Free Radical Reactions
Allylic Bromination
Last updated: May 28th, 2026 |
Allylic Bromination and Benzylic Bromination: What Is It, And How Does It Work?
- In previous articles on radicals, we’ve seen how bromine (Br2) can selectively react with tertiary C-H bonds (bond strength 93 kcal/mol) over secondary (96 kcal/mol) and primary (100 kcal/mol) C–H bonds (See: Selectivity in Free Radical Reactions – Bromination vs. Chlorination)
- If you recall that bond dissociation energies (BDE’s) are a good guide for predicting radical stability, then you won’t be surprised to learn that “benzylic” and “allyllic” C–H bonds can also be brominated selectively. These C-H bonds are particularly weak because the free radical formed through the homolytic breaking of the allylic C-H is stabilized by resonance.
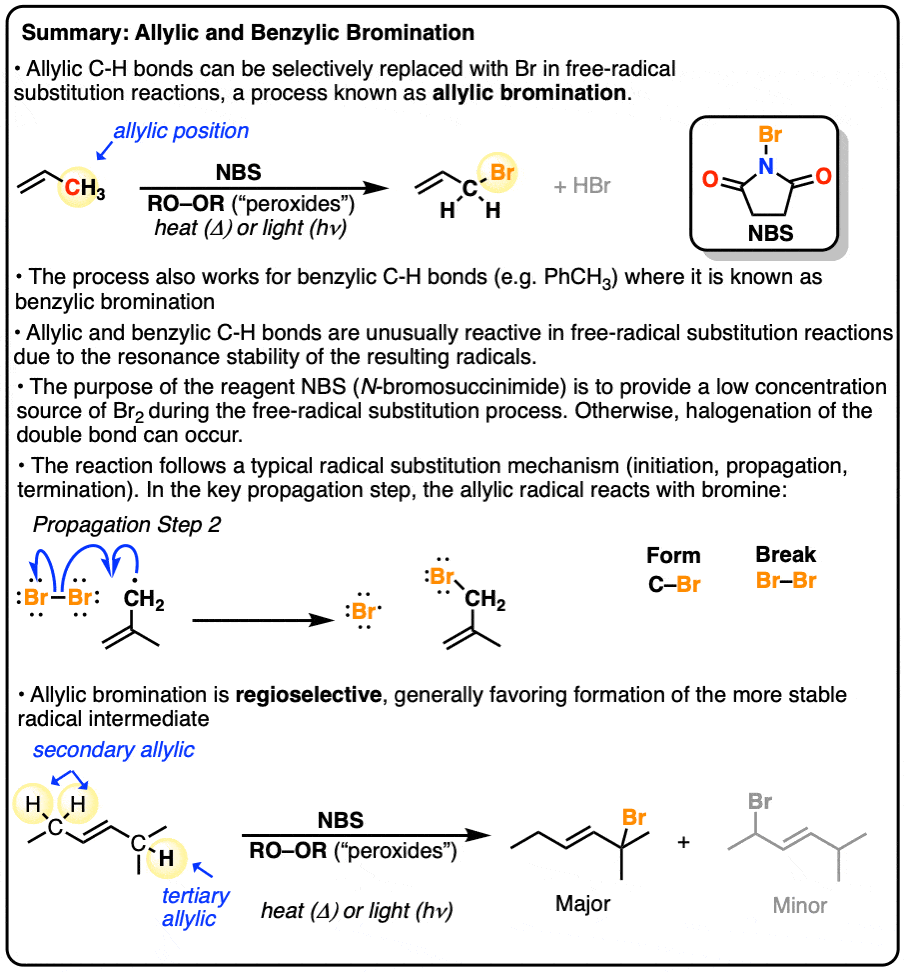
Table of Contents
- Allylic and Benzylic Bromination: Examples
- Free Radicals Are Stabilized By Resonance
- The Mechanism of Benzylic Bromination With Br2 and Light/Heat
- Br2 Is Not An Appropriate Reagent For Allylic Bromination Since It Forms Vicinal Dibromides
- Using N-Bromo Succinimide (NBS) Ensures A Low Concentration Of Br2
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Allylic and Benzylic Bromination: Examples
So what is allylic and benzylic halogenation, anyway? Here’s an example of each.
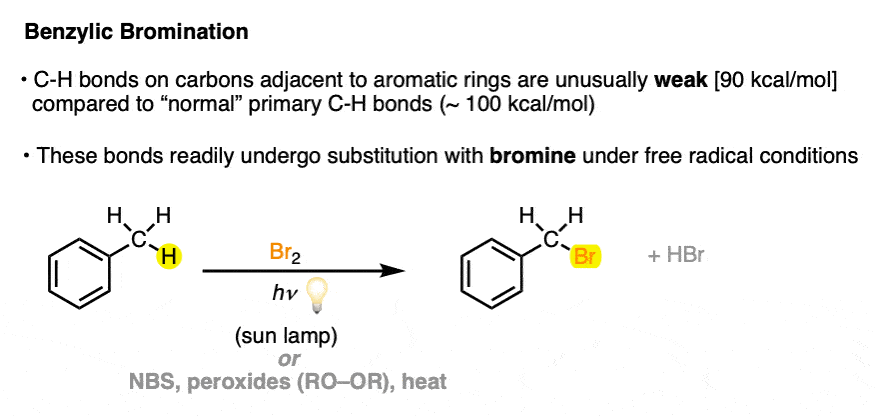
Take toluene and treat with either Br2 in the presence of light, as per this procedure, or N-Bromosuccinimide (NBS) in the presence of a radical initiator + heat (or light) and one of the benzylic C–H bonds is replaced with C–Br.
(Note that the “benzylic” position is the carbon attached to the benzene ring; the C-H bonds of the benzene ring itself are the “phenyl” C-H bonds).
