Alcohols, Epoxides and Ethers
Making Alkyl Halides From Alcohols
Last updated: May 28th, 2026 |
Making Alkyl Halides From Alcohols
In today’s post we show that treating alcohols with HCl, HBr, or HI (which all fall under the catch-all term “HX” where X is a halide) results in the formation of alkyl halides.
- Primary alcohols tend to proceed through an SN2 mechanism
- Tertiary alcohols tend to proceed through an SN1 mechanism
- Watch out for rearrangements in reactions where a secondary carbocation may be formed
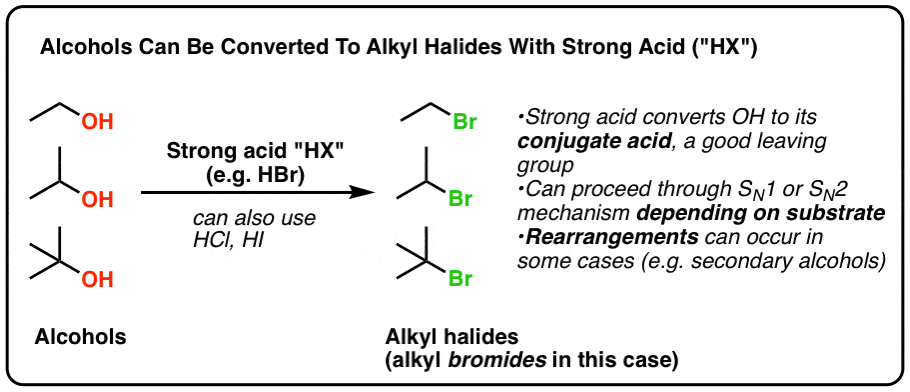
Table of Contents
- Adding Acid To Alcohol Produces A Good Leaving Group (H2O)
- Alkyl Halides From Methyl and Primary Alcohols via the SN2 Reaction
- Alkyl Halides From Tertiary Alcohols Proceeds Through The SN1 Pathway
- A Good Rule Of Thumb For Secondary Alcohols With HX: Assume SN1
- Rearrangements In The Formation of Alkyl Halides From Alcohols
- Rearrangement Example #1: Hydride Shift
- Rearrangement Example #2: Alkyl Shift
- Rearrangement Example #3: A Special Case of Alkyl Shift – Ring Expansion and Contraction
- What Doesn’t Work?
- Summary: Alkyl Halides From Alcohols
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Adding Acid To Alcohol Results In A Good Leaving Group
We’ve said many times in this series of posts that alcohols are poor substrates for SN1 and SN2 reactions. That’s because the hydroxyl ion (HO-) is a poor leaving group, and therefore not likely to either 1) depart of its own accord, leaving behind a carbocation (SN1 pathway) or 2) to be displaced by an incoming nucleophile (which would be an SN2 reaction).
However we’ve also seen that treating an alcohol with acid leads to an interesting “personality adjustment”: the alcohol (R-OH) is converted to its conjugate acid, (R-OH2+) which now possesses a decent leaving group (the weak base we know as water, H2O). (See post: How to Make Alcohols More Reactive)
We saw, in a previous post, how this allows for formation of (symmetrical) ethers from alcohols, either via SN2 pathway (with primary alcohols) or an SN1 pathway (tertiary alcohols).
We might ask: can this be extended to form other functional groups besides ethers?
Absolutely. Treating alcohols with HCl, HBr, or HI (which all fall under the catch-all term “HX” where X is a halide) results in the formation of alkyl halides.
This occurs in a two step process:
- first, the alcohol is protonated to give its conjugate acid.
- Secondly, a substitution occurs.
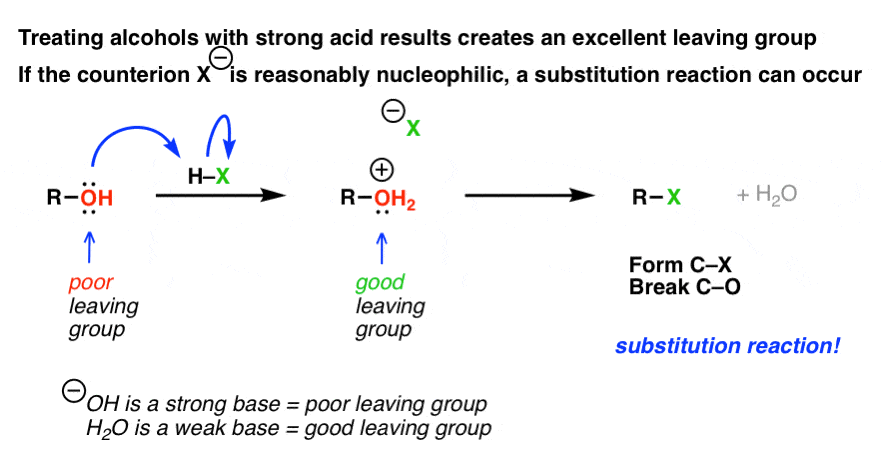
Notice how that second step (substitution) was left vague in the diagram above. That’s because, as we’ve seen, the type of substitution pathway depends on the substrate.
