Organic Chemistry Tips and Tricks
The Most Annoying Exceptions in Org 1 (Part 1)
Last updated: January 23rd, 2024 |
One of the common complaints you hear a lot from people learning English is that there are just so many exceptions. The plural of “goose” is “geese”, but the plural of “moose” is “moose”. Why is that? Who knows. You just have to memorize it. (Edit: well, when learning English, you do have to memorize it. But that’s not going to be the point of this post. Maybe this wasn’t the best example to choose).
Organic chemistry is also rife with many exceptions (real and apparent) to the beginning student. It isn’t long after you learn about Markovnikoff’s rule, for instance, that you learn when this “rule” is broken. And that’s just the first of several exceptions that come up in the course.
So one common approach is to simply memorize these exceptions, the way you might memorize “i before e, except after c”. While a memorization approach could be effective for exams that test the ability to regurgitate book knowledge, however, any instructor looking to test problem-solving ability could easily design a test that will render such pure memorization efforts ineffective.
Behind every exception there is a deep reason why things occur the way they do, and these reasons illustrate deeper principles of organic chemistry. The purpose of today’s post is to illustrate the key concepts behind some of the most common “exceptions” in Org 1.
Annoying exception #1 : Hydroboration. Not long after you learn about Markovnkoff’s rule, you learn that when you add a borane to an alkene, it adds the “opposite way”. What’s going on here? Interestingly, it isn’t as much of an exception as it seems. Things make a lot more sense if you examine the relative electronegativities of the atoms being added. In all cases – whether adding HBr, HCl, H3O(+), or BH3, the most electronegative atom always adds to the most substituted carbon, because that’s the carbon that best stabilizes positive charge. In BH3, it just so happens that the most electronegative atom is hydrogen. It’s not really that weird after all.
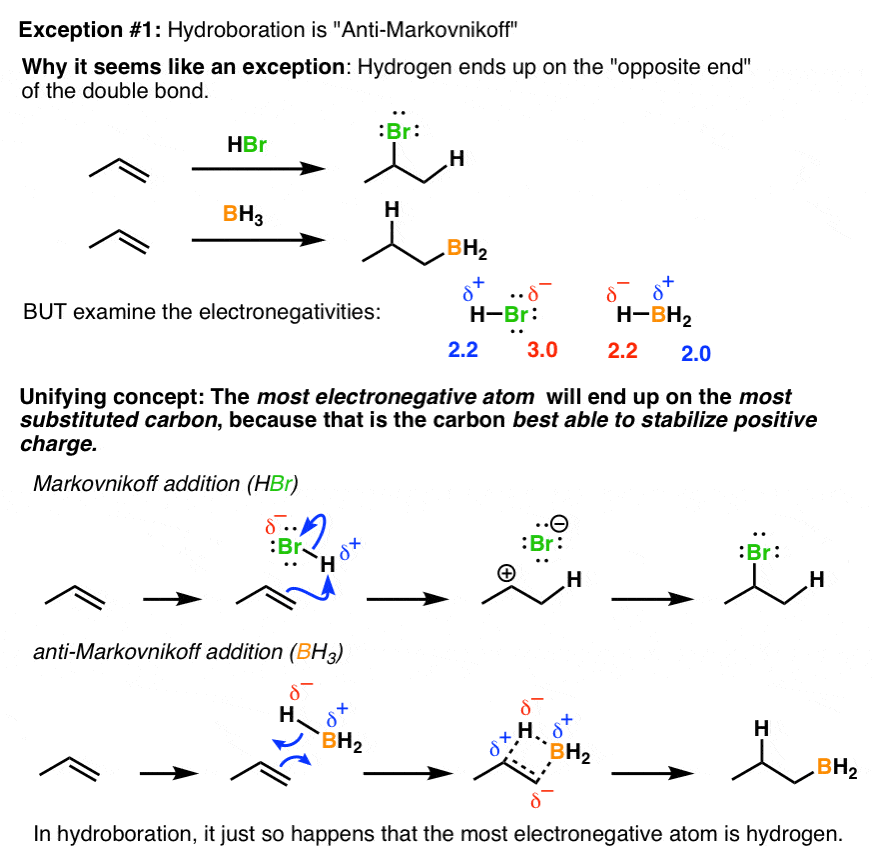
Annoying exception #2 – “Peroxides”. HBr by itself does Markovnikoff addition to alkenes but if peroxides are present, it adds the opposite way. Again, one approach here is to just memorize that if you see “peroxides”, it goes the other way. However, it isn’t much more effort to understand that what is going on in both cases is exactly the same: just as in the above example, an electron-deficient intermediate (carbocation or radical) ends up on the most substituted carbon.