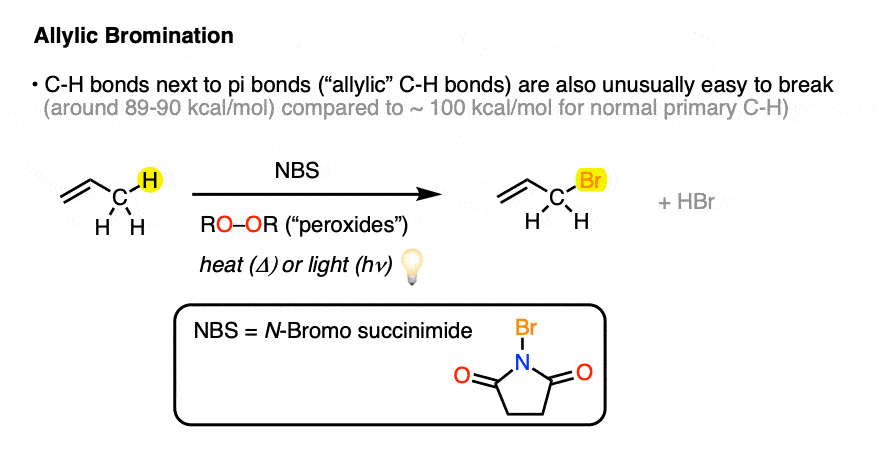
Here’s an example of the allylic bromination of propene, this time using NBS and peroxides (or heat)
We’ve spent a considerable amount of time discussing the stability of free radicals, particularly that primary radicals are particularly unstable.
So what’s different in this case? And what’s this “NBS” stuff?
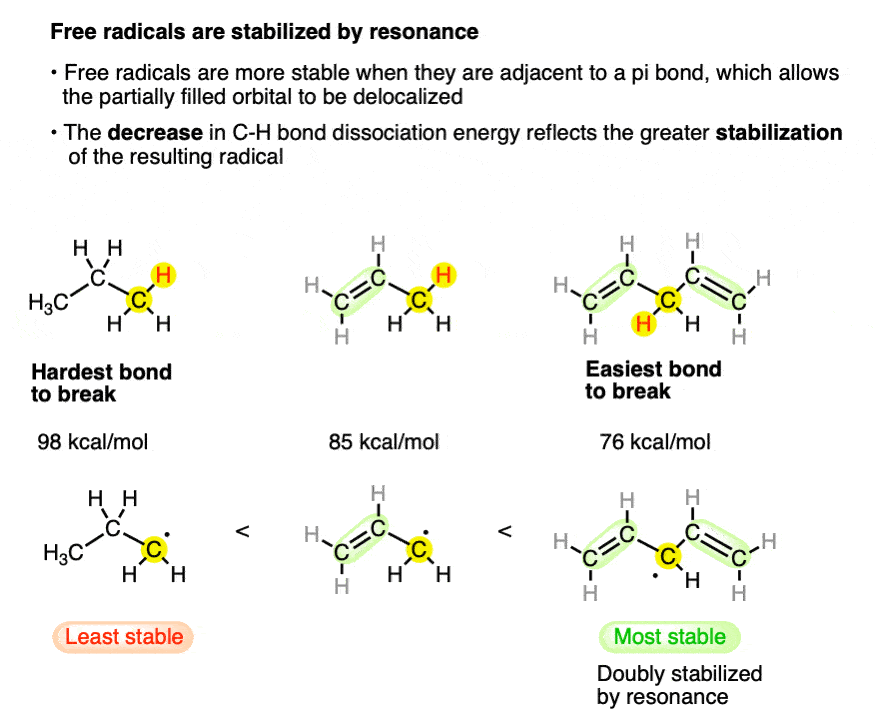
2. Free Radicals Are Stabilized By Resonance
Recall that free radicals are stabilized by resonance, and bond dissociation energies (BDE’s) aka “bond strengths” measure homolytic bond cleavage.
So the methyl groups (above) aren’t ordinary methyl groups – the resulting radicals are greatly stabilized by resonance!
Therefore when we look at bond dissociation energies for benzylic and allylic C–H bonds it should not be surprising to find that these bond strengths are quite weak (89-90 kcal/mol for a primary allylic or benzylic radical) relative to tertiary C-H bonds (93 kcal/mol).
So how might allylic or benzylic bromination work?
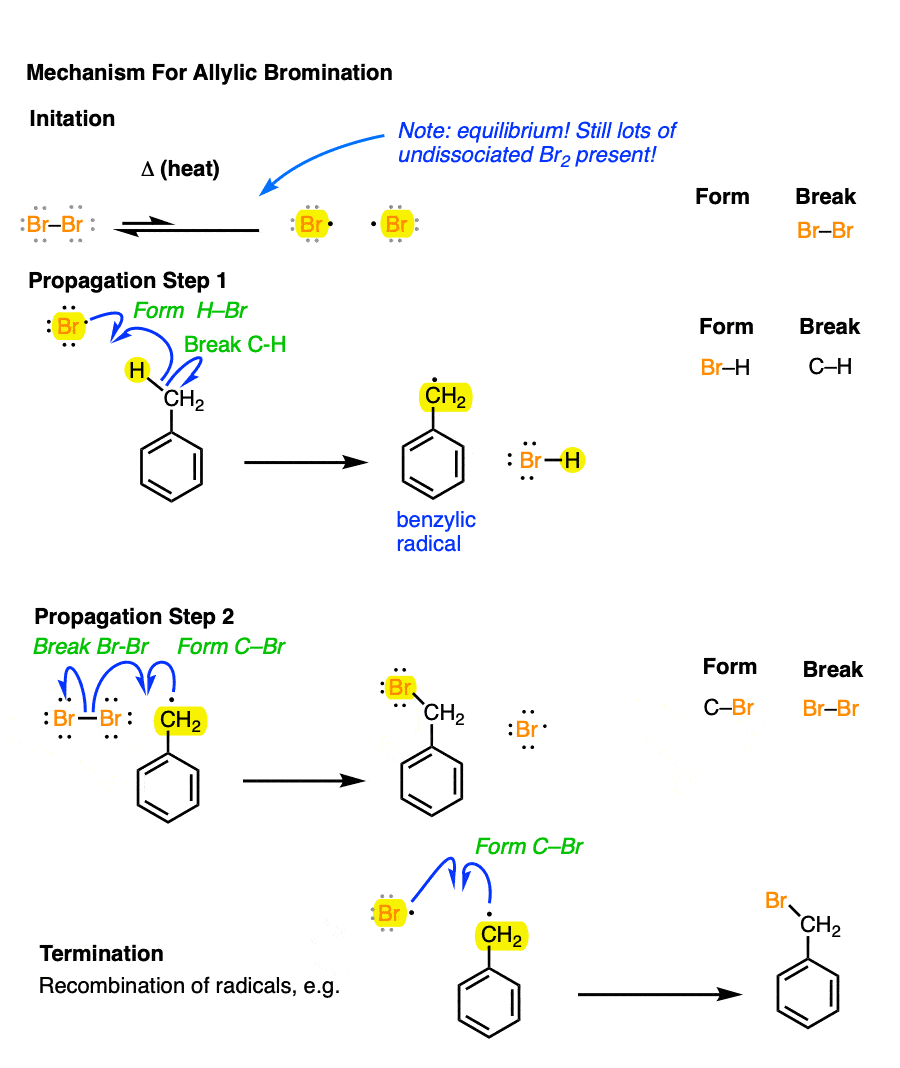
3. The Mechanism of Benzylic Bromination With Br2 and Light/Heat
For benzylic bromination, hopefully imagining the mechanism will be straightforward: after initiation (by heat or light), bromine radical then breaks the C-H bond (forming the benzylic radical) [propagation step #1] and the benzylic radical attacks Br2 to re-generate bromine radical [propagation step #2]. These two steps repeat until the concentration of Br2 runs low, whereupon radical chain termination will occur.