2. Alkyl Halides From Methyl And Primary Alcohols Via SN2 Reaction
Knowing how sensitive the SN2 reaction is to steric hindrance, we should expect that for methyl alcohol and for primary alcohols, the SN2 pathway dominates. And it does! (See post: The SN2 Mechanism)
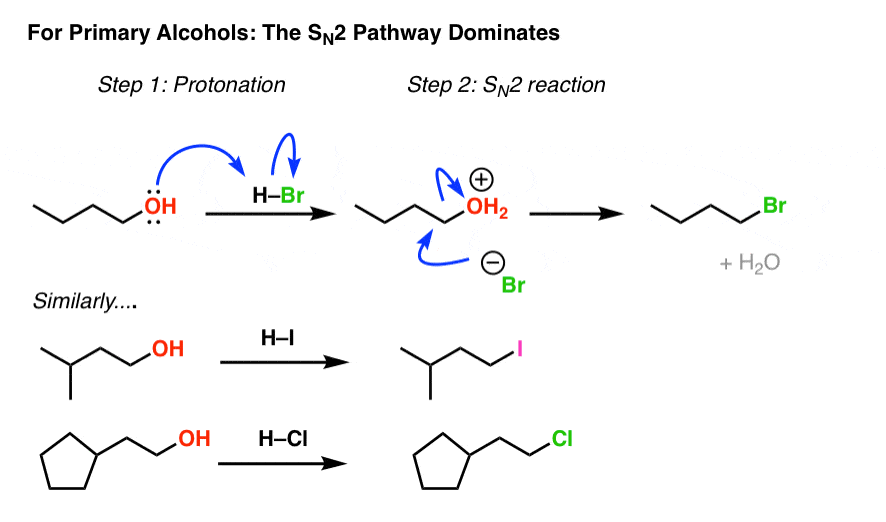
Note how in each case we begin by protonating the alcohol, creating a good leaving group which is then displaced by the conjugate base of the acid. (See post: What Makes a Good Leaving Group?)
Alkyl chlorides, bromides, and iodides can each be made this way.
3. Alkyl Halides From Tertiary Alcohols Proceeds Through The SN1 Pathway
Likewise, understanding the trends of carbocation stability, we should expect that conversion of tertiary alcohols to alkyl halides proceeds through an SN1 pathway. And it does. (See post: The SN1 Mechanism)
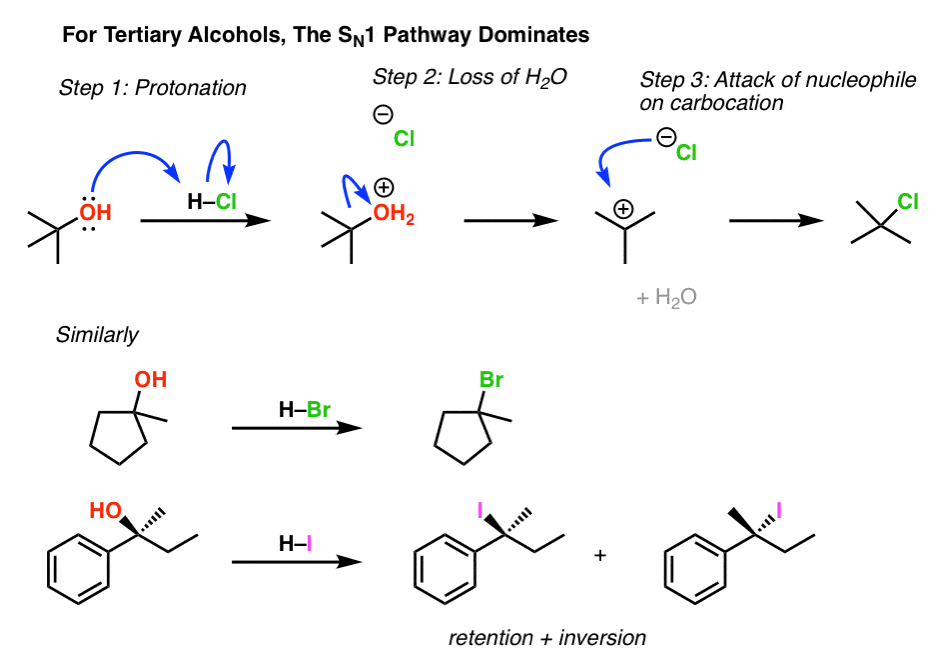
Note in the last example that beginning with a chiral starting material will lead to a mixture of inversion and retention (often called, “racemization“) because it goes through the (flat) intermediate carbocation.
4. A Good Rule of Thumb For Secondary Alcohols With HX: Assume SN1
Methyl, primary, and tertiary alcohols all represent pretty straightforward cases.
“So what about secondary alcohols?” you might ask. Ah yes. This is where things get interesting – and is, therefore, the stuff of which exam questions are made.
In the lab, treatment of secondary alcohols with HX leads to a mixture of products from SN1 and SN2 pathways. For practical purposes it is generally not a useful process, especially if you care about preserving stereochemistry.
