Alkyne Reactions
Hydrohalogenation of Alkynes
Last updated: May 6th, 2026 |
Alkyne Hydrohalogenation – Addition of HX To Alkynes – HCl, HBr, and HI
In the previous three posts on alkynes we’ve introduced some new reactions that are specific to alkynes (versus alkenes):
- deprotonation (and subsequent substitution) (See Article: Acetylide Formation and Alkylation)
- partial reduction to alkenes (See Articles: Lindlar Reduction, Reduction of Alkynes With Na/NH3)
- formation of aldehydes and ketones through net “hydration”. (See Post: Hydroboration and Oxymercuration of Alkynes)
In this post we’ll go back to a key reaction mechanism pattern we observed with alkenes: the so-called, “carbocation pathway” that includes addition of HX and H3O+ and explore how many of the reactions of alkenes we’re familiar with can also be used with alkynes.
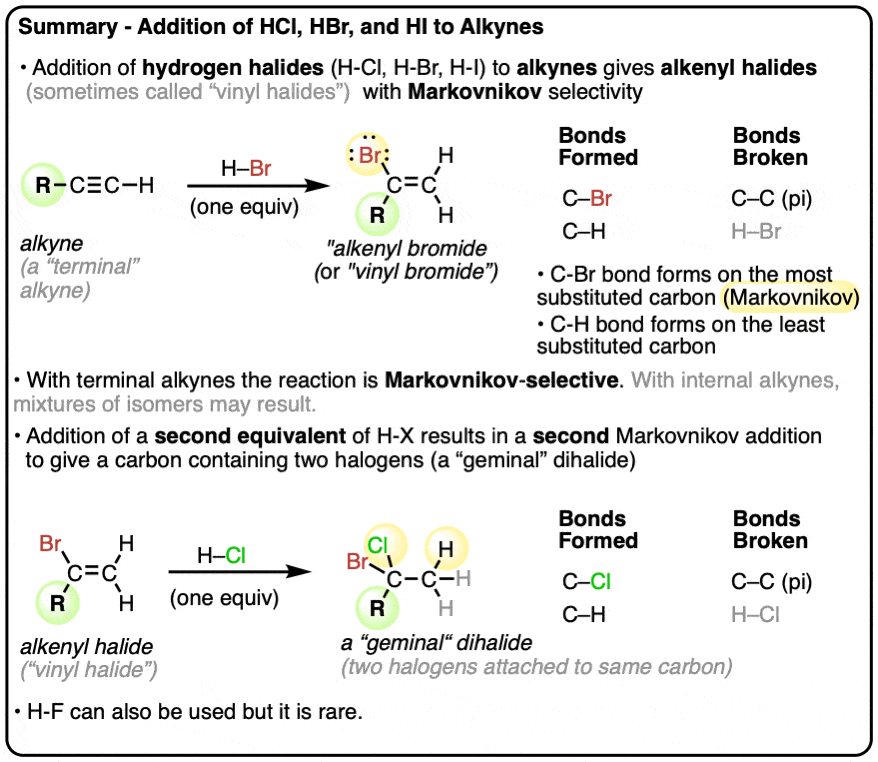
Table of Contents
- Addition of Hydrogen Halides (HCl, HBr, HI) To Alkynes – Once
- Addition Of A Second Equivalent Of HX To A Vinyl Halide Gives A Geminal Dihalide
- Addition of Hydrogen Halides To Alkynes – The Mechanism
- Comparing Alkenes and Alkynes In The “Carbocation Pathway”.
- Summary: Addition of Hydrogen Halides To Alkynes
- Notes (+ Termolecular Mechanism!)
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Addition of Hydrogen Halides To Alkynes (Once) – Hydrohalogenation
With all the focus on the ways in which alkyne chemistry can differ from alkene chemistry, it’s helpful to be reminded of all the ways in which they are similar.
The three major examples in this category are the reaction of hydrohalic acids (H-Cl, H-Br, and H-I) with alkynes. If you recall, when added to alkenes, these reagents were:
- attacked by the π bond of the alkene to give a carbocation on the most substituted carbon, giving “Markovnikov” regioselectivity (See Post: Markovnikov’s Rule) followed by
- attack of halide ion on the carbocation.