[See Initiation, Propagation, Termination]
4. Br2 Is Not An Appropriate Reagent For Allylic Bromination Since It Forms Vicinal Dibromides
Let’s turn to allylic bromination. Do you see any reason why treating the molecule below with Br2 might lead to problems?
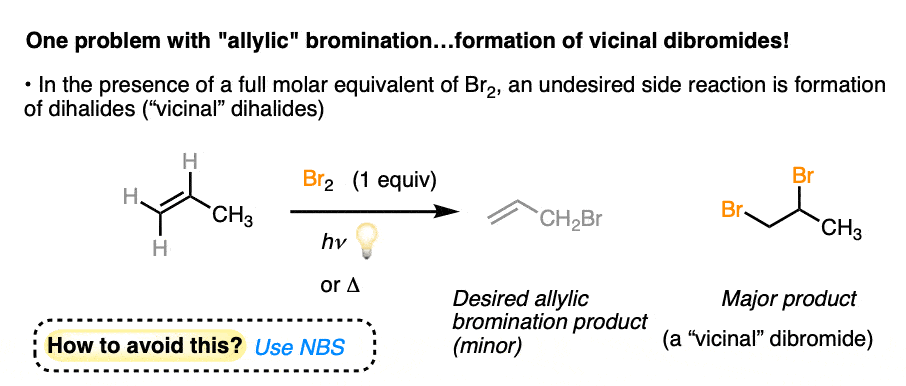
You might recall that alkenes react with Br2 to form vicinal dibromides, and that is in fact exactly what would occur if we were to just dump in 1 equivalent of Br2 with an alkene.
At issue here is the fact that we have 1 equivalent of Br2 swimming around, of which only a small proportion at any given time will exist as bromine radical [due to the initiation step].
How might we solve this problem and favour the radical substitution over the dibromination?
Imagine, if you will, we had a very low concentration of Br2. If the concentration of Br2 is kept low, not only will the rate of dibromination be lower, the relative concentration of bromine radical relative to Br2 will increase. Therefore, the rate of C-H abstraction relative to dibromination will increase, which will allow our allylic bromination product to be formed in a higher yield. [Note 1]
Of course, this creates a new question: how do we generate a low, constant concentration of Br2?
5. Using N-Bromo Succinimide (NBS) Ensures A Low Concentration Of Br2
As it turns out, there’s a way. If the bromonium ion source N-bromosuccinimide (NBS) is present with a trace amount of acid (HBr), HBr will react with NBS to give succinimide and Br2.[Note 2]
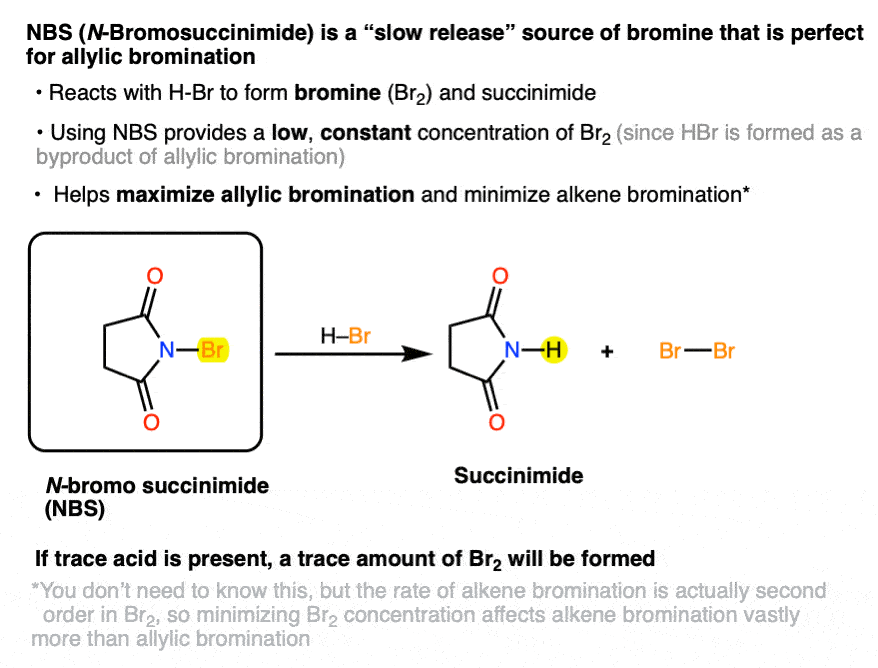 Since one equivalent of HBr generates one equivalent of Br2, Br2 will be generated only after the completion of Propagation Step #1.[Note 3].This keeps the concentration of Br2 low and allows the free-radical reaction to out-compete the alkene addition reaction.
Since one equivalent of HBr generates one equivalent of Br2, Br2 will be generated only after the completion of Propagation Step #1.[Note 3].This keeps the concentration of Br2 low and allows the free-radical reaction to out-compete the alkene addition reaction.