However, your introductory textbook and course notes are not “the lab”. The purpose of a course is to introduce you to important concepts in organic chemistry. And from an instructor’s standpoint, it so happens that the conversion of secondary alcohols to secondary alkyl halides by HX is an excellent opportunity to bring up the subject of carbocation rearrangements. This falls under the purview of the SN1 pathway.
So a good rule of thumb is to assume – for the purposes of your course – that secondary alcohols treated with HX will proceed through an SN1 mechanism.
5. Rearrangements In The Formation Of Alkyl Halides From Alcohols
We’ve covered rearrangements (hydride and alkyl shifts) before in the context of SN1 reactions. But it’s worth touching on again.
The basic premise is this. Carbocations are unstable, electron-poor species. Their stability generally increases with the number of attached carbons, which serve to donate electron density. Hence, the stability of carbocations increases in the direction methyl < primary < secondary < tertiary. We also saw that carbocations are stabilized by resonance.
As we saw in a previous series of posts – consult this if you need more hand holding! – carbocations can undergo 1,2 shifts of C-H and C-C bonds, resulting in new carbocations.
Such rearrangements are most likely to occur if they can result in a more stable carbocation. For example, the rearrangement of a secondary to a tertiary carbocation, is a favoured (energetically “downhill”) process, whereas a rearrangement from a tertiary to a secondary carbocation (energetically “uphill”) is unlikely.
Anytime a reaction proceeds through a carbocation intermediate, we need to be on the lookout to see if it is adjacent to a carbon which can generate a more stable carbocation through a shift of a C-H or C-C bond.
There are three cases in particular to watch out for.
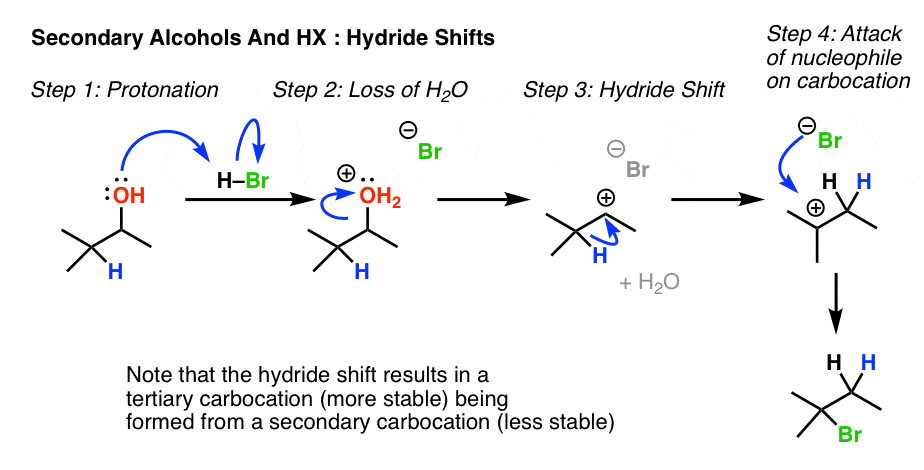
6. Rearrangement Example #1: A Hydride shift
Look for a secondary alcohol adjacent to a tertiary carbon. Note this common example, where protonation leads to loss of water, followed by a hydride shift and then trapping of the carbocation by the halide ion.
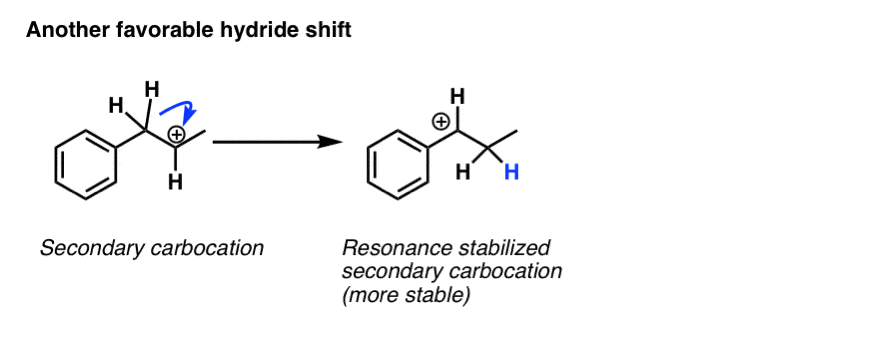
Another example of a favourable rearrangement is when a secondary carbocation is adjacent to an “allylic” or “benzylic” hydrogen. Rearrangement results in a secondary carbon which is stabilized by resonance.
7. Rearrangement Example #2: Alkyl Shift
Look for a secondary alcohol adjacent to a quaternary carbon (i.e. a carbon attached to 4 other carbons). Note how this is essentially the exact same process as the hydride shift above, except that CH3 is migrating, not H.
![]()
8. Rearrangement Example #3: A Special Case of Alkyl Shifts – Ring Expansions And Contractions
Look for a secondary alcohol that is adjacent to a strained ring (cyclobutane in the classic case). Once the secondary carbocation is generated, a bond in the strained ring migrates, leading to expansion of the ring by one. This is particularly favourable in the case of cyclobutane to cyclopentane since cyclobutane is highly strained (about 26 kcal/mol) whereas cyclopentane has minimal ring strain.