Since alkynes merely differ from alkenes in the addition of a second π bond, we would expect that these reactions would also work for alkynes as well – and they do!
If we treat an alkyne with a single equivalent of H–Cl [note – we’ll just use H-Cl in all of these examples, but HBr and HI work in exactly the same way] we end up forming an alkenyl chloride.
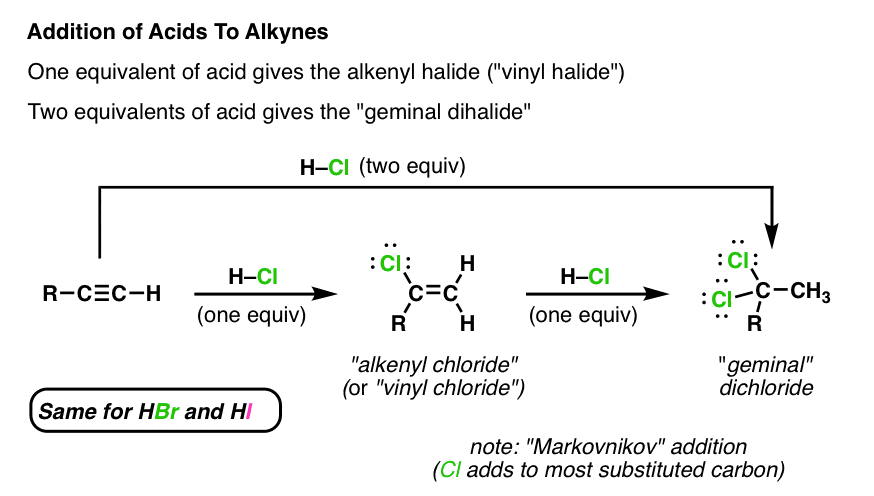
Note that the chlorine atom ends up attached to the most substituted carbon of the alkene [“Markovnikov” regioselectivity].
If we just use one equivalent of HX, we can get the reaction to stop at the alkenyl halide stage.
2. Addition Of A Second Equivalent Of HX To An Alkyne
You might be wondering if it’s possible to for this π bond to react with a second equivalent of H-Cl. The answer is yes. [Note – it is possible to just “stop” the reaction at this stage if we use just one equivalent, because the product (alkenyl chloride) is less reactive towards HCl than the starting alkyne].
Indeed, if we add a second equivalent of H-Cl, it adds to either side of the C-C π bond, giving us the product where two chlorine atoms are on the same carbon. By the way, we call this a “geminal” dichloride (think Latin – “gemini” = twins).
We can also get this product if we simply add two equivalents of H-Cl to the starting alkyne.
3. Addition of Hydrogen Halides To Alkynes – The Mechanism For Hydrohalogenation
So how might this reaction work? In a very similar fashion to how H-Cl adds to alkenes.
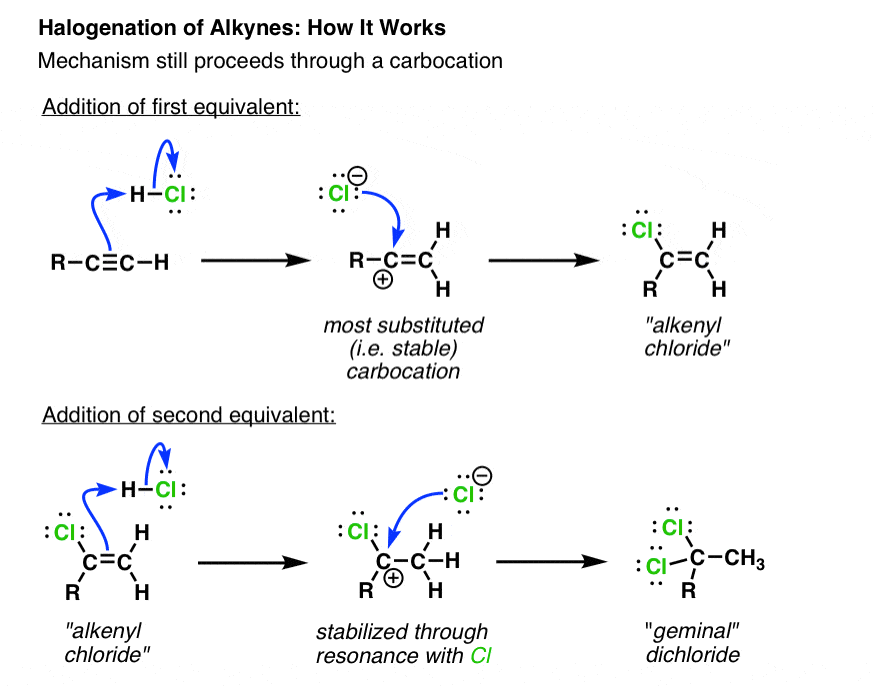
The first step is protonation of the alkyne with H-Cl in such a manner as to give the most stable carbocation intermediate.
