Elimination Reactions
E1cB: Elimination (Unimolecular) Conjugate Base
Last updated: May 29th, 2026 |
The E1cB (E1, Conjugate Base) Elimination Mechanism
- The E1cB (Elimination, Unimolecular, Conjugate Base) mechanism is a third mechanistic pathway for elimination reactions.
- In many ways it is the exact opposite of the E1 mechanism, as the first step is deprotonation to form a carbanion, followed by elimination in the second step.
- It does occasionally come up in introductory organic chemistry courses, particularly in the mechanism of the aldol condensation, aryne formation, and elimination of alkenyl halides to give alkynes.
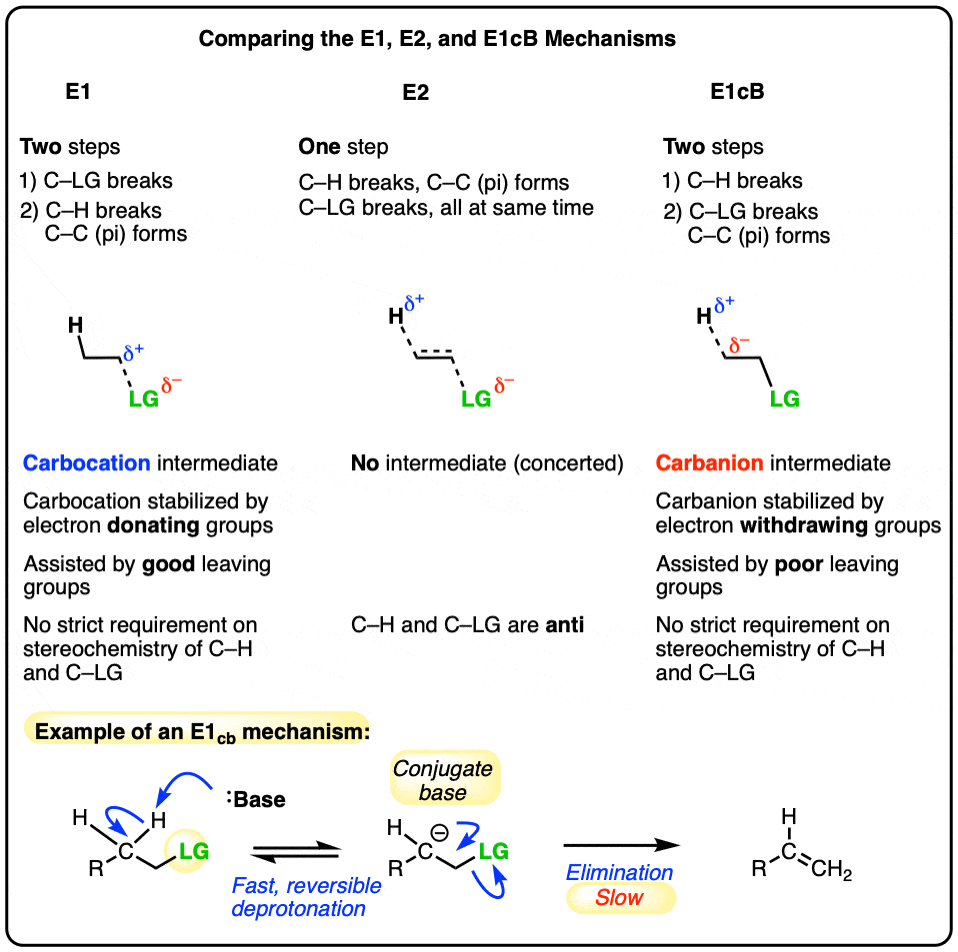
Table of Contents
- Elimination To Give Alkenes: The E1 and E2 Pathways
- The E1cB Mechanism
- The E1cB Mechanism And The E1 Mechanism Are Two Extremes, With The E2 Mechanism Lying In The Middle
- Examples Of The E1cB Mechanism In Introductory Organic Chemistry: The Aldol Condensation
- Another Example: Formation Of Alkynes From Alkenyl Halides
- Formation Of Arynes From Phenyl Halides
- The Reaction Coordinate Diagram Of The E1cB Reaction
- Summary: When Is a Reaction Likely To Go Through An E1cb Mechanism?
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
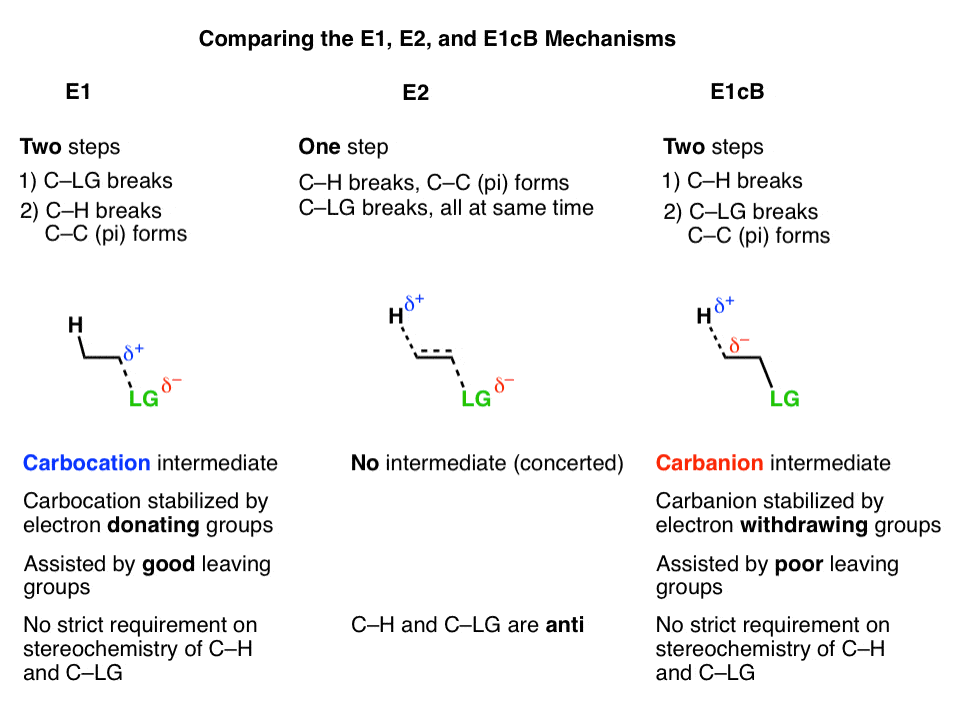
1. Elimination To Give Alkenes: The E1 and E2 Pathways
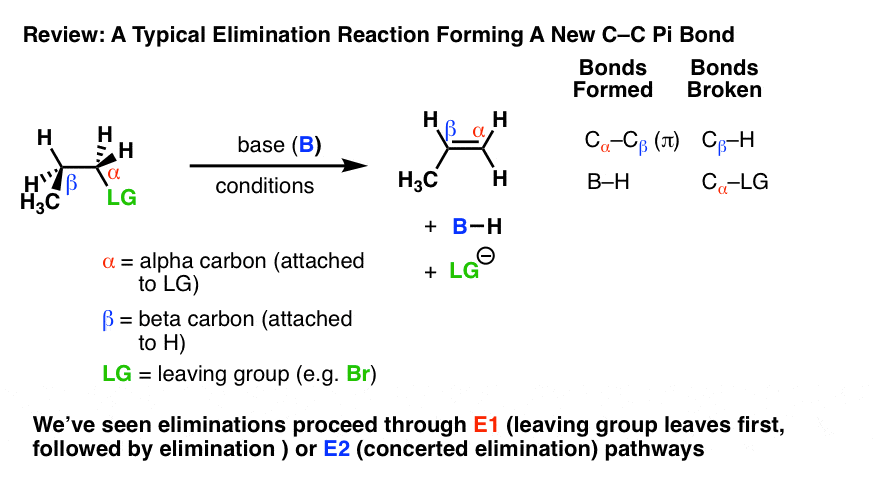
Elimination reactions are one of the most common and powerful methods we’ve learned to make alkenes (carbon-carbon pi bonds) from alkyl halides.
In elimination reactions giving new C-C pi bonds, the same general pattern of bond-forming and bond-breaking is always observed!
- A single bond between the carbon and a leaving group breaks (C-LG). The carbon attached to the leaving group (LG) is referred to as the “alpha“(α) carbon.