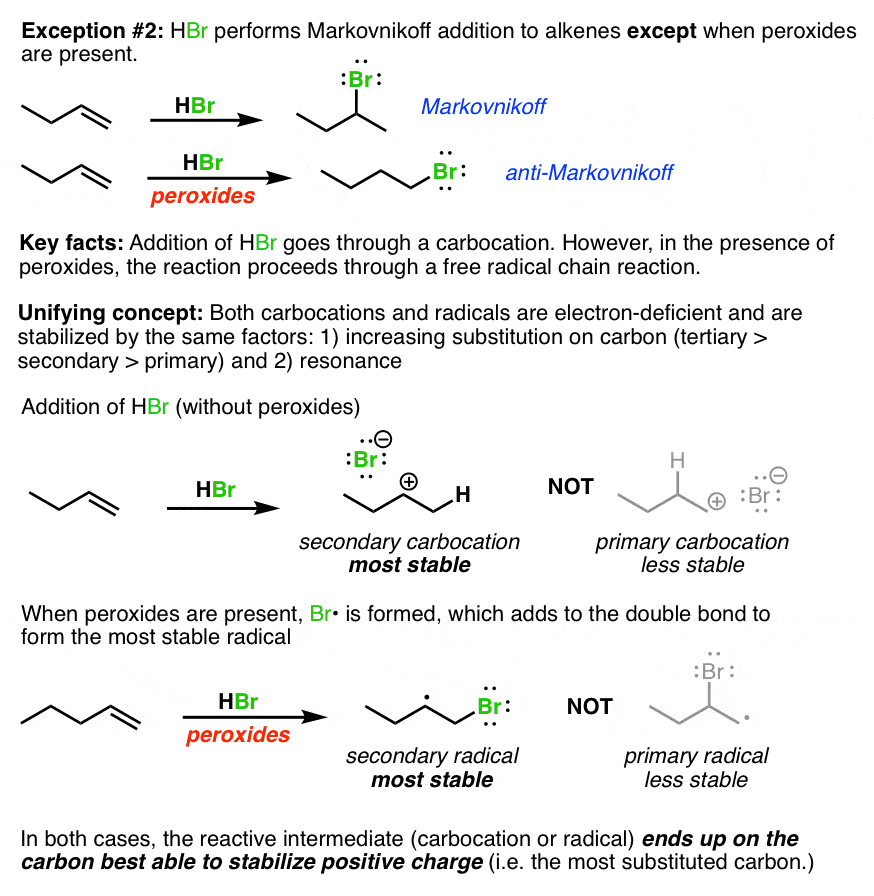
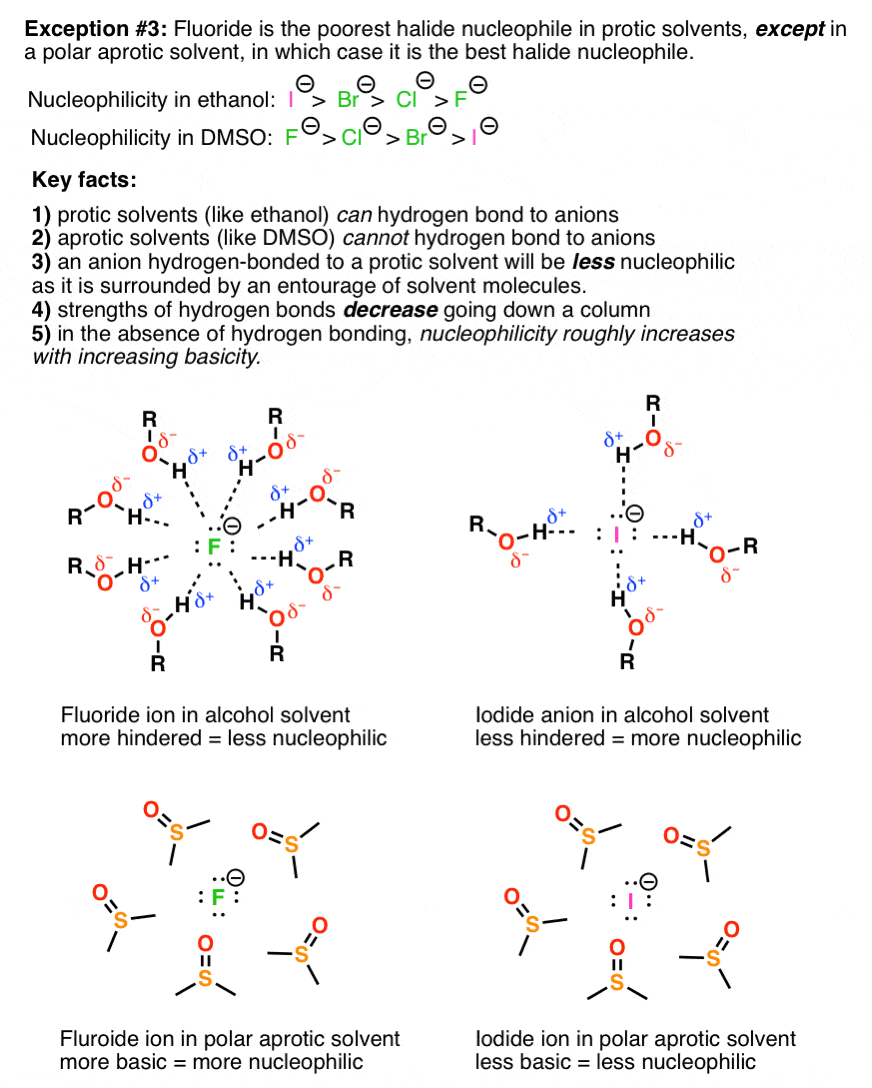
Annoying exception #3 – the nucleophilicity of halides in polar protic vs. polar aprotic solvents. Iodine is a better nucleophile than F(-) in polar protic solvents, but fluoride is a better nucleophile than I(-) in polar aprotic solvents.
What’s the key lesson here? There are really three key trends here. 1) nucleophilicity is decreased by hydrogen bonding – and a nucleophile in a protic solvent will be surrounded by solvent molecules it is hydrogen-bonded to 2) hydrogen bonding ability decreases as one goes down a column in the periodic table 3) in the absence of hydrogen bonding, nucleophilicity increases with basicity.
In fairness, thiis example is tough, because the key trends oppose each other and the relative nucleophilicities are not something that could have easily been predicted from first principles, but result from actual measurement of reaction rates. Furthermore, basicity and nucleophilicity are measured by different yardsticks – one (basicity) is measured according to equilibrium, and the other (nucleophilicity) is measured according to reaction rate. There’s no getting around having to memorize these trends, but understanding why they operate is key.
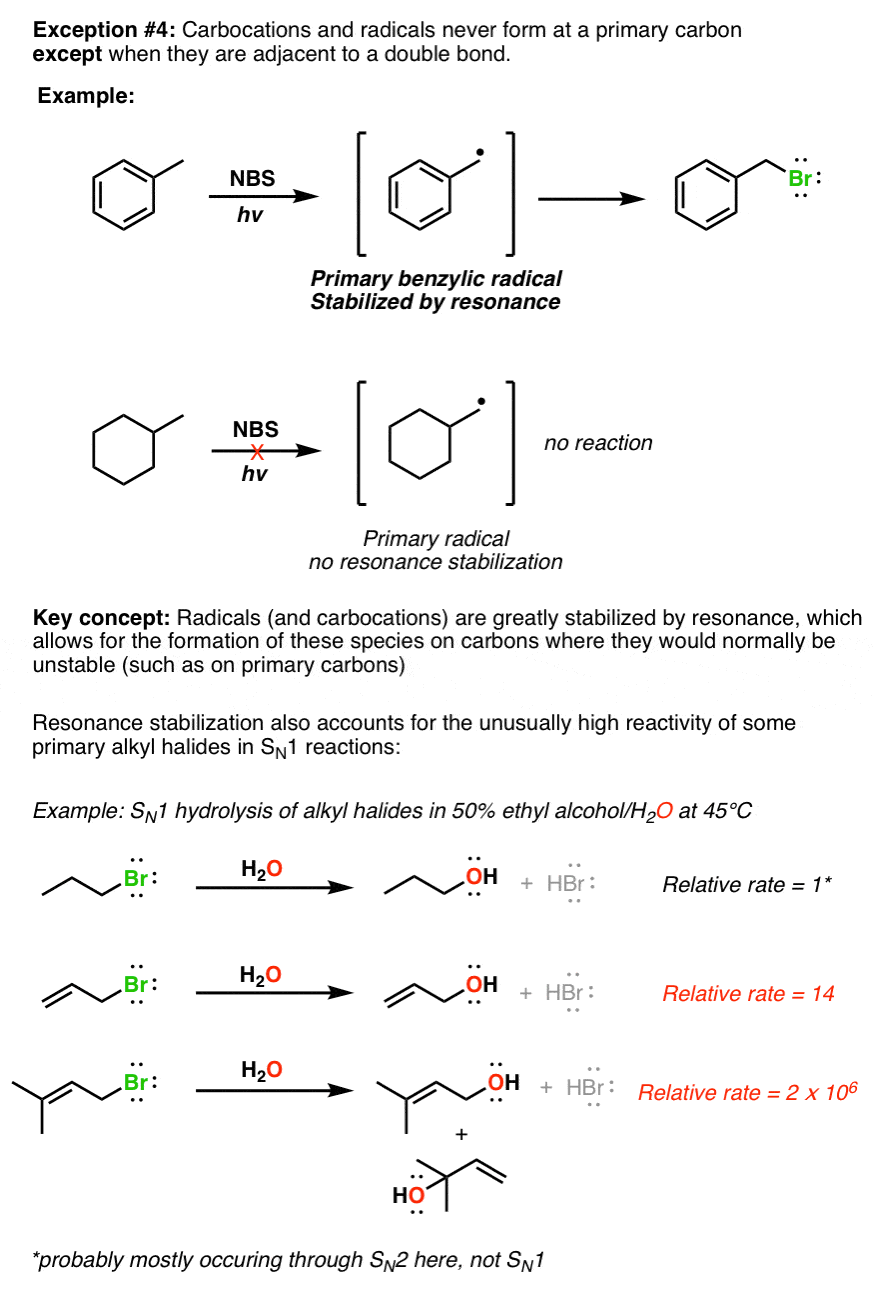