![]()
This type of alkyl shift commonly gives students a hard time, which of course makes it a favourite exam problem of instructors. Although the curved arrow drawn is no different than that for the previous two cases, I think the main difficulty is in mapping the product from the starting material. In this respect I recommend two things:
- Number the carbons (not necessarily IUPAC – just number to keep track of them). For instance here the arrow in Step 3 (the alkyl shift) shows us breaking C2-C3 and forming C1-C3. Applying the rules of curved arrows implies that C1 will then be neutral and C2 will become a carbocation. That’s all that’s happening. No other bonds are formed or broken in this step. It takes practice to get this right.
- Draw the ugly version first. THEN redraw to make it look good.
Ring contractions are also possible, although are not as favourable as the opening of strained rings. The same principles apply.
9. Alkyl Halides From Alcohols: What Doesn’t Work?
It’s always helpful to know what doesn’t work in the formation of alkyl halides from alcohols.
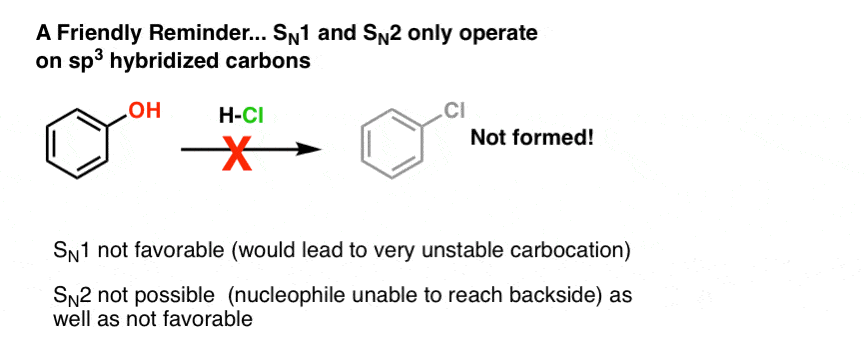
First of all, since we’re dealing with substitution reactions here, some familiar rules apply. Only alkyl alcohols (alcohols on sp3 hybridized carbons) will undergo SN1 and SN2 reactions. Both the SN1 and SN2 pathways involve buildup of positive charge on carbon, and sp2 and sp hybridized carbocations are extremely unstable. This attempted SN1 of phenol, for example, will fail miserably:
You might also wonder if we can use reagents like HCN, HOAc, or HN3 to convert alcohols to nitriles, esters, and azides respectively. Generally, no. The problem is that each of these are fairly weak acids (pKa 4 and above) so these will only give a low concentration of the protonated alcohol. Since the reaction rate is proportional to concentration, formation of these products will be slow. [With azides, there are also potential complications with a different type of rearrangement, but as a curtiusy we’re not going to deal with that schmidt right now : – ) ]
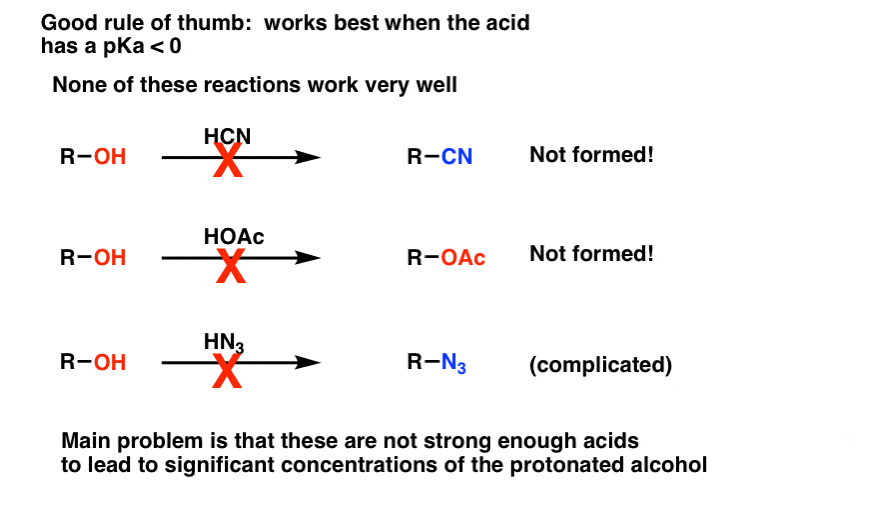
For our purposes, conversion of alcohols to other substitution products using strong acid is limited to HCl, HBr, HI, and the special case of H+/ROH which gives symmetrical ethers. A good rule of thumb is that the conjugate acid of the nucleophile should have a pKa of 0 or less in order for the reaction to occur.
10. Summary: Making Alkyl Halides From Alcohols
So alcohols can be converted to alkyl halides. You might ask, “why should we care?”. The answer is that, as we said, converting an alcohol (which has a poor leaving group) into an alkyl halide (which has a great leaving group) now allows us to do all kinds of functional group interconversions that were not previously possible. The SN2 is a very useful and powerful reaction, for example. Once a primary alcohol has been converted to a primary alkyl halide, we can then treat it with all varieties of nucleophiles to make a multitude of functional groups.