Since carbocations are stabilized to a greater extent by electron releasing alkyl substituents than by hydrogen, the new carbocation will form at the end of the alkyne bearing the carbon substituent.
In the next step, the carbocation is attacked by the chloride ion to give the alkenyl chloride.
What about the second equivalent of H-Cl ? Given the fact that the geminal dichloride is the product here, the most reasonable mechanism for its formation is merely a repeat of the steps from the first reaction (as shown).
However it’s worth pointing out one interesting feature. Note that the carbocation in this case bears a chlorine substituent. Since carbocations are electron poor, and chlorine is quite an electronegative element, it’s interesting to point out that the electron releasing ability of the alkyl group [and the ability of chlorine to donate a lone pair to the carbocation] “win out” here over the electron-withdrawing character of the chlorine.
[If you go on to second-semester organic chemistry and cover the reactions of aromatic rings, you’ll see that Cl and other halide ions act as pi-donors toward adjacent carbocations. See post: Why Are Halogens Deactivating ortho-para Directors?]
As mentioned above, the reactions of alkynes with HBr and HI (as well as HF, just in case you’re curious) follow the exact same pathway.
[Note: there is considerable evidence to suggest that this reaction in fact proceeds not through a carbocation intermediate, but through a “termolecular” reaction incorporating two equivalents of H-X and the alkyne. This is covered inconsistently in courses and textbooks. I strongly suggest you double check your textbook to verify how it is taught in your course. See Note 1].
4. Comparing Alkenes and Alkynes In The “Carbocation Pathway”
It’s probably worth tying back this post to the post on alkenes and the carbocation pathway, noting the similarities and differences between the chemistry of alkenes and alkynes. Hopefully this table will prove useful:

5. Summary: Addition of Hydrogen Halides To Alkynes (Hydrohalogenation)
As with alkenes, reactions that follow this pathway proceed through a carbocation intermediate and provide the “Markovnikov” products as major.
The key difference in this pathway is that hydration of alkenes gives alcohols, whereas hydration of alkynes gives carbonyl derivatives (i.e. ketones/aldehydes) after keto-enol tautomerism of the intermediate enol.
In the next post, we’ll explore the “3-membered ring” pathway with alkynes.
Next Post: Alkynes – The 3-Membered Ring Pathway
Notes
Note 1. The “termolecular” pathway for hydrohalogenation of alkynes
The intermediacy of vinyl carbocations in addition to alkynes seems to belong in the bucket of “mechanisms that are oversimplified for an introductory audience”, with the hope that textbooks will reach consensus in the future.
Prof. Hilton Weiss of Bard College writes:
I’ve done a lot of work on this and, of course, I believe my own results. My initial paper denying the vinyl cation intermediate (before doing any research) was in JChemEd 1993, p 873… You might look at Maitland Jones’s textbook or Carey and Sundberg. Actually my current belief is that the vinyl cation is EXTREMELY RARE in additions to alkynes. In Stang’s paper on the rearrangement of the “t-butyl vinyl cation” by solvolysis of the corresponding triflate, he made the triflate ester by adding trifluoromethane sulfonic acid to t-butylacetylene. This addition occurred with NO rearrangement. If triflic acid (pKa =-10) won’t protonate an alkyne, nothing will. Conjugated alkynes (e.g. phenylacetylene) can form conjugated vinyl cations but only in strong acids. Aqueous acids are not even close. (H3O+ =-1.7, HBr = -9, HCl = -7). I would not be surprised if the strongest acids add via a short-lived ion pair but even that is rare. Most textbooks say that alkenes and alkynes react by the same mechanism: it’s easier for students as long as you don’t look too close. By the way, the termolecular mechanism does not involve a proton and a halide ion attacking the alkyne at the same time; too improbable. First there is a reversible pi complex between acid and alkyne followed by a halide attached anti periplanar at the more positive carbon.