- A single bond between the carbon and a hydrogen breaks. The carbon bearing this hydrogen is referred to as the “beta” (β)carbon.
- A new pi bond forms between the alpha carbon and the beta carbon.
Everyone agrees on what happens. The key question we’d really like to know, however, is “how did it happen?”.
[Screenwriters know this. They give away the endings to movies all the time – often in the first 5 minutes. But what keeps you watching is trying to understand how something happened. Why else would people sit through a six hour movie trilogy if they already know Anakin is going to become Darth Vader in the end?]
The sequence in which these bonds form and break is the “mechanism” of a reaction, or if you will, the “story” of how this reaction happens.
We’ve seen two of these patterns so far for elimination:
- In the E1 mechanism, elimination happens in two steps. The C-LG bond breaks first, leaving behind a carbocation. This is the slow step. In the second step, a base deprotonates the beta carbon, breaking C-H. The lone pair of electrons from the C-H bond forms the new pi bond with the empty p orbital from the carbocation. The rate law is unimolecular (hence E1) since it depends only on the substrate.
- In the E2 mechanism, the C-H bond and C-LG bond break at the same time that the new C-C pi bond is formed. This is a concerted mechanism. The rate law is bimolecular (hence E2) since the reaction rate depends both on the concentration of base and of substrate.
There is actually a third possibility, and that’s what we’re going to talk about today.
2. The E1cB Mechanism
The third possibility is that the beta carbon is deprotonated first (form B–H, break C–H). In the limiting case, this would give an anionic intermediate, the “conjugate base” of the substrate. [See: Acid Base Reactions In Organic Chemistry]
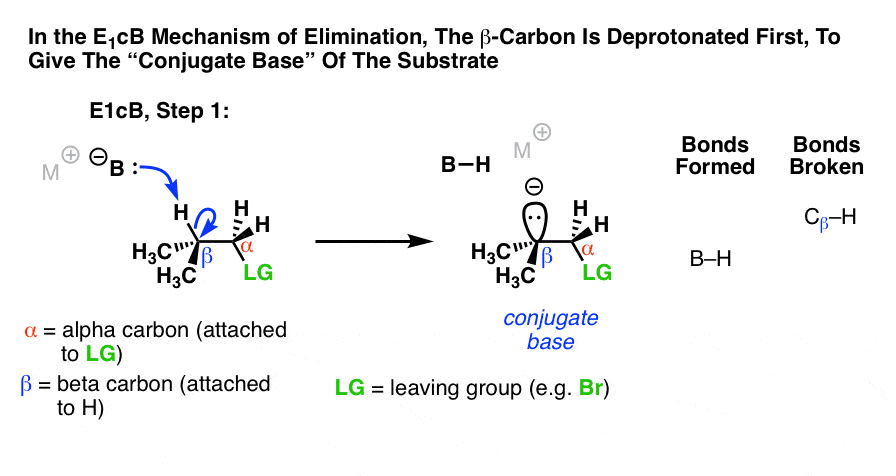
Since the first step is deprotonation to give an anion, then you would be right to guess that this mechanism is more likely to occur when the C–H bond is relatively acidic, as when the alpha carbon is adjacent to an electron withdrawing group like a ketone, nitrile (CN), or nitro group (NO2). These groups stabilize negative charge. [See: Factors That Stabilize Negative Charge]
In the second step, the pair of electrons from the conjugate base then displaces the leaving group, forming a pi bond [form C–C pi, break C–LG].
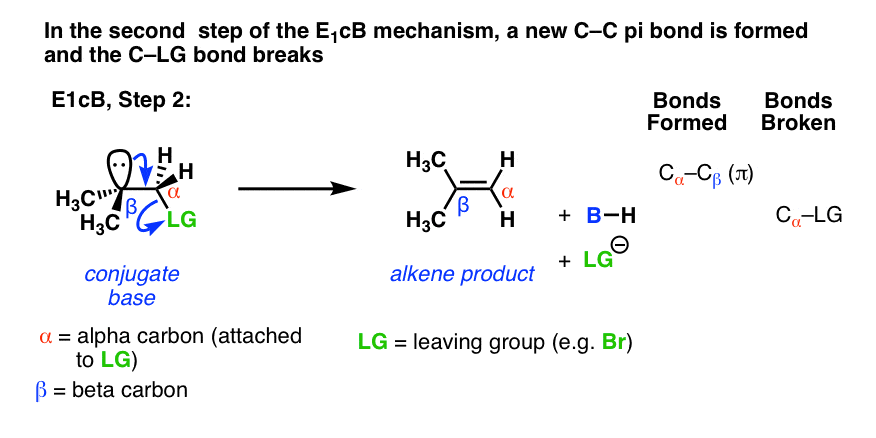
In the (many) cases of the E1cB where the second step is the slow step, you can imagine that this step would be made slower by a relatively poor leaving group like (HO-), (CH3O-) or even (believe it or not!) fluorine [Note 1].
3. The E1cB Mechanism And The E1 Mechanism Are Two Extremes, With The E2 Mechanism Lying In The Middle
It’s helpful to think of the E1cB mechanism and the E1 mechanisms as two extremes, or mirror images, and the E2 cutting exactly in between. It’s kind of like the continuum between “quitting” and “being fired”, with “mutually agreeing to part ways” somewhere in the middle.
- The first step in the E1 mechanism is loss of a leaving group (break C-LG) to give a carbocation intermediate, and this step is promoted when there are electron-donating groups adjacent to the alpha carbon (making the carbocation more stable).
- The first step in the E1cB mechanism is deprotonation (break C-H) to give a carbanion intermediate, and this step is promoted when there are electron-withdrawing groups adjacent to the alpha carbon (making the carbanion more stable).
- The E2 mechanism is in between these two extremes, where everything happens at once.
So does this mechanism actually occur in reactions we see in introductory organic chemistry? Yes it does!