However nice it is to be able to do this, though, it’s far from ideal. We have to use strong acid, which can often cause complications if we have acid-sensitive functional groups on our molecule. Furthermore, all those pesky rearrangements on secondary carbons are a hassle. They can screw with our stereochemistry and lead to undesired products. You might ask, “isn’t there some way to get around that?”.
Yes! We’ll talk about a very nice way around this dilemma in the next post!
Next Post – Tosylates And Mesylates
Notes
Quiz Yourself!
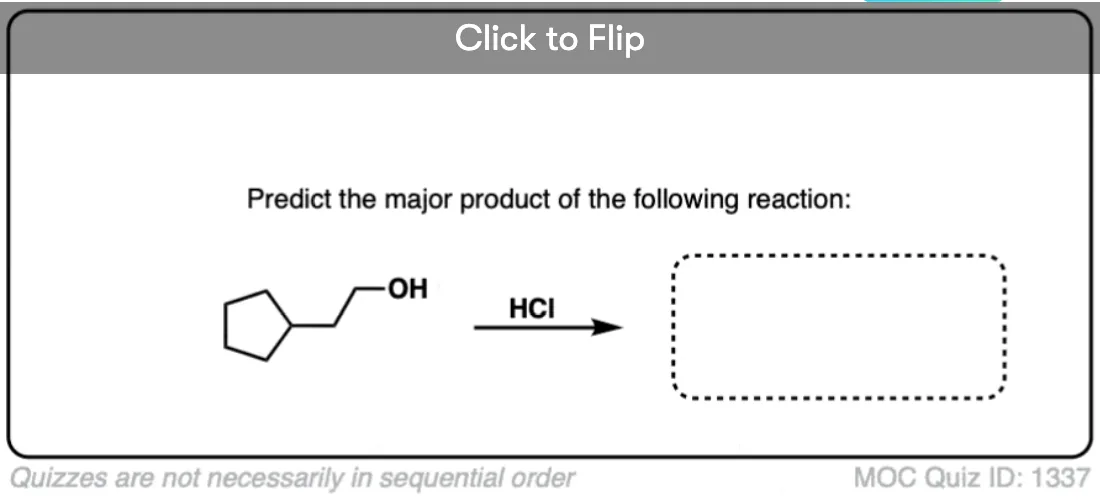
Become a MOC member to see the clickable quiz with answers on the back.
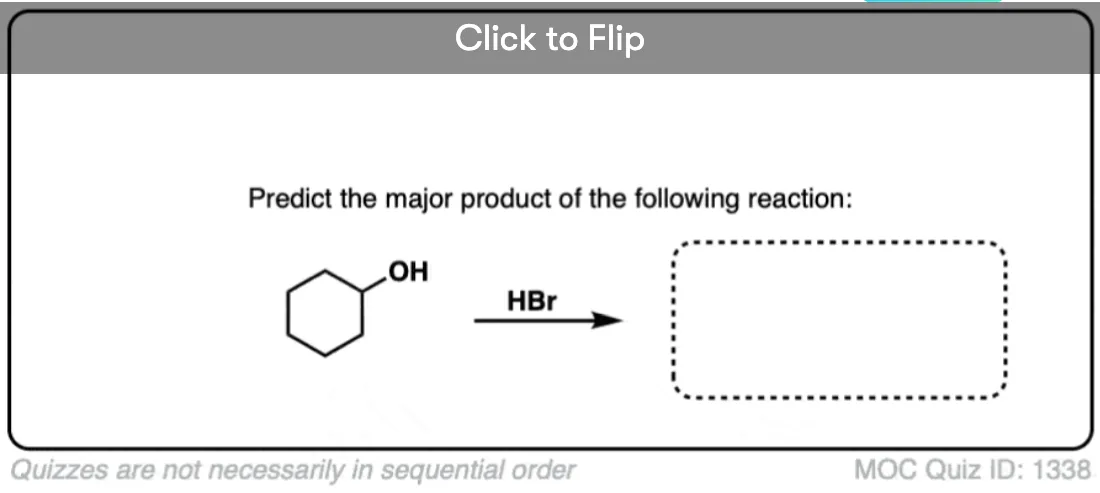
Become a MOC member to see the clickable quiz with answers on the back.