Annoying exception #4 – Primary carbocations and radicals are unstable, unless they are stabilized by resonance. This example illustrates the importance of resonance, which is one of the key stabilizing factors in organic chemistry. In general, any factors which allow a charge (or unpaired electron, in the case of the radical) to be distributed over a larger area tends to be stabilizing, which makes up for the otherwise unstable situation of having a carbocation on a primary carbon.
{kind=link}
That takes care of some of the common “electronic” effects. Next post, we’ll look at some of the “steric” effects that lead to weird exceptions in Org 1.
When HBr adds to an alkene to initially yield a secondary carbocation, this can rearrange to a tertiary carbocation at a neighboring carbon if there is an opportunity to do so via hydride shift or alkyl shift. When HBr/H2O2 adds to an alkene to initially yield a secondary radical, can this also rearrange to a tertiary radical at a neighboring carbon via a radical hydride or alkyl shift?
No, generally there are no rearrangements in free-radical chemistry analogous to the rearrangements of carbocations.
There are such things as intramolecular hydrogen atom transfers, which can sometimes result in more stable radicals, but these are generally outside the scope of what we cover here.
I completely agree about the nucleophilicity order in polar apotti solvent. But, this makes me doubtful of the reaction mechanism of the Conant Finkelstein reaction in which I(-) substitutes Cl or Br of a chloro or bromo alkane. Please clarify my doubt. (Maybe I am asking a silly question as a high school student. But, sir please clarify my doubt.)
The reason the finkelstein proceeds to completion is not because of the nucleophilicity of iodine versus other halides, it is that NaI is soluble in acetone whereas NaCl and NaBr are not. This drives the equilibrium toward formation of the alkyl iodide product.
Hope this helps
James
these are probably the topics that are gonna show up on the exam! thank you. very insightful..
I really can’t thank you enough for producing this site….. Taking ochem 1 right now. Seems like professors gloss-over the “why” of everything in favor of speedy teaching. Nice to find a place that can tell me why things happen the way they do on a molecular level. I’m big on minutiae. I have to know “why” before my brain can accept new information. If it doesn’t add up, it’s quickly rejected and calls everything associated with it into question. So thanks for helping me out the pieces together! Confidence in your knowledge is key with such a complex subject. ✌
Thanks! Now I understand why Nuc decreases in polar protic solvents. But one question, and it seemed to stump some fellow classmates: suppose you have a strong Nuc/weak base in a polar protic solvent. If the C is secondary, will it prefer to undergo Sn1 or Sn2? What if the beta carbon has a double bond, would that increase the likeliness of an Sn1?