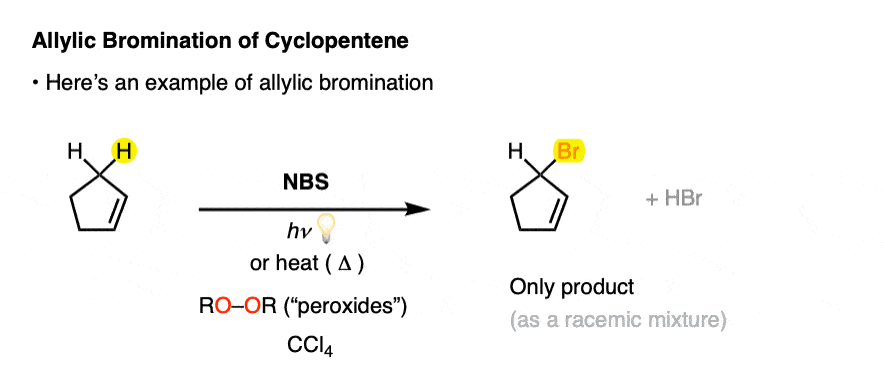
In all other respects the allylic bromination reaction is identical to the benzylic bromination reaction mechanism shown above. However in *some* situations there is an extra twist of allylic rearrangement, which we will briefly discuss in the next post.
Next Post: Allylic Rearrangement
Notes
Note 1. If this explanation sounds somewhat unsatisfactory, you’re on to something. After all, doesn’t this decrease the concentration of Br• as well? One additional complication is that theoretical studies show strong evidence for a termolecular rate determining step in formation of the bromonium ion (that is, involvement by the alkene and two molecules of Br2). Decreasing the concentration of Br2 would therefore vastly decrease the rate of bromonium ion formation (and thus dibromination) relative to free radical halogenation.
Note 2. As it happens, NBS which has been left out for awhile will usually be contaminated with trace HBr (as well as Br2, which gives it a yellow or orange colour). For this reason if doing a radical reaction with NBS it’s common to avoid recrystallizing it before use, as it will remove the trace acid that jump starts the reaction in the first place.
Note 3. As long as the initial concentration of HBr is low (i.e. it is used as a catalyst)
Quiz Yourself!
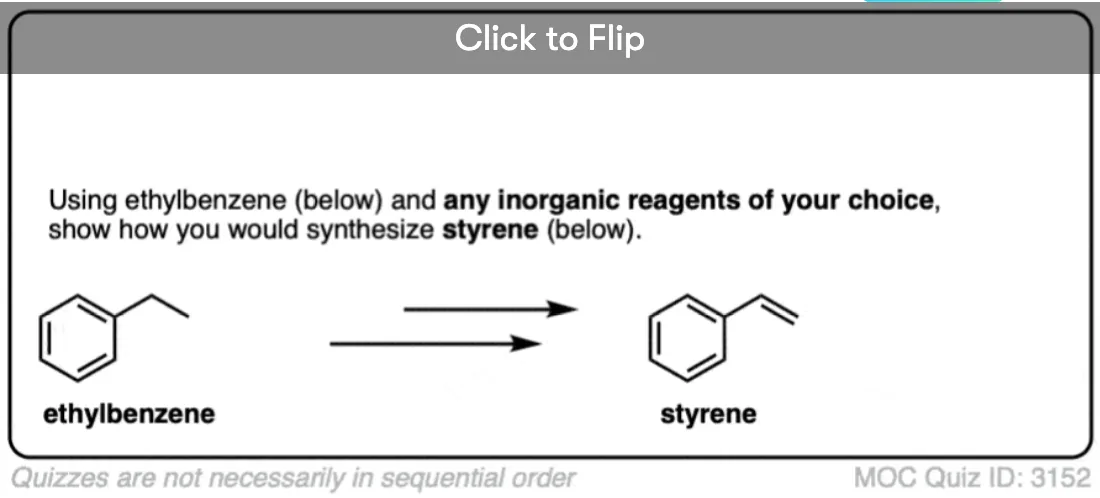
Become a MOC member to see the clickable quiz with answers on the back.
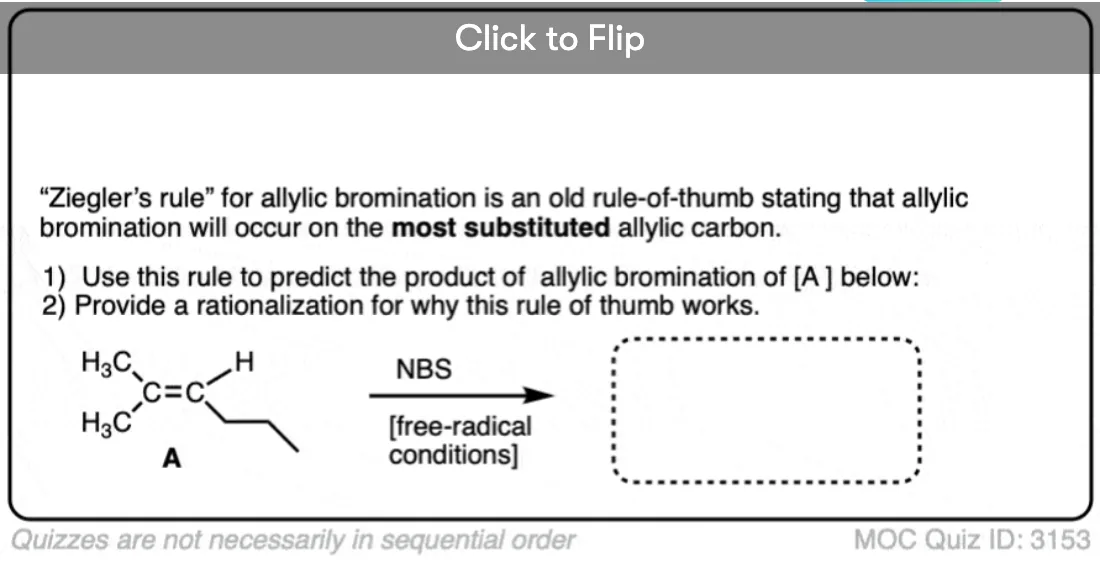
Become a MOC member to see the clickable quiz with answers on the back.