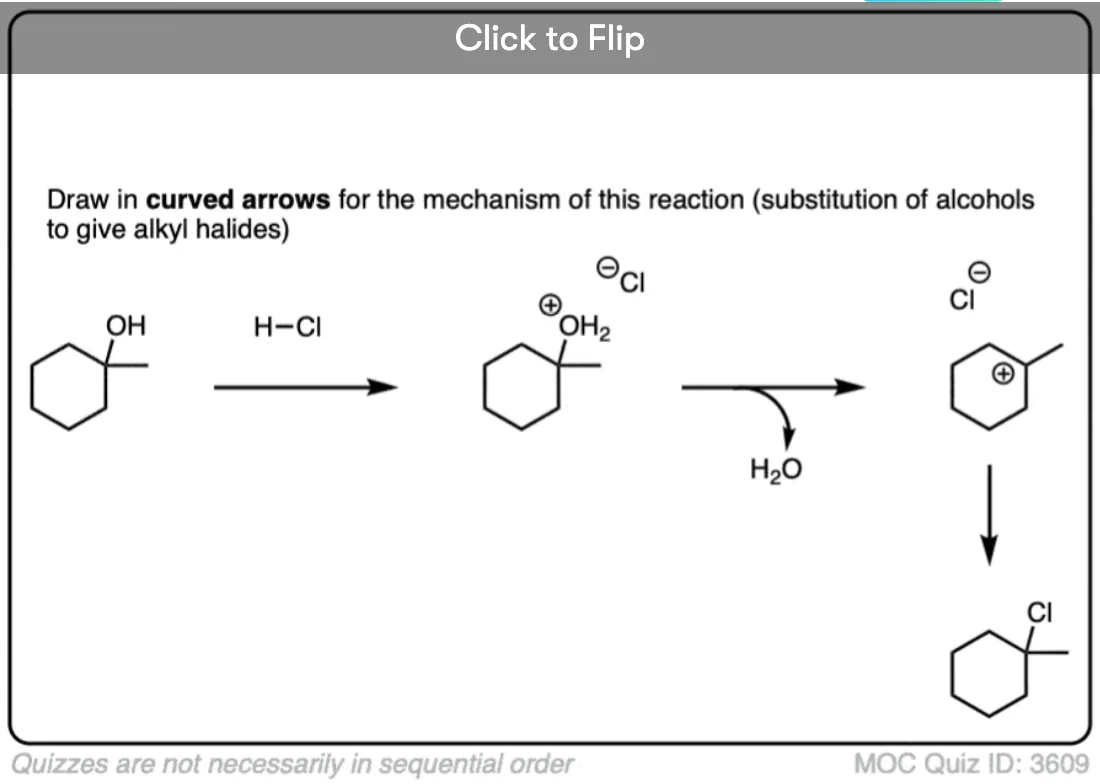
Become a MOC member to see the clickable quiz with answers on the back.
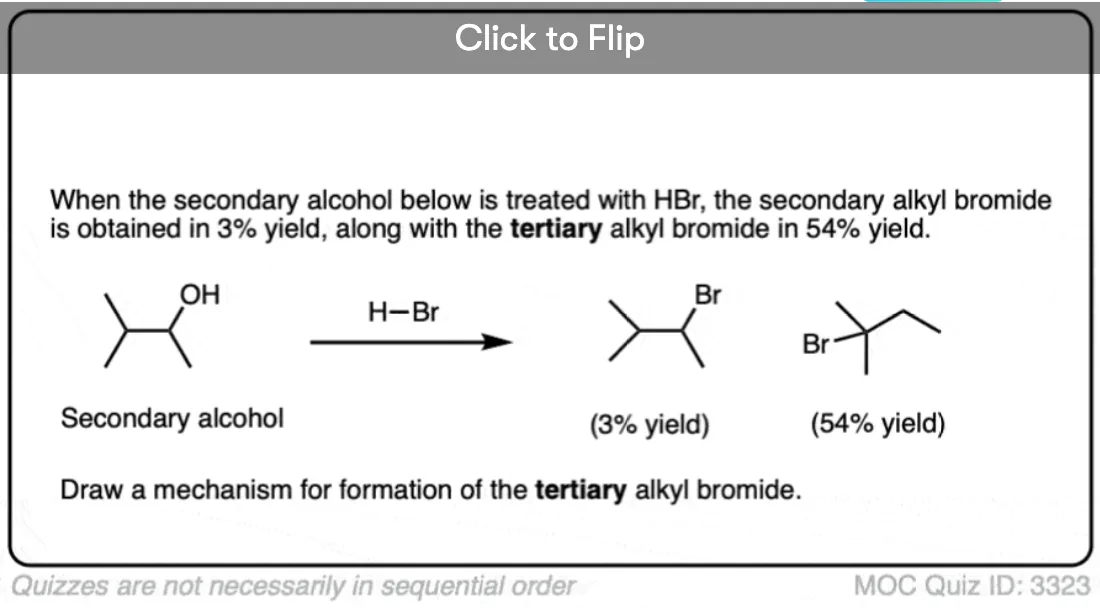
Become a MOC member to see the clickable quiz with answers on the back.