Thank you to Prof Weiss for writing. A link to the J. Chem. Ed. article is here.
A few years later, Prof. Thomas T. Tidwell wrote a rebuttal, stating the reasons why the vinyl carbocation pathway is valid. Read it here.
There is also a rebuttal to the rebuttal (1996) by Weiss. Read here.
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
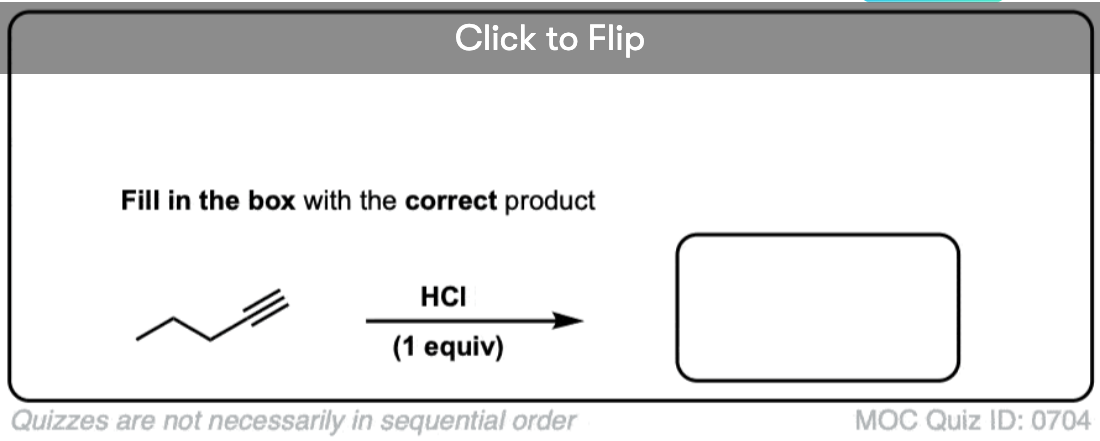
Become a MOC member to see the clickable quiz with answers on the back.
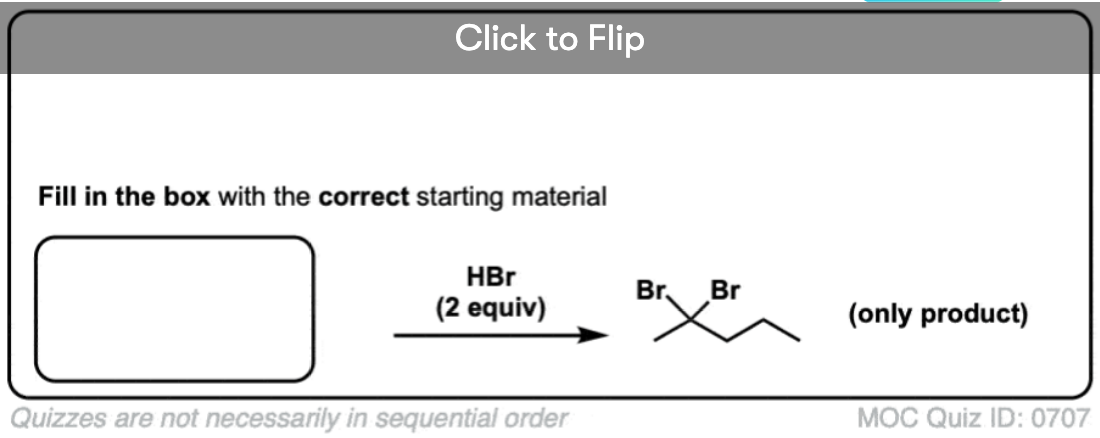
Become a MOC member to see the clickable quiz with answers on the back.