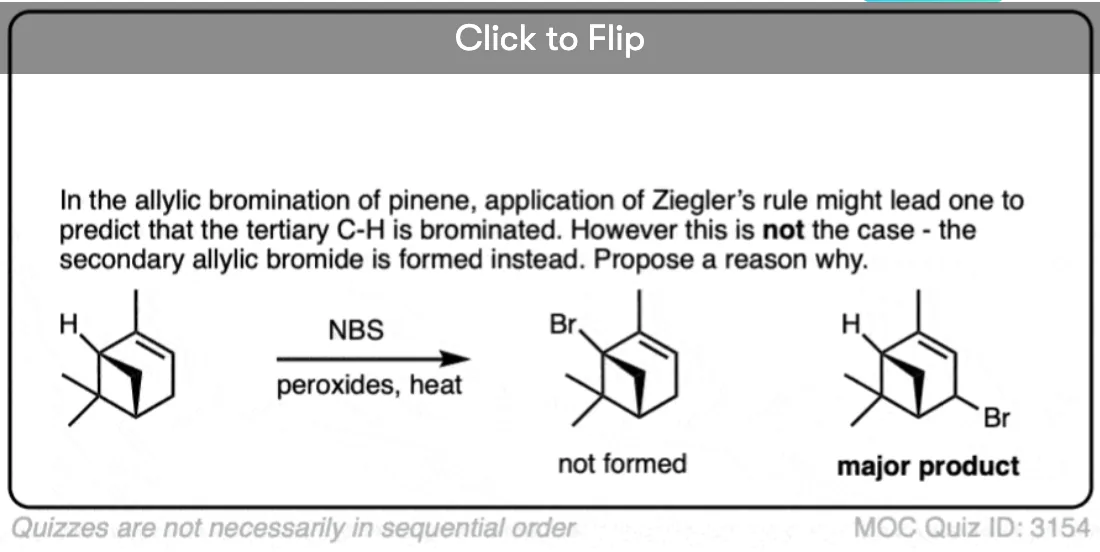
Become a MOC member to see the clickable quiz with answers on the back.
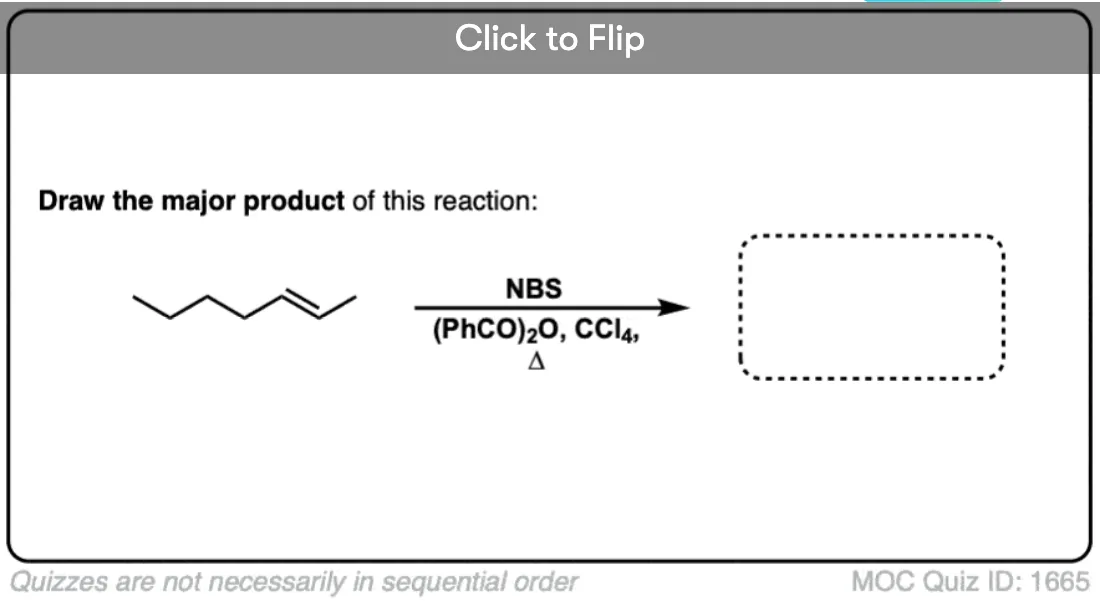
Become a MOC member to see the clickable quiz with answers on the back.
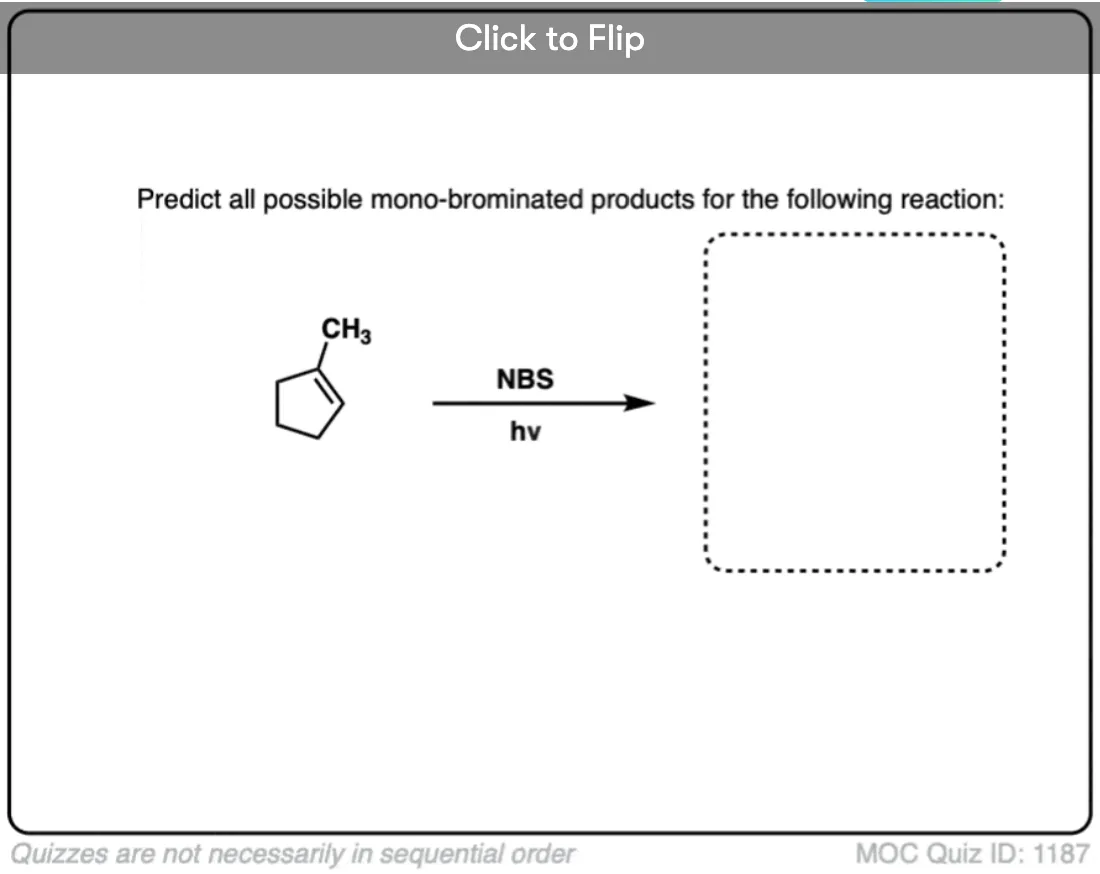
Become a MOC member to see the clickable quiz with answers on the back.