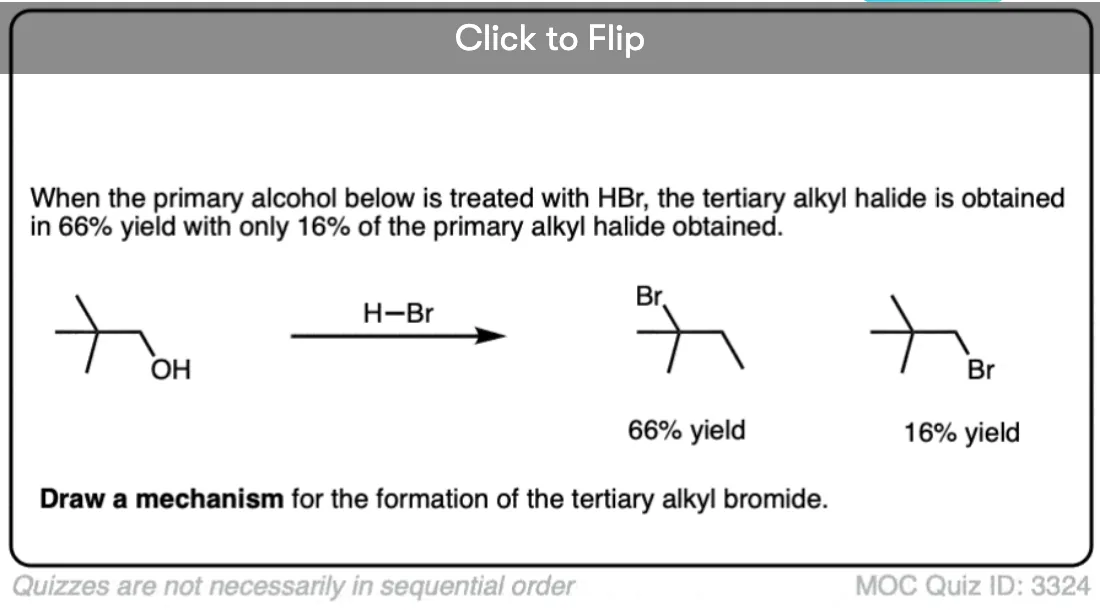
Become a MOC member to see the clickable quiz with answers on the back.
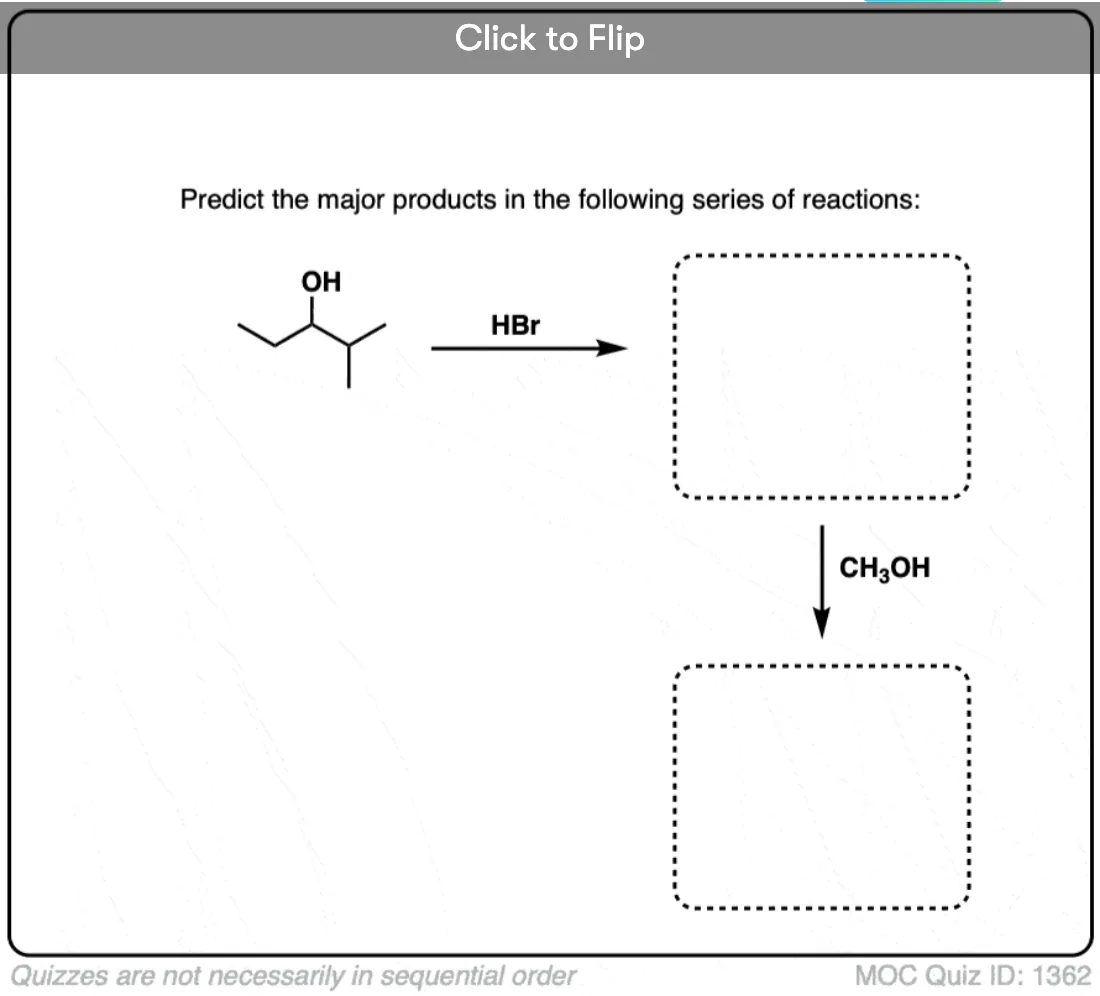
Become a MOC member to see the clickable quiz with answers on the back.