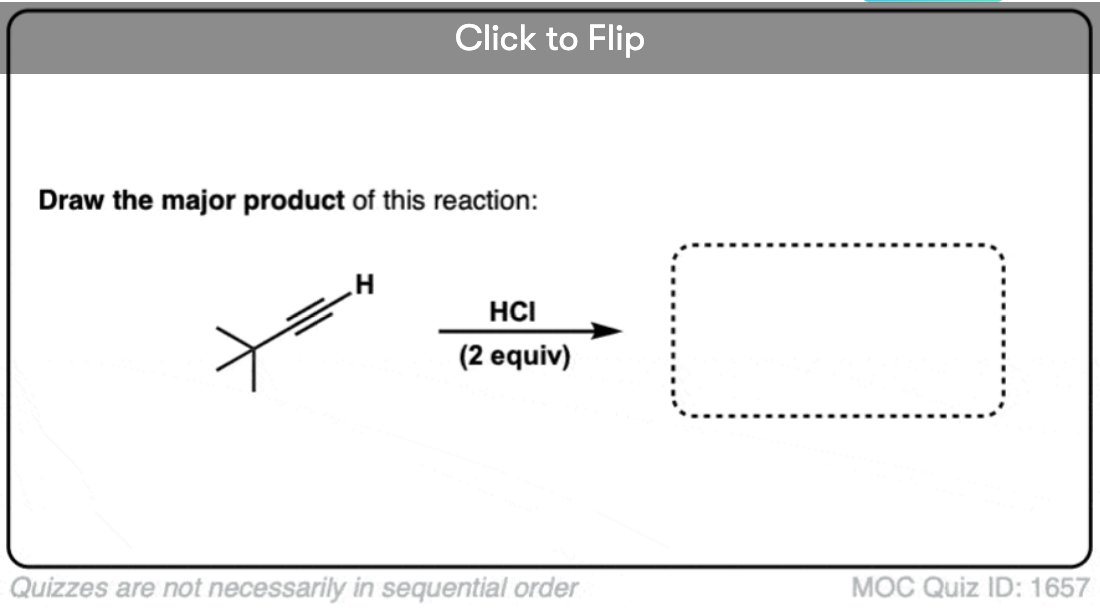
Become a MOC member to see the clickable quiz with answers on the back.
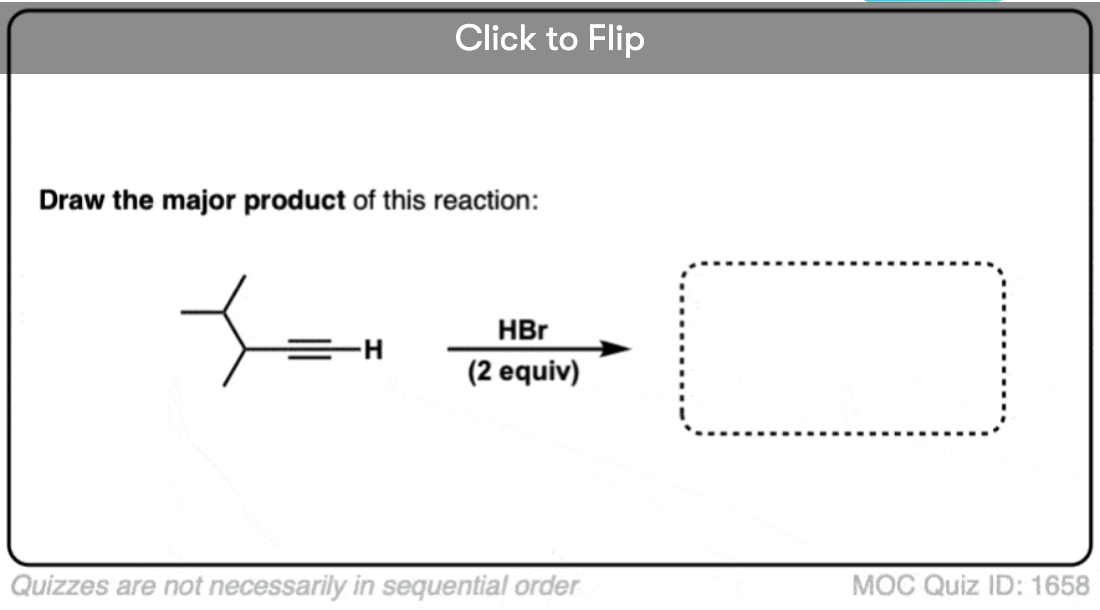
Become a MOC member to see the clickable quiz with answers on the back.