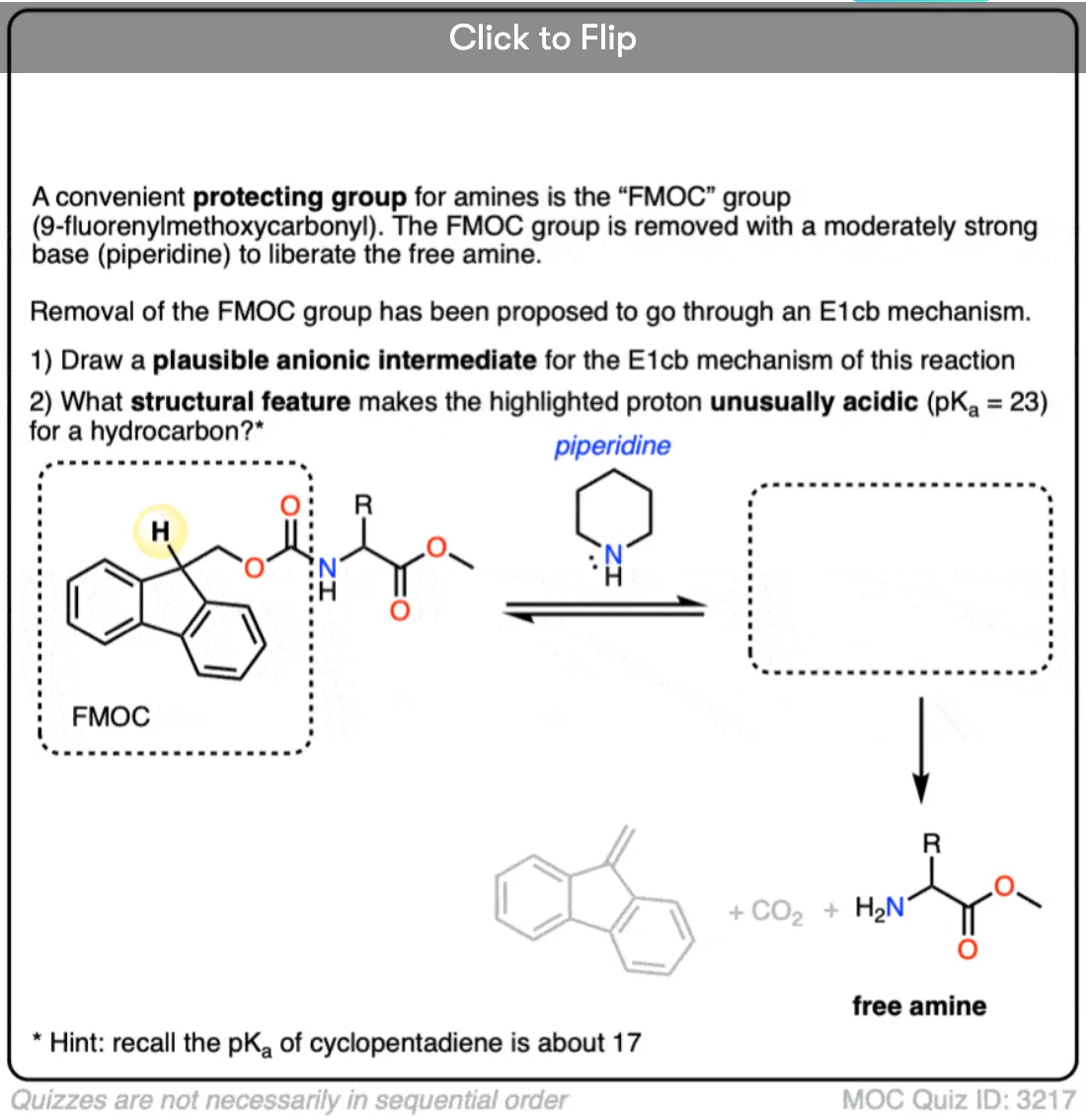
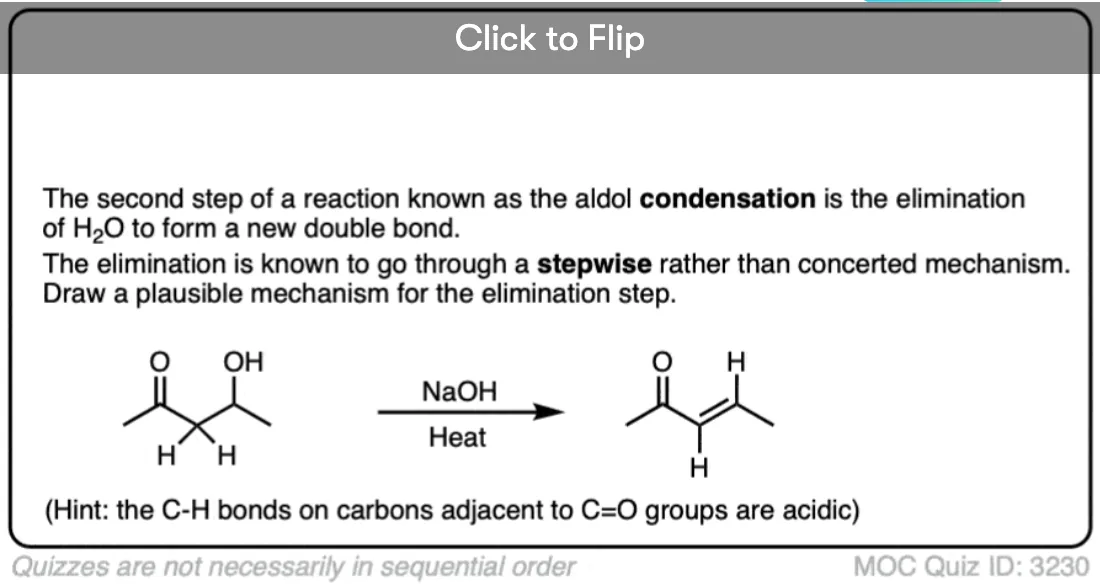
4. Examples Of The E1cB Mechanism In Introductory Organic Chemistry: The Aldol Condensation
Probably the most commonly encountered example of the E1cB mechanism in introductory organic chemistry is in the aldol condensation reaction, specifically the last step where the new C-C double bond is formed.(See post: The Aldol Addition and Condensation Reactions]
Below, left, is the product of an alcohol addition reaction. If this is heated with base (like NaOCH3), it loses water, giving the alkene. (Loss of water = “condensation”)
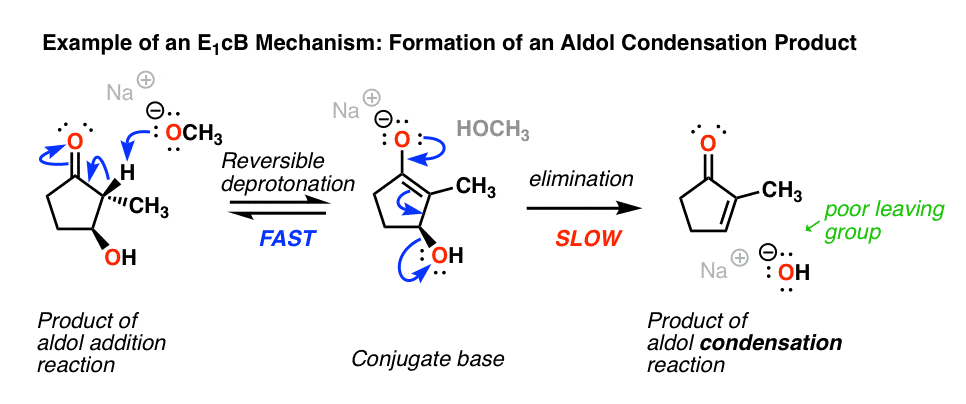
Wait a minute, you may say. “I thought the hydroxyl ion (HO-) was a poor leaving group? What’s going on here?”
Exactly! If there was a really good leaving group (like OTs or Br) present instead of OH, an E2 (or even E1) mechanism would be more likely. [Note 2] The bad leaving group (HO-) means the elimination step is slow, which is largely responsible for this being a two-step mechanism.
Another (very subtle) point is stereochemistry. If you look really closely you’ll notice in the drawing above that the C–H bond and the C–OH bond are not anti to each other (a requirement for the E2) but instead are syn.
So unlike the E2, the E1cB mechanism doesn’t have a strict requirement that the C-H and C-LG bonds be anti to each other.
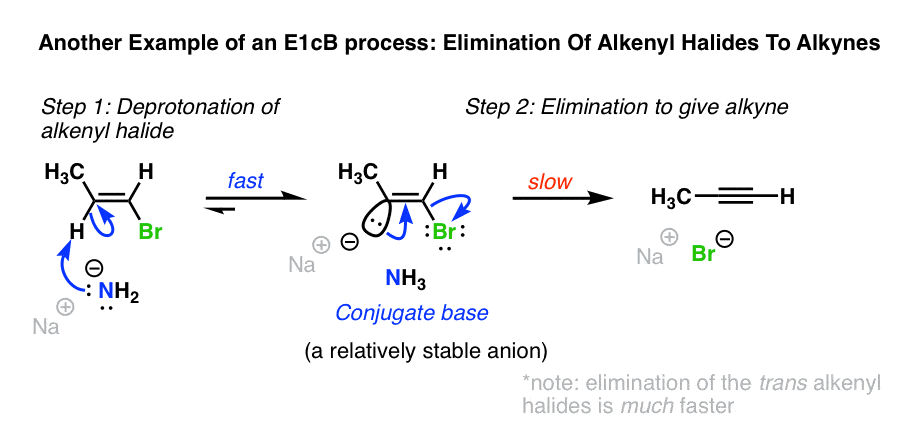
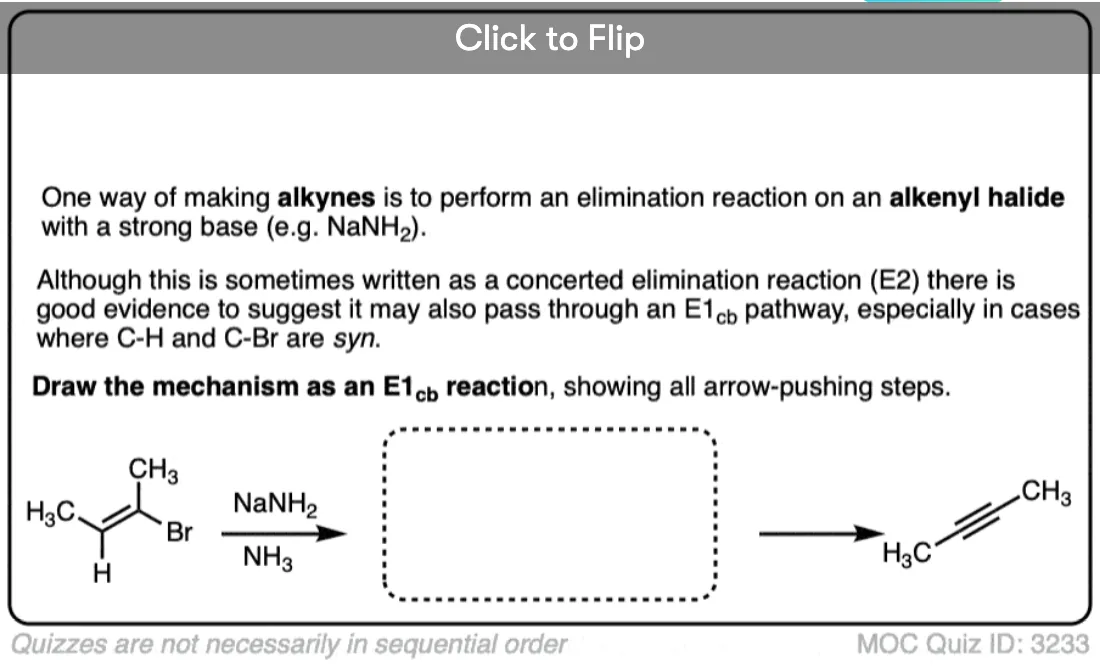
5. Another Example: Formation Of Alkynes From Alkenyl Halides
Another example of the E1cB is in the formation of alkynes from alkenyl halides with NaNH2 in liquid ammonia (NH3). [See: Alkynes via Elimination Reactions]
When alkenyl halides are treated with NaNH2 in NH3 as solvent, elimination to give the alkyne occurs. This reaction works even if the H and Cl are cis to each other, which would seem to rule out the E2 mechanism. [The elimination when H and the halide are trans is much faster, though! [Note 3]
NaNH2 is an extremely strong base. Being sp2 hybridized, the C-H bond of the alkenyl halide is reasonably acidic (pKa about 40) and able to stabilize negative charge.