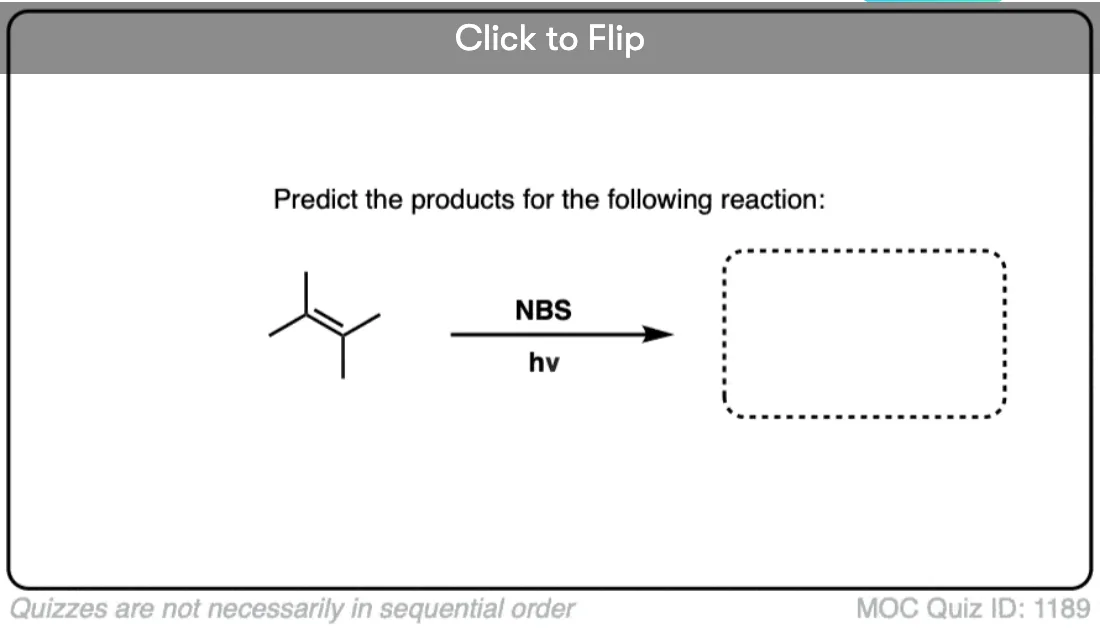
Become a MOC member to see the clickable quiz with answers on the back.
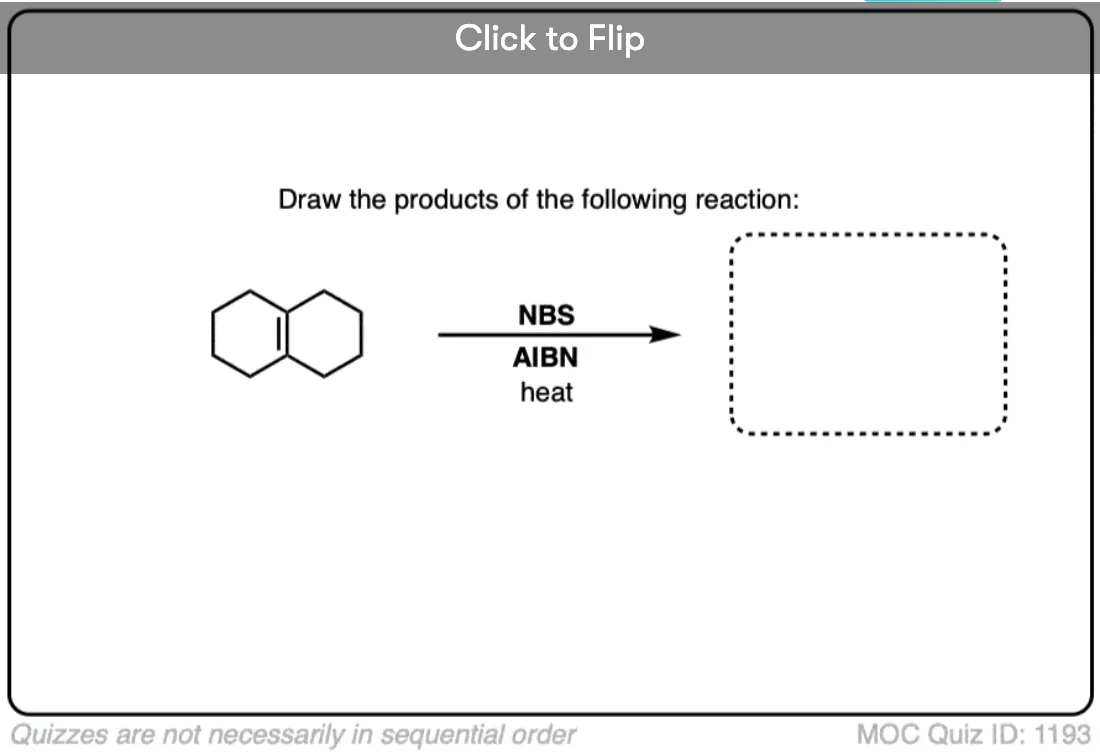
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
Allylic bromination with NBS is sometimes called the Wohl-Ziegler reaction.