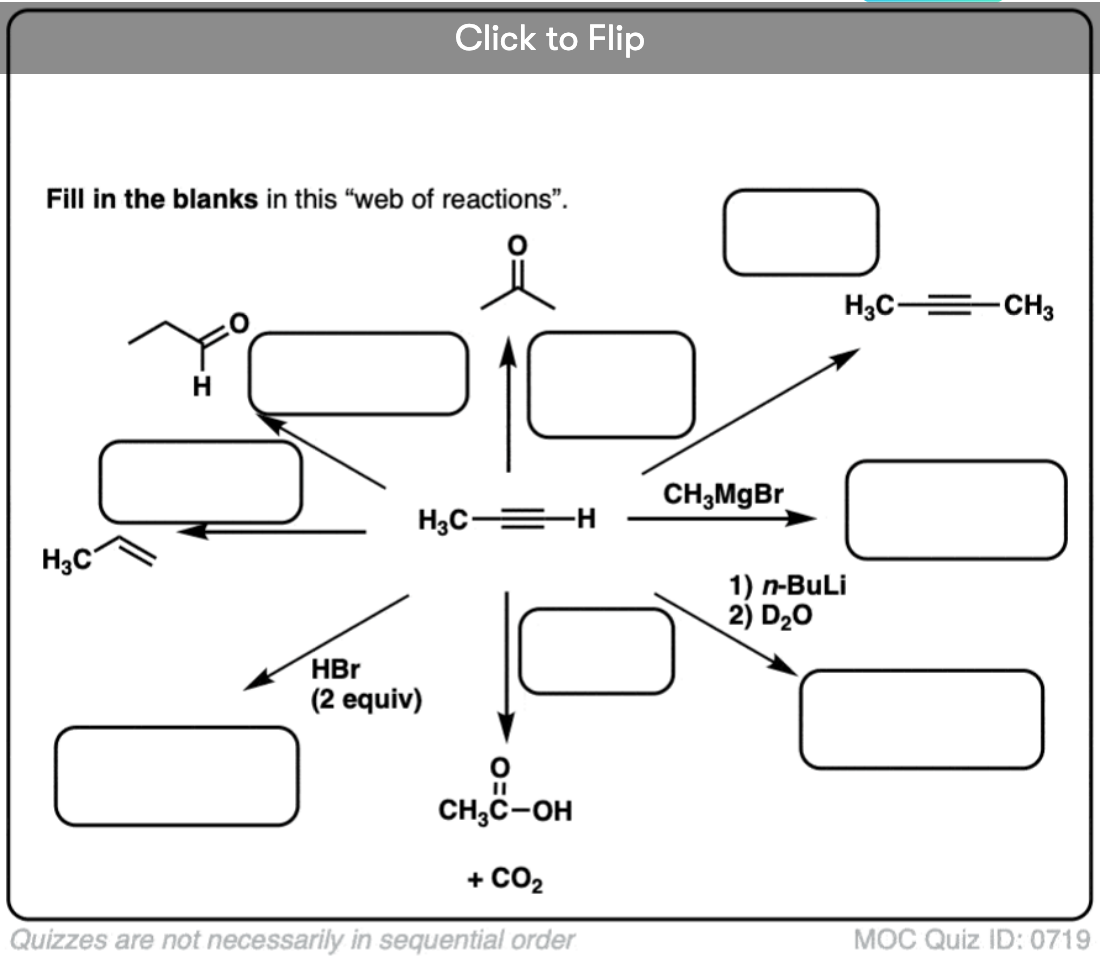
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
The addition of HX to alkynes is believed to go through a vinyl cation after initial protonation of the alkyne.