In this case a two-step mechanism likely operates, where 1) a carbanion intermediate is formed by deprotonation, followed by 2) loss of the leaving group.
6. Formation Of Arynes From Phenyl Halides
A related situation to alkenyl halides where the E1cB mechanism can arise is in the formation of benzyne and other “arynes” [See: Nucleophilic Aromatic Substitution – The Benzyne Mechanism]. Although benzyne does not have a “true” pi bond, many of the same principles of the E1cB still apply.
Deprotonation of the aryl fluoride by the strong base (NaNH2) occurs first, giving a carbanion. In this specific case, the second step is loss of the bad leaving group, fluorine. [Note 1]
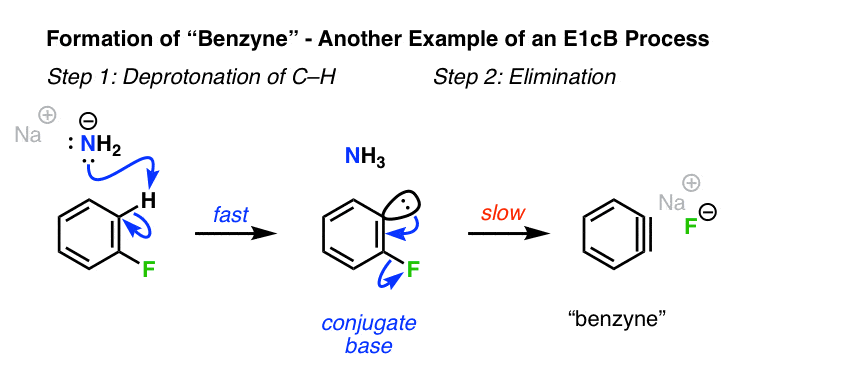
[Note – apparently, when F or Cl is the leaving group, deprotonation is the slow step. However, when the leaving group is Br, or I, deprotonation is rate-determining and breakage of C-X is fast. Note that both of these still qualify as E1cB so long as the reaction proceeds through the carbanion intermediate! ]
7. The Reaction Coordinate Diagram Of The E1cB Reaction
So what does the reaction coordinate diagram of the E1cB look like? If we assume that the first step is fast and the second step is slow (rate-determining), and there is an anion intermediate, then the reaction coordinate diagram of the E1cB should look roughly like this:
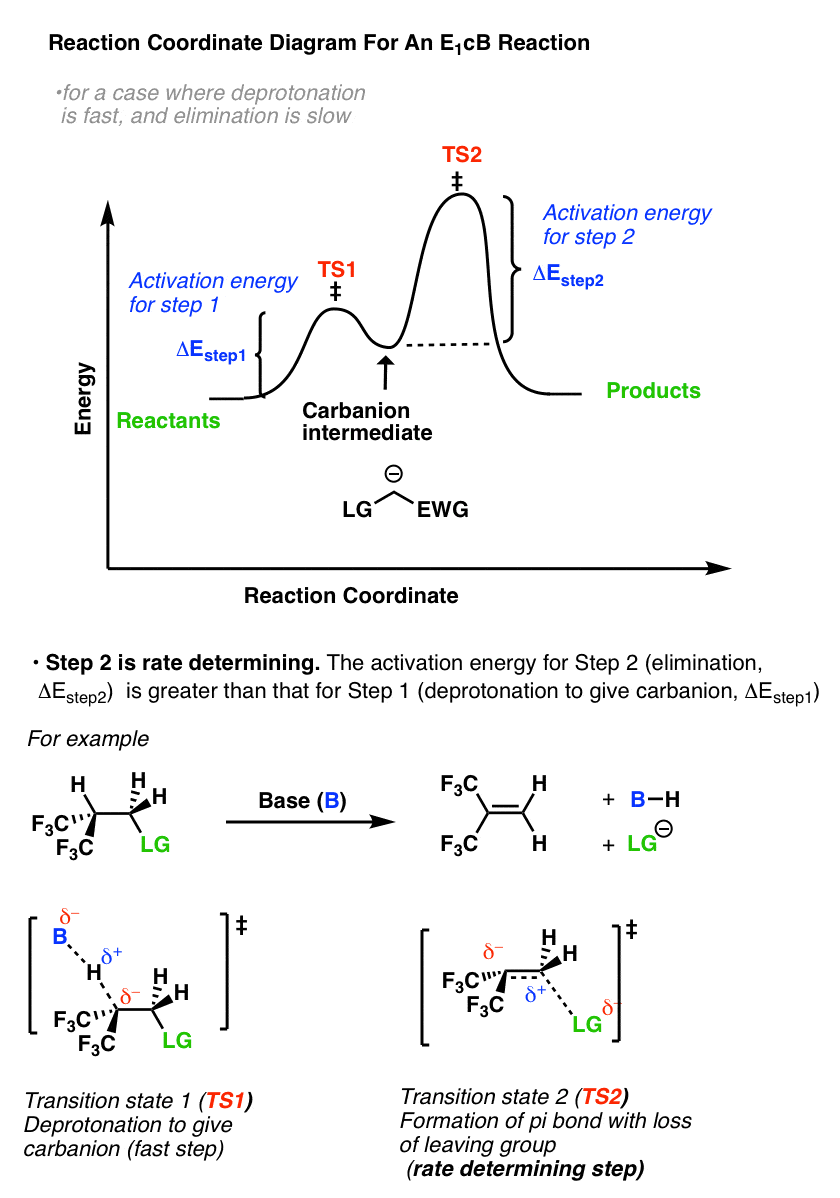
- We’ve drawn a small hump going from reactants to TS1 with activation energy (ΔEStep1 ) equal to [Δ E TS1 – Δ E reactants].
- This then proceeds to the anionic intermediate (the “well” in the middle)
- Which in turn proceeds through TS2 with activation energy (Δ EStep2 ) equal to [Δ E TS2 – Δ E intermediate]
- Since ΔEStep2 > ΔEStep1, the second step is rate determining
Compare that to the reaction coordinate of the E1 elimination, which is the mirror image.
