society 1966 88 (15), 3522-3527
DOI: 10.1021/ja00967a011
Interesting study of decarboxylation of a beta-keto acid in a bicyclo[3.2.1]octane system, where it is found that the rate of decarboxylation correlates with the dihedral angle between the departing CO2H and the pi-bond of the ketone (ideal dihedral angle = 90° as expected).
Hello!
In §8 you discuss a Curtius rearrangement in which an acyl azide is heated and rearranged into an isocyanate, losing N2 in the process.
However, in the corresponding diagram, you draw a molecule subtitled “Acyl acid”… Is this an oversight or is there another explanation?
Hi – yes, that’s a typo! Should be “azide” not “acide” :-)
Let’s say I plan out a synthesis that involves the formation of beta keto carboxylic acid as a product (like in Doebner modification of Knoevenagel condensation), what are the methods to make sure that this acid doesn’t decarboxylate? Will low temperature (like room temperature) do the trick? Or will I need to choose a specific solvent system?
Depends on structure, but at the end of it, there are lots of mild room-temperature protocols for basic ester hydrolysis (LiOH, H2O/THF) that will give you the carboxylic acid at the end. Acidic hydrolysis generally involves heat and with heat comes decarboxylation.