- Polar additions to olefins and acetylenes. V. Bimolecular and termolecular mechanisms in the hydrochlorination of acetylenes
Robert C. Fahey and Do-Jae Lee
Journal of the American Chemical Society 1968, 90 (8), 2124-2131
DOI: 1021/ja01010a034
Hydrogen chloride adds to aryl acetylenes in acetic acid to give mixtures of a-chlorostyrenes and the corresponding vinyl acetate. - Reaction of acetylenes with hydrogen chloride in acetic acid. Effect of structure upon AdR2 and Ad3 reaction rates
Robert C. Fahey, Michael T. Payne, and Do-Jae Lee
The Journal of Organic Chemistry 1974, 39 (8), 1124-1130
DOI: 1021/jo00922a024
The preference for a mechanism depends on the individual structure of the alkyne and the overall reaction conditions. - Solvolysis of vinyl triflates. Effect of alkyl substituents, solvents, and added nucleophiles
Richard H. Summerville, Carol A. Senkler, and Paul v. R. Schleyer
Journal of the American Chemical Society 1974, 96 (4), 1100-1110
DOI: 1021/ja00811a026 - Stereochemistry of vinyl cations and vinylic substitutions
H. Summerville and Paul v. R. Schleyer
Journal of the American Chemical Society 1974, 96 (4), 1110-1120
DOI: 10.1021/ja00811a027
Alkynes react when heated with trifluoroacetic acid to give addition products. Mixtures of syn and anti addition products are obtained, and similar reactions occur with trifluoromethanesulfonic (triflic) acid. These reactions presumably also proceed through a vinyl cation intermediate. - Theoretical investigations on carbocations. Structure and stability of C3H5+,C4H9+(2-butyl cation), C5H5+,C6H7+(protonated benzene), and C7H11+(2-norbornyl cation)
Hans Joachim Koehler and Hans Lischka
Journal of the American Chemical Society 1979, 101 (13), 3479-3486
DOI: 1021/ja00507a009
One mechanism that has been proposed for this reaction is initial protonation of the alkyne via a bridged intermediate. This paper shows that this hydrogen-bridge structure is not energetically feasible. Various MO calculations place the bridged ion 30-45 kcal/mol above the vinyl cation in energy. - Kinetics of the acid-catalyzed hydration of allene and propyne
Paul Cramer and Thomas T. Tidwell
The Journal of Organic Chemistry 1981, 46 (13), 2683-2686
DOI: 1021/jo00326a016
Solvent isotope effects are indicative of a rate-determining protonation. - Substituent effects on the acid hydration of acetylenes
Annette D. Allen, Yvonne Chiang, A. J. Kresge, and Thomas T. Tidwell
The Journal of Organic Chemistry 1982, 47 (5), 775-779
DOI: 1021/jo00344a006
Alkyne reactivity increases with addition of electron-donating substituents. The reactivity of alkynes is somewhat more sensitive to substituent effects than is the case for alkenes. - 2-Butyne and hydrogen chloride cocrystallized: solid-state geometry of Cl-H••p hydrogen bonding to the carbon-carbon triple bond
Dietrich Mootz and Axel Deeg
Journal of the American Chemical Society 1992, 114 (14), 5887-5888
DOI: 10.1021/ja00040a077
The short length of this JACS communication belies the difficulty of this experimental work! This paper describes an X-ray structure of the addition complex between HCl and an alkyne, with the HCl perpendicular to the C-C p bond. - The electrophilic addition to alkynes
Hilton M. Weiss
Journal of Chemical Education 1993, 70 (11), 873
DOI: 1021/ed070p873
This paper argues that vinyl cations are too unstable and therefore cannot be intermediates in electrophilic additions to alkynes. This is not entirely correct, as vinyl cations have been observed in superacid media under the right conditions. - Bromide assisted addition of hydrogen bromide to alkynes and allenes
Hilton M. Weiss and Kim M. Touchette
J. Chem. Soc. Perkin Trans 2 1998:1523
DOI: 10.1039/A703569A
The reaction of 4-octyne with TFA in CH2Cl2 containing 0.1-1.0 M bromide ion proceeds mainly via anti addition. The presence of bromide ion greatly accelerates the reaction as compared to reaction with TFA alone, indicating the involvement of Br– in the rate-determining step.
- Electron transmission study of the splitting of the p* molecular orbitals of angle-strained cyclic acetylenes: implications for the electrophilicity of alkynes
Lily Ng, Kenneth D. Jordan, Adolf Krebs, and Wolfgang Rueger
Journal of the American Chemical Society 1982, 104 (26), 7414-7416
DOI: 1021/ja00390a005
Another possible explanation for the lower reactivity of alkynes relative to alkenes has to do with the availability of the unfilled orbital in the alkyne. It has been shown that a p* orbital of bent alkynes (e.g. cyclooctyne) has a lower energy than the p* orbital of alkenes, and it has been suggested that linear alkynes can achieve a bent structure in their transition states when reacting with an electrophile.