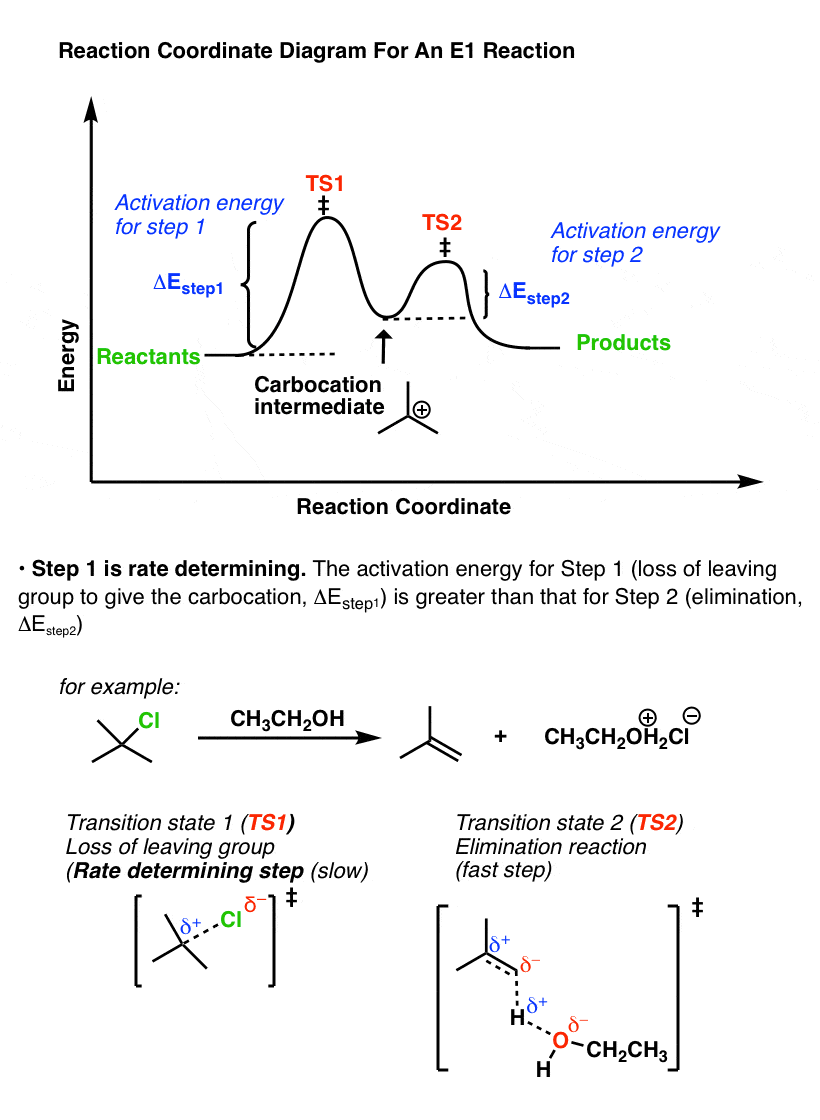
In the E1 mechanism, the energy barrier (activation energy, ΔEStep1 ) for the first step (loss of leaving group) is higher than the energy barrier (ΔEStep2) for the second step (elimination).
The reaction coordinate in the E2 case shows both of these steps happening at the same time.
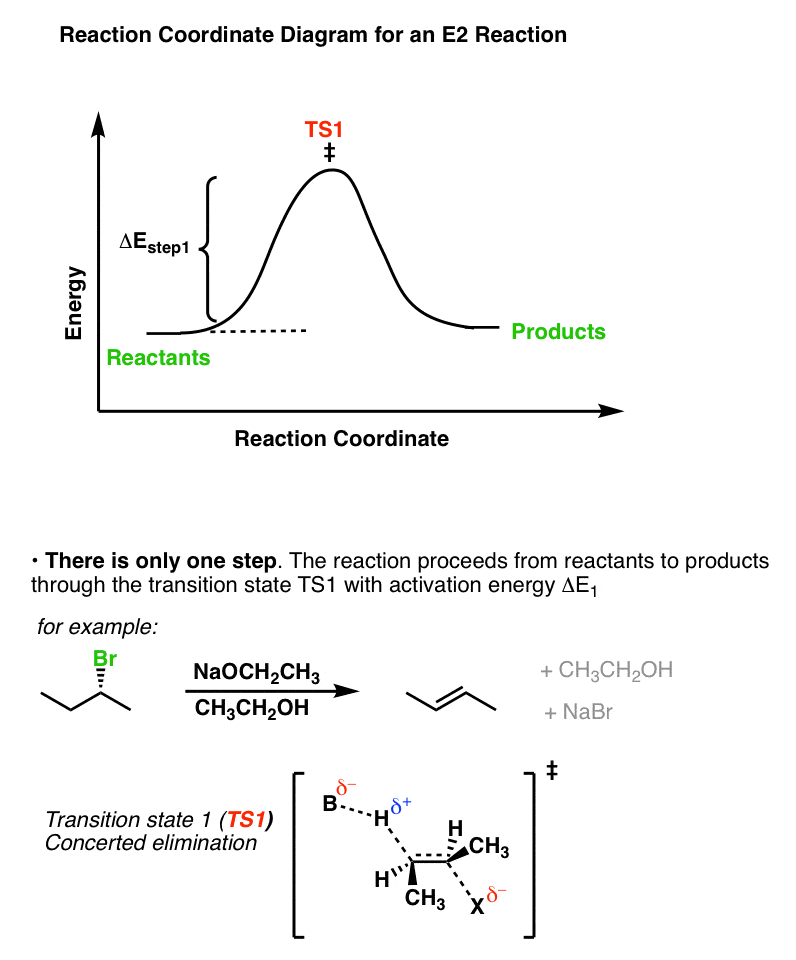
8. Summary: When Is a Reaction Likely To Go Through An E1cB Mechanism?
So, to summarize, in the E1cB mechanism, elimination occurs in two steps:
- Deprotonation of the substrate to give an anion (its conjugate base), followed by
- Loss of a leaving group to give a new C-C pi bond.
The E1cB mechanism is not very commonly encountered in introductory organic chemistry, but when it is observed, expect to see several of these characteristics:
- The presence of an electron-withdrawing group which helps to stabilize an intermediate anion
- A relatively poor leaving group (which will slow down the second step)
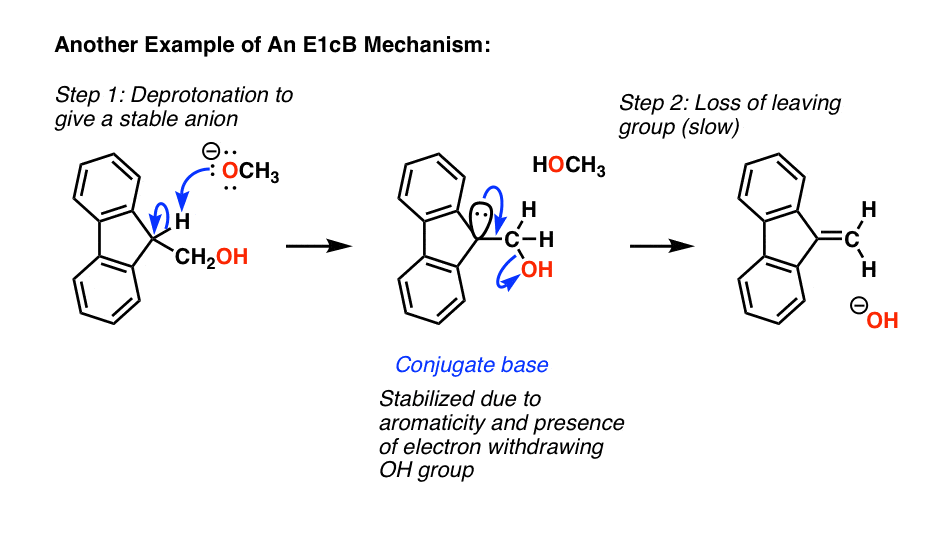
Here’s one last classic example of an E1cB, where deprotonation leads to a very stable anion (it’s aromatic) which slowly loses the poor leaving group (hydroxide ion) forming a new C-C pi bond.
Notes
Note 1. We never consider F(–) as a leaving group for SN2 reactions, but under certain conditions it can be a leaving group in various types of substitution and elimination reactions. One example is in nucleophilic aromatic substitution, another example of a mechanism that proceeds through a relatively stable anion.
Note 2. For more on this see reference 7, which suggests that in eliminations with a good leaving group, E2 is followed even when a strong electron withdrawing group is present.