- Die Synthese des Cantharidins.
Ziegler, K., Schenck, G., Krockow, E.W., Siebert, A., Wenz, A. and Weber, H. (1942),
Justus Liebigs Ann. Chem., 551: 1-79.
DOI:10.1002/jlac.19425510102
Ziegler’s report on using NBS for allylic bromination, from 1942. - Brominations with N-Bromosuccinimide and Related Compounds. The Wohl-Ziegler Reaction.
Carl. Djerassi
Chemical Reviews 1948 43 (2), 271-317
DOI: 10.1021/cr60135a004
An early but useful review of allylic bromination by Prof. Carl Djerassi (Stanford), who made many important contributions to steroid chemistry. - Laws of Addition and Substitution in Atomic Reactions of Halogens
ADAM, J., GOSSELAIN, P. & GOLDFINGER, P. .
Nature 171, 704–705 (1953).
DOI: 10.1038/171704b0
The “Goldfinger Mechanism” for allylic bromination, which proposed that NBS serves to provide a low concentration of Br2. Earlier proposed mechanisms involved the succinimidyl radical as the chain carrier, which is incorrect. It should be noted that addition of Br• to alkenes *does* occur to some extent, but the reaction is reversible, and if Br2 and HBr concentrations are kept low, any addition product will revert to the starting alkene. - THE HIGH-TEMPERATURE CHLORINATION OF OLEFIN HYDROCARBONS
FREDERICK F. RUST and WILLIAM E. VAUGHAN
The Journal of Organic Chemistry 1940 05 (5), 472-503
DOI: 10.1021/jo01211a002
An early, key observation. Rust and Vaughn found that addition of Cl2 to alkenes is favored at low temperatures, whereas at higher temperatures allylic substitution dominates. - The Ratio of Substitution to Addition in the Reaction of Chlorine with Olefins in Dilute Carbon Tetrachloride Solution
T. D. Stewart, Kenneth Dod, and George Stenmark
Journal of the American Chemical Society 1937 59 (9), 1765-1766
DOI: 10.1021/ja01288a507
An important piece of the puzzle to figuring out the mechanism for allylic halogenation, from 1937. Allylic substitution is favored over addition at lower concentrations of halogen (Cl2). - Mechanisms of Benzylic Bromination
Glen A. Russell, Charles. DeBoer, and Kathleen M. Desmond
Journal of the American Chemical Society 1963 85 (3), 365-366
DOI: 1021/ja00886a040
Benzylic bromination follows the same mechanism as allylic bromination, as this paper explains.NBS (N-Bromosuccinimide) is a convenient reagent for free-radical bromination, and the following papers are mechanistic studies involving NBS: - The Mechanism of Benzylic Bromination with N-Bromosuccinimide
E. Pearson and J. C. Martin
Journal of the American Chemical Society 1963 85 (3), 354-355
DOI: 10.1021/ja00886a029
These papers by Prof J. C. Martin (UIUC) were early in his career, before he did the work that he is most well-known for (studies on ‘hypervalent’ molecules, including the development of the ‘Dess-Martin Periodinane’). - The Identity of the Chain-Carrying Species in Brominations with N-Bromosuccinimide: Selectivity of Substituted N-Bromosuccinimides toward Substituted Toluenes
E. Pearson and J. C. Martin
Journal of the American Chemical Society 1963 85 (20), 3142-3146
DOI: 10.1021/ja00903a021 - N-bromosuccinimide. Mechanisms of allylic bromination and related reactions
H. Incremona and James Cullen Martin
Journal of the American Chemical Society 1970 92 (3), 627-634
DOI: 10.1021/ja00706a034 - Radical Bromination of Cyclohexene in CCl4 by Bromine: Addition versus Substitution
W. McMillen and John B. Grutzner
The Journal of Organic Chemistry 1994 59 (16), 4516-4528
DOI: 10.1021/jo00095a029
This paper describes careful kinetic studies that demonstrate that a low concentration of Br2 (such as that provided by impure NBS) will favor radical substitution over a polar addition reaction. - 4-BROMO-2-HEPTENE
L. Greenwood, M. D. Kellert, and J. Sedlak
Org. Synth. Vol. 38, p.8 (1958).
DOI: 10.115227/orgsyn.038.0008
A reliable procedure for allylic bromination with NBS in Organic Syntheses. - o-XYLYLENE DIBROMIDE
Emily F. M. Stephenson
Org. Synth. Vol. 34, p.100 (1954)
DOI: 10.15227/orgsyn.034.0100
A reliable procedure for benzylic bromination with Br2 in Organic Syntheses. - The evolution of free radical chemistry at Chicago
Frank R. Mayo
Journal of Chemical Education 1986 63 (2), 97
DOI: 1021/ed063p97
For those interested in the history of science, this retrospective by Mayo tells the story of how he and Kharasch discovered the ‘peroxide effect’ and thereby a new area of organic chemistry.
i am doing bromination reaction with use of dibromo dimethyl hydantoin 0.54 mole but i found dibromo impurity level well above to 10.5%,so how i can reduce di bromo impurity to below 3.0%
Why can’t you use Br2 and peroxides for benzylic cases?
You can, as in this procedure: http://www.orgsyn.org/demo.aspx?prep=CV4P0984
If the NBS you are using happens to be significantly contaminated with HBr/Br2, is it then possible to add more than one bromine?
It is standard procedure in many reactions to add a slight molar excess (e.g. 10%) of a reagent just to ensure completion of the reaction. I have not seen analyses of “bad” NBS to quantify just how high the Br2 content can get, but I would not expect it to be more than 10%.
So my short answer is no, not to any significant extent that it would impair the yield by more than 10%.
If there *is* a lot of Br2 present, two things could happen. First, if the impure NBS is just added to solution and dissolved, without adding alkene first, the Br2 that has been liberated can re-combine with succinimide (this is, after all, how NBS is formed).
If a relatively high concentration of Br2 persists, then what is more likely is that one would start to see dibromination of the alkene (i.e. addition of Br2) across the double bond, since bromination tends to be 3rd order overall, and low concentrations of Br2 avoid this side reaction.
In this case, then there would again a lower yield of the desired product, but not double addition of bromine.