Isn’t a vinyl carbocation usually highly unstable for it to form appreciably?
There’s controversy about how likely the vinyl cation is in this mechanism. At the bottom of the post there is discussion about a termolecular mechanism.
why halo alkenes are less reactive towards addition than corresponding alkenes ? is it some what regarding to the electron withdrawing effect of the halogen on the pi bond deecreasing its electron density ? similar to deactivating nature of halogen in aromatic rings ?
It is due to the electron-withdrawing nature of the halogens.
Hello,
What would the reaction look like for an internal alkyne, where there is equal substitution if the alkyne? How many constitutional isomers and products would be formed?
Well, if the internal alkyne isn’t symmetrical, then there would be two constitutional isomers (alkenes). Each of those constitutional isomers would be present as a mixture of stereoisomers (E/Z) since these reactions are not very stereoselective.
So four, at least.
I have. Apriciation of this side. Alot
What catalyst is best for the hydrohalogenation process??
The reaction uses stoichiometric (not catalytic) acid.
Can rearrangement take place in Hydrohalgenation of Alkynes like it happens in Alkenes.
I have questions. What happen if a molecule contain both double bond and triple bond? Do both will be reacted with HBr? And how if only 1 mol of HBr is used? I really confused because i thought maybe one of them will react since only one HBr molecule will react for one reactant molecule.
Alkenes will be more reactive toward HBr than alkynes. A molecule with both an alkene and an alkyne will react with HBr at the alkene. When an alkene is protonated, the resulting carbocation is sp2 hybridized (33% s character) . When an alkyne is protonated the resulting carbocation is sp hybridized (50% s character). The more s-character present in a carbocation, the harder it is to form (less stable) since the empty orbital will be closer to the nucleus.
When 3methyl butyne is reacted with excess HBr then what we get 2,2dibromo 3methyl butane ?
Why we don’t get 2,3dibromo 3methyl butane even allylic carbocation is more stable then vinylic carbocation formed as intermediate ?
That is a great question. If you look at the alignment of orbitals that would have to happen, in order for a 1,2-hydride shift, the C-H bond must be aligned with the empty sp2 orbital. After that occurs, you’re left with an empty p orbital that is not aligned with the p-oribtals of the double bond, so is not actually resonance stabilized!
It’s similar to what would happen upon the protonation of allene at the central carbon. You end up with a carbocation that *looks* like it is allylic but the transition state won’t have the empty p-orbital aligned with the pi bond, so it’s not actually a delocalized carbocation the way an allyl carbocation is.
james does rearrangement of carbocation take place when the first carbocation ie, the vinyl carbocation is formed
why the videos are not playing
What videos? Could you be more specific?
What about the evidence for the termolecular process that avoids the unfavourable vinyl cation and would account for the stereoselectivity ?
Yes. That deserves coverage. Thank you for bringing that up.
Hello, when ethynilcyclohexane reacts with one equivalent of HBr is there a cation rearrangment so Br “touches”the ring un the final product?
Thanks a lot!
What will be the reaction when alkyne react with 2HBr in presence of ROOR
Great post! Worth noting that the vinyl cation produced after protonation by HCl is linear (as drawn), not bent. The LUMO of the linear cation is higher in energy than the LUMO of the hypothetical bent cation. I got burned by this back in the day… :-)
James, do you know of an experiment using DCl that shows a preference (or lack thereof) for syn or anti addition of D and Cl to the alkyne?
Good to point out, thanks!
Re: DCl, I assume a mixture of both would be obtained. Don’t see a direct reference to it in March , although there is a footnote referencing mechanistic discussions of HX addition to alkynes in Stang, P. J, et. al. in “Vinyl Cations” Academic Press NY 1979 p. 24. My version of March is not the newest though.
“Bromide Assisted Addition of Hydrogen Bromide to Alkynes and Allenes” Journal the Chemical Society, Perkin II, 1998, 1523.
HBr adds to terminal alkynes almost exclusively anti in some cases. With more acidic solutions, mixtures arise, in part due to isomerization of the alkene product.
Thank you!