Note 3. See reference 5 – the reaction is much easier when H and Br are trans to each other (can be done with NaOH!) but requires NaNH2/NH3 when the Br and H are cis.
More on the E1cB
[1-How do we determine E1cB vs E2 – 2 Kinetic Isotope Effects – 3 E1CBreversible– 4 E1CBIRR – 5 E1CBAnion – 6 Summary – Summarizing the 3 Main Flavors of E1CB – 7 – Stereochemistry. ]
1. How might we determine that some reactions follow an E1cB mechanism versus an E2 mechanism? How do you know the difference?
That is a great question.
These studies always begin with experimental observations, followed by applied curiosity. For example, consider the reaction below.[See reference 9] We might naively begin by considering this an E2 reaction. After all, there’s a strong base, a leaving group (bad though it may be) and the stereochemistry of the C-H and C-OH bonds are capable of being anti to each other.
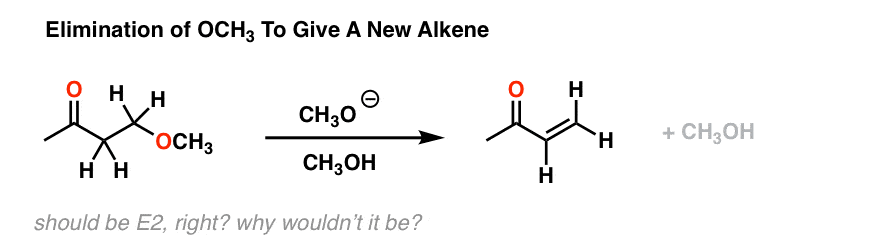
E2, right? However, it turns out that if you run this reaction in a deuterated solvent, and stop it halfway, you end up with a lot of this:
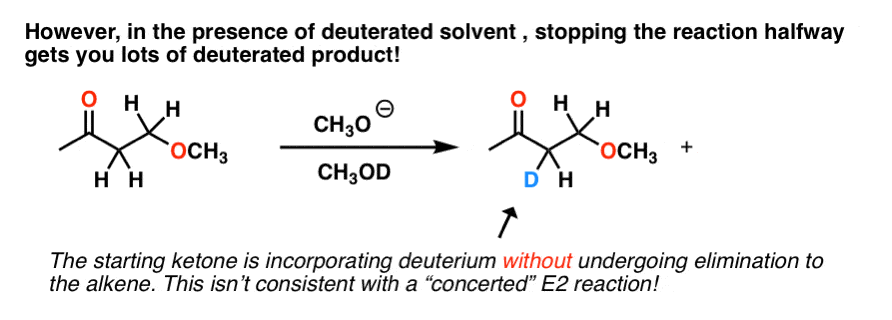
We don’t see this in E2 reactions, since the C-H bonds can only break when they are anti to the leaving group.
In this example, C-H bonds are clearly breaking, but elimination does not always happen. So what is going on?
The best explanation for what’s happening here is that the base forms the conjugate base of the substrate, and this can react with the deuterated solvent (as an acid), resulting in deuterium exchange at that position.
It turns out that exchange is 220 times faster than elimination.
That’s very interesting. But, how important is the conjugate base in the elimination? Just because we know it’s being formed doesn’t prove it’s involved in the elimination.
Maybe formation of the anion is just a “dead end”, and a slower E2 reaction is responsible for formation of the alkene?
Perhaps. It actually takes quite a lot of work to answer that question (See Reference 2), but the clear evidence that an anion is formed should at least plant the seeds of doubt that the E2 is the only possibility here.
2. Kinetic Isotope Effects
In theory, one way to tell the difference between the E1cB and the E2 is by studying rate laws. It turns out that this is generally difficult in the case of the E1cB [See Reference 1] , so chemists have had to resort to other methods.
A key strategy for studying organic reaction mechanisms is to measure kinetic isotope effects, which take advantage of the subtle differences in bond strengths between isotopes. For example C-D (deuterium) bonds are slightly shorter (and stronger) than comparable C-H bonds. If C-H bond breaking occurs in the rate determining step, then the rate for the reaction of the C-H compound (kH) should be slightly faster than the rate of the deuterium labelled analog with a C-D bond (kD). This difference, termed the primary kinetic isotope effect, has been experimentally determined to have a range kH/kD of about 2-8 in most cases for the E2.
So if C-H is broken in the rate determining step, then we should see a primary kinetic isotope effect in the range of 2-8. If it is not, then there should be no difference between the two.
Right?
3. E1cBreversible
In the case of the reaction above, the kinetic isotope effect is about 1.0 . That is a clear indication that the C-H bond is not broken in the rate determining step, which rules out the E2 (concerted) mechanism.
These types of E1cB reactions are referred to as E1cBreversible indicating a reversible deprotonation step followed by (slow) loss of leaving group.
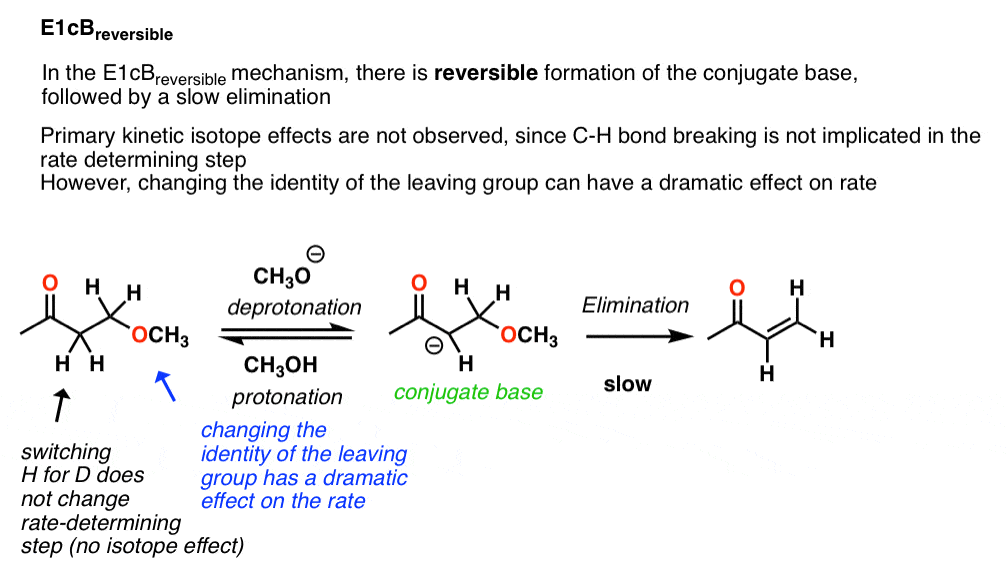
Since loss of the leaving group is slow, one should expect to see big effects on the reaction rate by tweaking the identity of the leaving group. For example, leaving group ability can be greatly modified by the identity of the group X on the phenyl group (shown). Changing the X group to a strong electron withdrawing group like NO2 should make the leaving group much less basic (and a better leaving group) and hence increase the rate. That’s exactly what happens.
4. E1cB IRR
So is that it? Just measure kinetic isotope effects, and we can tell the difference between E2 and E1cB?
Not necessarily!
For example, this reaction below shows a large primary kinetic isotope effect, meaning that C-H is broken in the rate determining step.
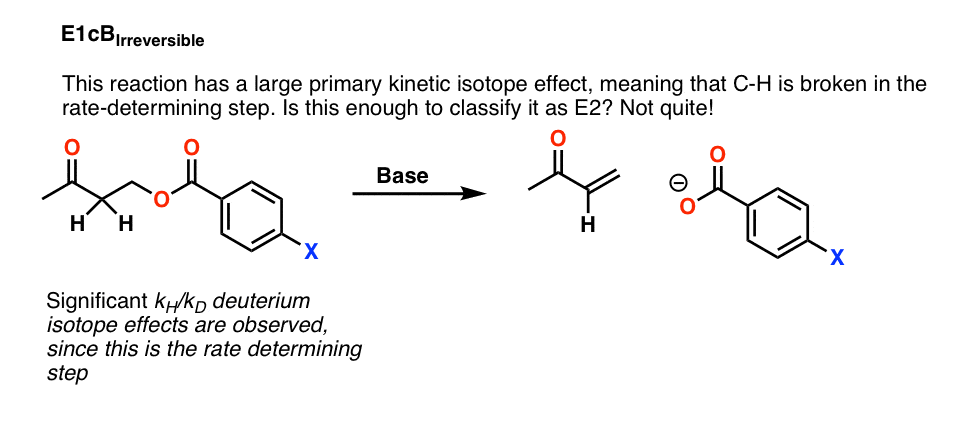
Does that mean that we can safely classify this reaction as E2? Not yet.