I have the same confusion. Of course whenever I look no one seems to be able to answer this problem clearly if anyone bothers to answer it at all besides just saying “I- is a strong nucleophile so SN2.”-gee thanks. However, the answer in your scenario where it’s polar protic would definitely go SN1. Why? Because the transition state of the SN1 reaction is stabilized by the high dielectric constant of the polar protic solvent. Stabilization is particularly pronounced in a protic one due to H bonding that develops as the LG becomes negatively charged. My problem is very similar to yours but slightly different. What do you do if you’re looking at a secondary substrate and an I- is the nucleophile but the solvent is not stated. What do you do in this case? I have a ton of examples where this is the case in my problem sets and they’re extremely confusing. According to logic, and this article, the I- is too stable to act as a good nucleophile if the the solvent is aprotic, and so one would assume the mechanism to be SN1 but I don’t know, this is my question. On the other hand if the solvent is polar protic the I- all of a sudden becomes a strong nucleophile so you’d think SN2, but because the solvent is polar protic the key feature is transition state stabilization making this scenario to be SN1 without a doubt. It almost makes me wonder why we are even bothered being taught the trend where nucleophilicity is inversely related to basicity going down a group on the periodic table. It seems like a waste of time and extremely confusing. Realistically what we should be taught is that nucleophilicity follows basicity, while also knowing that if you’re in a polar protic solvent, despite the reverse trend becoming the case, it doesn’t matter. Because polar protic will go SN1 in a secondary substrate. If anyone can PLEASE answer me that question where it’s secondary and no solvent is stated and your nucleophile is I-, I’d greatly appreciate it.
And sorry, the double bond for the beta part of your question I don’t know.
All the confusion you guys are having is because you think that the author said that I- is a strong nucleophile. (For conclusion/summary, read the last para)
No. He never said that I- is a strong nu- ; only that it becomes a ‘better’ nucleophile with protic solvents. In general, the foremost requirements for SN1 reactions are:-
1. A good leaving group attached to the carbon chain.
2. More steric hinderance, so reactivity of carbon follows the order 3° > 2° > 1°.
3. Polar protic solvent, so that the good leaving group has lots of protons flowing around to bond to.
4. A weak nucleophile : This one is more much of a common sense fact. Because we are already choosing a carbon chain containing a good/excellent leaving group then why would we need a strong nucleophile? And in fact, if we choose a strong nucleophile then it will bond to the protons of the protic solvent well before the leaving group could bond to it. So, an SN1 reaction would never occur.
And that’s why I- is a “better” nu- in protic solvents (not stronger) because in protic conditions, a weaker nu- is more favorable for the SN1 reaction.
And to clarify, I- is a weak nu- because HI is a strong acid, so its conjugate base (I-) would be very weak.
In example of exception 4 there should be
‘ Unstable because of no resonance ‘ not ‘ because of resonance
I changed it to, “no resonance stabilization”
This is helpful! Thanks for taking the initiative and wanting to teach, regardless!
Hi, this is a great website!
I just noticed, though, that the McMurry 8th ed. textbook gives a different explanation for the Anti-Markovnikov hydration reaction with borane. On page 224, it says that the hydroboration reaction does not occur with a carbocation intermediate, but instead proceeds as a concerted reaction in which Markovnikov regiochemistry of the boron atom is disfavored by steric hindrance. This mechanism is supported by the fact that hydroboration results in the syn addition of water.
Actually, on looking at your mechanism again, the concerted step appears to be present. However, I think that the syn stereochemistry is best explained by sterics. Otherwise, it leaves the question of racemization hanging.
Thanks for maintaining this website!
Hi!
Just wanted to point out that in this case, sterics explains the anti-Markonikov addition in hydroboration, NOT the syn stereochemistry. (Of course, the explanation provided in point #2 of the post is also a sensible explanation for anti-Markonikov addition!)
The concerted addition is what explains the syn stereochemistry.
Racemization does occur actually, since the concerted addition can occur either above or below the plane of C=C.
I’m a Korean student studying Organic Chemistry for entering medical school. I’ve studied Organic Chemistry for a while but always thought that I’m missing something very important. But since I found your website coincidentally, I can understand the things that I just memorized before. I’m very appreciate that. Everyday I come here and I see myself not just memorizing reactions but just feeling them..
Excellent. If you learn just one thing, I hope it’s that there are key patterns and rules in organic chemistry, and it’s not just arbitrary.
Very helpful, I must say! Awesome! Just one point. The point #3.. In the diagram, the last two images both show fluoride being surrounded by DMSO. I think one of them should be Iodide.
I’m somewhat confuse… just memorize? I can do that.
Thanks for pointing that out… the example in the first paragraph was probably not the best.