If a large excess of NBS is added, then yes, it could start to be an issue.
What will be the product when 3- methylene cyclohexene react with HBr? Explain
You will need to identify what will be the kinetic and thermodynamic product. https://www.masterorganicchemistry.com/2017/03/22/reactions-of-dienes-12-and-14-addition/
I’m a little bit confused. It seems like your explanation to achieve a low concentration of Br2 to avoid dibromination is to react a trace amount of HBr with NBS to get a small amount of Br2, and then react Br2 in the presence of a solvent and with light to end up with an allylic brominated product. Why can’t we just start with a small amount of Br2 and react that with light in the first place? Why do we have to start with HBr and NBS?
If On product 3 bromo cyclo pentene further addition of Br2, is done then what will be the product ?
Bromination is possible ??
Yes, if you added Br2 to 3-bromocyclopentene, you would brominate the double bond.
Hi James
I just wanted to know why we are using CCl4 as a solvent?
Nice and high boiling. If it takes heat to initiate the reaction it’s useful to choose a solvent which has a relatively high boiling point. There’s also no C-H bonds which can potentially react with free radicals.
Hi James
I’ve a question about para ethyltoluene,(4-ethyltoluene) what will happen when it reacts with HBR, i can’t figure out where the bromation will take place,
what happens if chlorobenzene reacts with KI ? is there any reaction? if yes where does it take place? i’ve an orgnic examen ,and i really don’t know how to handle those kinds of reactions.
Thanks
Bromination will not happen using HBr as a reagent. In that case all that will happen is that you will (transiently) protonate the ring.
Chlorobenzene plus KI under (extraordinarily) forcing conditions MAY yield iodobenzene via nucleophilic aromatic substitution, but there are much better ways to do it.
how can one selectively brominate one benzylic site out of two different sites……say if i want to slectively brominate the benzylic -CH2- group of p-ethyltoluene instead of the benzylic -CH3 group?
The secondary radical should be more stable, so you should at least get some selectivity for that position.
If you were to have a molecule with a t-butyl substituent and a methyl substituent, with which group would the Br radical be more likely to react with?
The particular molecule that brought up this question is 1-methyl-3-tert-butylbenzene.
Methyl. There’s no benzylic C–H hydrogen for the bromine radical to remove from the t-butyl.
Hi james
why wouldn’t Br• attack on the vinylic carbon instead of allylic carbon just like it does in the peroxide effect ?
And for the reason u give why would be it not applicable in peroxide effect?
Gamma should be nu.
Yes, fixed, thank you!
Hi James,
Would the same reasoning for allylic bromination apply to allylic chlorination as well?
Yes.
Hi,
Can I use DCM (dichlormethan) as a solvent instead of PhCl? Radical reactions are often not very selective. Why can brominate anyway selective just at the benzylposition and not somewhere else?
Many thinks
Usually you want a solvent that has a reasonably high boiling point since the initiation step requires heat. Dichloromethane is pretty low boiling. You could go for dichloroethane instead. (83 C° versus 40 C°)
Hi James–
As I understood, allylic means the molecule two away from the double bond, not directly attached, and vinylic means it is attached to the double bond. Therefore wouldn’t the radical on the 3rd carbon of 1-propene be vinylic?
A vinylic radical would be directly attached to the double bond – i.e. sp2 hybridized . For example a radical on the 1 or 2 carbon of 1-propene would be vinylic. The adjacent carbon (carbon 3) is allylic. If there was a 4th carbon that position would be called, “homoallylic”.
Dear james,
Can you please tell me what will be the product when 3 methyl cyclohexene reacts with NBS/br2.
Thank you
At least two different allylic bromination products are possible.
James,
Is there any other solvent except of CCl4 that can be used in benzylic halogenations?
Many thanks
I’d try chloroform or 1,2-dichloroethane.
wouldn’t Br• attack on the vinylic carbon just like it does in the peroxide effect and the resulting radical would further start a chain reaction by attacking on Br2
The vinyl C-H bonds have a bond dissociation energy of around 111 kcal/mol and the allyl C-H bonds have a bond dissociation energy of around 90 kcal/mol so it’s easier to break the allyl C-H bond and form the radical there.
Hey James,
Would I be correct in assuming that unless strict conditions are employed such as 1 equivalent of NBS, a second bromine addition will take place? Or will the EWG nature of the first bromide affect a second addition?
Also the physics student in me would like to point out that light is represented by h(nu) not h(gamma). Quite trivial I know.
Bromonium ion formation is reversible so as long as no strong nucleophiles are present to open it, it’s possible to get away with an excess of NBS. What often happens is that you need to keep adding the radical initiator (like AIBN or peroxides) over time if the reaction stops.
Thanks for the correction!
James,
In the allylic bromination, after the initiation step, bromine radicals are formed. They have one electron less than their requirement and so they want to pick one electron. Then why don’t they cleave the pi bond, which has loosely bound electron, and take it from there?
What you’re describing is addition of bromine radical to a pi bond, which is what happens when you treat an alkene with peroxides and HBr. In that reaction, once Br• adds to the pi bond, you obtain a radical on a carbon. If plenty of HBr is present, then the radical attacks the H of HBr and the cycle continues.
However in the case of allylic bromination, that reaction is (ideally) a cul-de-sac. Once Br• radical addds to the double bond, forming that carbon-based radical, there is (ideally) nothing for it to react with since we’re not adding HBr and we’re keeping the concentration of Br2 low. Therefore the addition will revert back to the double bond and Br radical, and eventually the Br• will abstract a hydrogen from the allylic position.
The same question came up in class today, and a quick bond energy calculation suggests the addition of Br2 across the double bond might be more favorable. The propagation step of allylic H abstraction is slightly endothermic, while the propagation step in “competing” radical addition to the C=C pi bond is slightly exothermic.
This had me confused for a while, but a trip to my old graduate physical organic text (Lowry & Richardson, 1981, pp. 717-726) suggested that the Arrhenius collision orientation factor for abstraction is much more favorable than that for pi-addition, even though the activation energy is a bit less. The net result is a difference in rate constants of a factor of ~5 at 300 K. At the typical reaction temperature (refluxing CCl4, ~350 K) the difference is about a factor of 13.
Thanks Dave. Does L&R point to the original literature reference?