It’s possible that this is a case of an E1cB reaction where deprotonation to give the conjugate base is the slow step, and loss of the leaving group is fast. This is called the E1cB irreversible
If that’s the case, there is a fairly simple way to test this: change the leaving group.
Breaking the C–LG bond is part of the rate determining step in the E2, but wouldn’t be a rate determining step in the E1cB.
As it turns out, changing the leaving group didn’t lead to any significant change in the reaction rate.
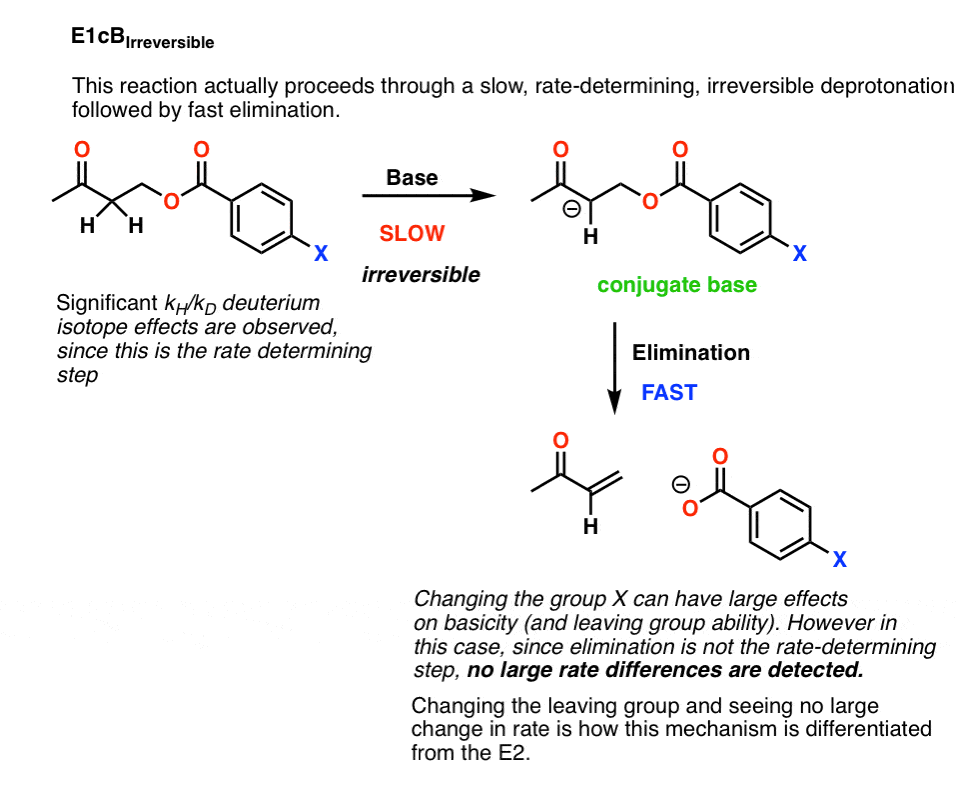
That would rule out an E2.
5. E1cbAnion
There’s one major possibility left, and it’s probably the easiest to visualize: fast, irreversible deprotonation, followed by slow elimination.
This would also not show a primary isotope effect, since C-H bond breaking is not in the rate determining step. The reaction rate would be very sensitive to the identity of the leaving group, however.
This flavor of the E1cB is called the E1cB anion. Here is an example (See Reference 8)
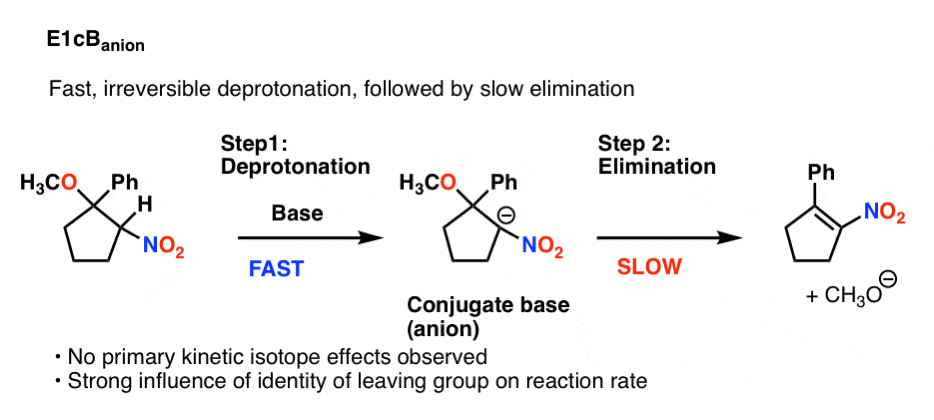
6. Summary: Three Flavors of E1cB
These three examples follow three possibilities for the three rate constants: k1 (deprotonation), its reverse (k-1), and elimination (k2)
- E1cB reversible (reversible, fast deprotonation followed by slow elimination)
- E1cB irreversible (irreversible, slow deprotonation followed by fast elimination)
- E1cB anion (irreversible, fast deprotonation followed by slow elimination)
The experimental differentiation of these different “flavors” of the E1cB from the E2 can be determined by studying kinetic isotope effects and making subtle changes to the identity of the leaving group.
There is also a variant of the E1cB called the E1cB ion pair which I haven’t discussed here, it’s discussed in this reference.
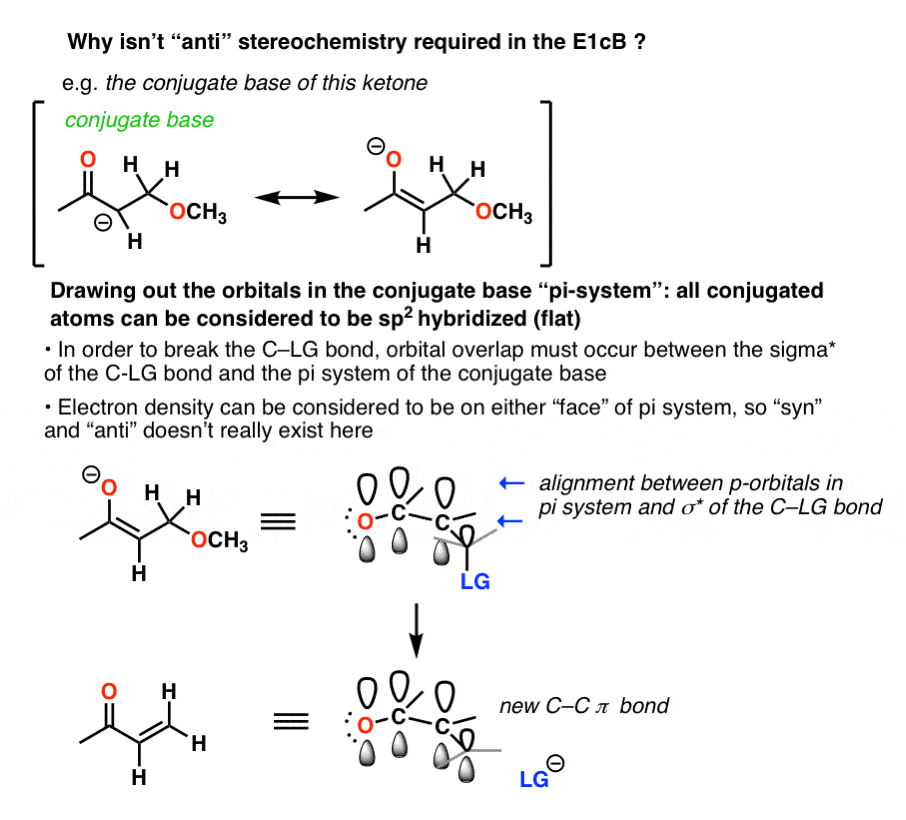
7. What About Stereochemistry?
The E2 has a strict stereochemical requirement that the C-H and C-X bond are anti to each other. The C-H bond must be anti-periplanar to the C-LG bond so that the pair of electrons from the breaking C-H bond is aligned with the C–LG sigma* (antibonding) orbital.
In layman’s terms, you need that C-H bond to be lined up with the C-LG so that its pair of electrons can push out the leaving group (while forming the new pi bond).
No orbital alignment? No deprotonation.
In the E1cB, however, both “syn” and “anti” elimination reactions can occur (also true of the E1, if you’ll recall)
The E1cB mechanism operates when the C-H bond is reasonably acidic. Although the substrate might be tetrahedral (sp3) to start, once the conjugate base (anion) is formed, it generally adopts a flat (trigonal planar, sp2) structure that has electron density on both “faces”. The orientation of the leaving group with respect to the anion does not matter at that point, so long as the C–LG antibonding orbital is aligned with the pi system.
That’s because the pair of electrons from the breaking C-H bond have to donate into the antibonding orbital of the C-X sigma * bond .
That isn’t the case in the E1cb. Nor is it the case in the E1.
In the case of the aldol dehydration you get an anion. lone pair in a p orbital. p orbital can be thought to have electron density on either face of the molecule, and since there is electron density on either face, the C-X bond can be broken no matter what.
Quiz Yourself!
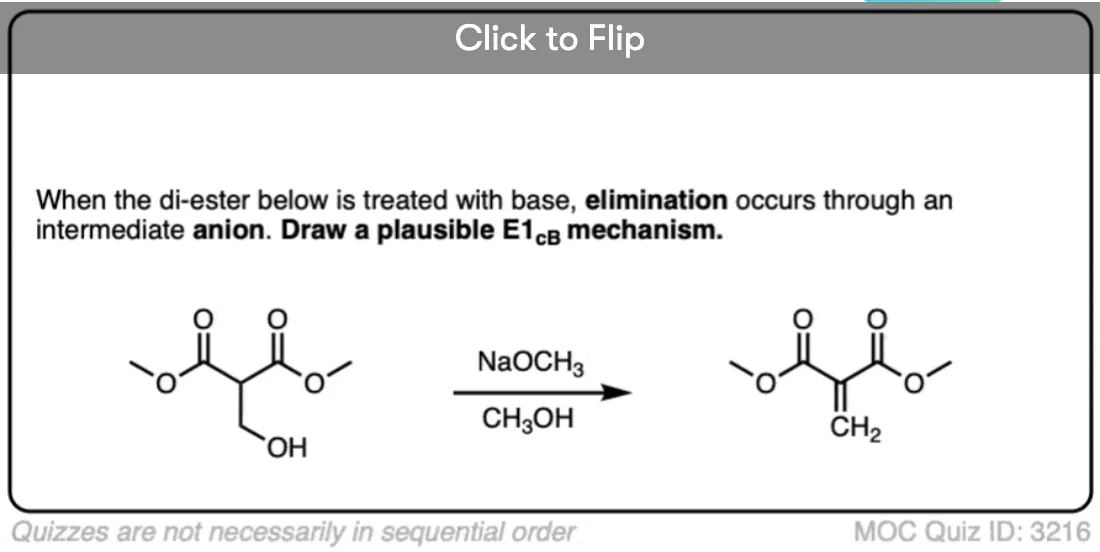
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
Understanding the differences between the “classic” E1, E2, and E1cB reactions is pretty straightforward. However, understanding how the differences between these mechanisms are determined is an adventure in hard-core physical organic chemistry. These are graduate-level papers!
For a general discussion of the E1cB, see Modern Physical Organic Chemistry by Anslyn and Dougherty, Chapter 10, and March’s Advanced Organic Chemistry.
-
- The Carbanion Mechanism Of Olefin-Forming Elimination
D. J. McLennan
Q. Rev. Chem. Soc., 1967,21, 490-506
DOI: 10.1039/QR9672100490
A broad overview of the E1cB mechanism with many examples. - β-Elimination of 9-Fluorenylmethanol in Aqueous Solution: an E1cB Mechanism
R. A. More O’Ferrall, S. Slae
J. Chem. Soc. B. B 1970, 260
DOI: 10.1039/J29700000260
A detailed mechanistic study of a classic E1cB reaction, the elimination of hydroxyl ion from 9-fluroenylmethanol. - Relationships Between E2 and E1cB Mechanisms of β-Elimination
R. A. More O’Ferrall
J. Chem. Soc. B. 1970 , 274
DOI: 10.1039/J29700000274
The first paper on the “More O’Ferrall-Jencks Plot”, a useful conceptual model for examining the relationship between E1, E2, and E1cB mechanisms. - Kinetics Of Basic Elimination Reactions Of The Dihaloethylenes And The Mechanism Of trans Elimination
Sidney I. Miller and Richard M. Noyes
J. Am. Chem. Soc. 1952, 629
DOI: 10.1021/ja01123a015
In this paper the authors determine the rate constants for the elimination of a variety of cis- and trans- dihaloethenes, finding that elimination of the cis dihaloethenes is considerably faster. The conclusion here is that elimination of the cis-dihaloethenes proceeds through a concerted process (E2) while elimination of the trans-dihaloethenes occurs through an anionic intermediate (remember: in the trans dihaloethenes, the H and Cl are syn to each other!) - Amines Derived From Dihalopropenes
A. T. Bottini, B. J. King, and J. M. Lucas
J. Org. Chem. 1962, 27, 3688-3690
DOI: 10.1021/jo01057a509
Another study where the authors show that alkynes can be formed from cis and trans alkenyl bromides. The reaction is much easier when H and Br are trans to each other (can be done with NaOH!) but requires NaNH2/NH3 when the Br and H are cis. -
Comparative mobility of halogens in reactions of dihalobenzenes with potassium amide in ammonia
Joseph F. Bunnett and Francis J. Kearley Jr.
The Journal of Organic Chemistry 1971 36 (1), 184-186
DOI: 10.1021/jo00800a036
This paper shows that that the rate determining step for formation of arynes depends on the nature of the halogen. Deprotonation is rate-limiting in the case of Br and I and departure of the leaving group is rate-limiting for F and Cl. These are both E1cB reactions.
(This paper is also unique, so far as I am aware, for being written in free verse) -
Elimination reactions: experimental confirmation of the predicted elimination of (.beta.-cyanoethyl)sulfonium ions through a concerted, E2 mechanism
Narinder S. Banait and William P. Jencks
Journal of the American Chemical Society 1990 112 (19), 6950-6958
DOI: 10.1021/ja00175a032
Study suggesting that in eliminations with a good leaving group, E2 is followed even when a strong electron withdrawing group is present. -
A first-order base-initiated .beta.-elimination reaction involving a carbanion intermediate
Frederick G. Bordwell, Kwok-Chun Yee, and A. C. Knipe
Journal of the American Chemical Society 1970 92 (20), 5945-5949
DOI: 10.1021/ja00723a022
Example of an E1cBanion mechanism. -
Base-catalyzed β-elimination reactions in aqueous solution. II. E1cB elimination from beta-methoxy ketones.
Leo R. FedorJournal of the American Chemical Society 1969 91 (4), 908-913
DOI: 10.1021/ja01032a019
Study on an E1cB(irreversible) mechanism.
- The Carbanion Mechanism Of Olefin-Forming Elimination
Where’s the reaction?
I changed the summary image to have a more concrete example up top. Fast (reversible) formation of an anion (the conjugate base) followed by slow elimination.
I understand in the aldol condensation the carbanion in the E1cB mechanism doesn’t have a strict stereochemical requirement (because the carbanion’s electrons are involved in resonance with the neighboring carbonyl) but can you assume in general that carbanions are sp2?
If there were no neighboring electron withdrawing group would the carbanion be sp3 — suggesting it requires an anti position to kick out the leaving group? Or do carbanions (like secondary/tertiary amines) tend not to be chiral because they can spontaneously invert? Or do almost all E1cb mechanisms require neighboring electron withdrawing groups (like carbonyls or NO2) which means they will all have sp2 character like the aldol reaction?
Lastly, where you write: “Note – apparently, when F or Cl is the leaving group, deprotonation is the slow step. However, when the leaving group is Br, or I, deprotonation is rate-determining and breakage of C-X is fast.” I think for F or Cl you might have meant to say the deprotonation is a “fast” (not slow) step.
Thanks!