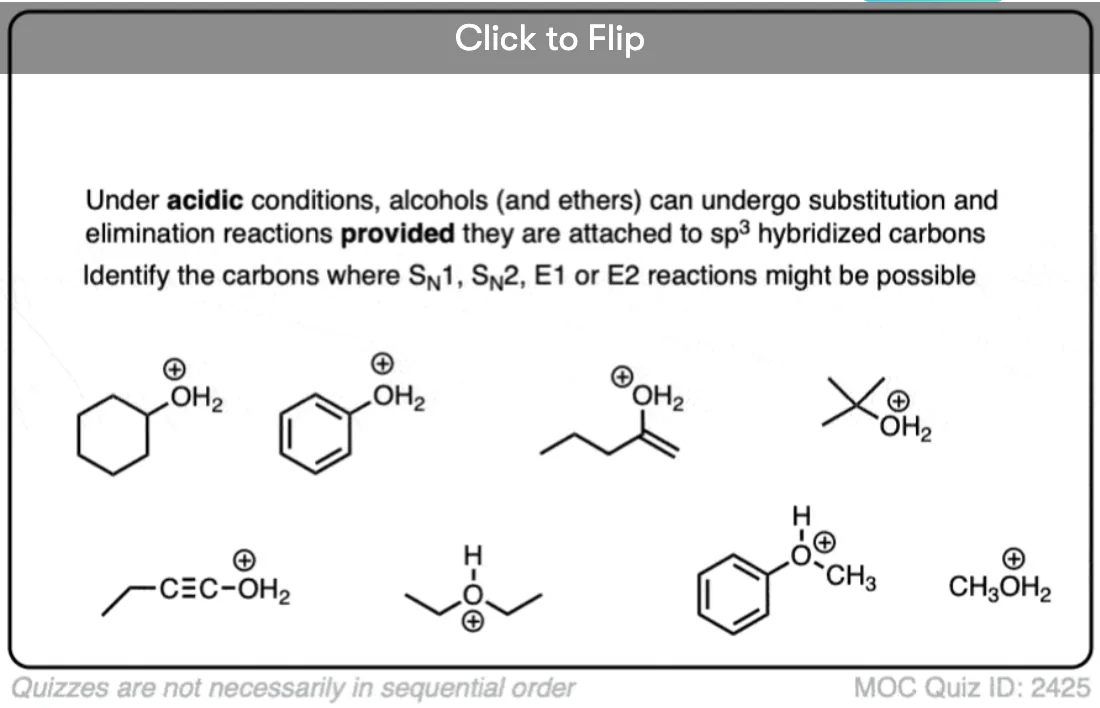
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
Alkyl chlorides from alcohols:
- -BUTYL CHLORIDE
James F. Norris and Alanson W. Olmsted
Organic Syntheses, Coll. Vol. 1, p.144 (1941); Vol. 8, p.50 (1928).
DOI: 10.15227/orgsyn.008.0050
An example of an SN1 conversion of tert-butanol to t-butyl chloride with HCl. This is from Organic Syntheses, a source of reliable and independently tested organic chemistry experimental procedures.Alkyl iodides from alcohols: - Reaction between unsaturated alcohols and potassium iodide in the presence of polyphosphoric acid
Richard Jones and J. B. Pattison
J. Chem. Soc. 1969, 1046
DOI: 10.1039/J39690001046
This paper uses KI + phosphoric acid to generate HI in situ, which converts alcohols to iodides. - 1,6-DIIODOHEXANE
Herman Stone and Harold Shechter
Synth. 1951, 31, 31
DOI: 10.15227/orgsyn.031.0031
A procedure from Organic Syntheses, a source of reliable and independently tested synthetic organic experimental procedures, for converting alcohols to iodides with KI + PPA (polyphosphoric acid). - Synthetic methods and reactions. 63. Pyridinium poly(hydrogen fluoride) (30% pyridine-70% hydrogen fluoride): a convenient reagent for organic fluorination reactions
George A. Olah, John T. Welch, Yashwant D. Vankar, Mosatomo Nojima, Istvan Kerekes, and Judith A. Olah
The Journal of Organic Chemistry 1979, 44 (22), 3872-3881
DOI: 1021/jo01336a027
Pyridinium poly(hydrogen fluoride), also known as PPHF or “Olah’s reagent” can be used as a Bronsted acid for converting alcohols to iodides, along with KI or NaI. - A Simple, Efficient, and General Method for the Conversion of Alcohols into Alkyl Iodides by a CeCl37H2O/NaI System in Acetonitrile
Milena Di Deo, Enrico Marcantoni, Elisabetta Torregiani, Giuseppe Bartoli, Maria Cristina Bellucci, Marcella Bosco, and Letizia Sambri
The Journal of Organic Chemistry 2000, 65 (9), 2830-2833
DOI: 10.1021/jo991894c
Lewis acids can be used for this reaction instead of a Bronsted acid, allowing for milder reaction conditions. - Direct conversion of alcohols into the corresponding iodides
Reni Joseph, Pradeep S. Pallan, A. Sudalai, T. Ravindranathan
Tetrahedron Lett. 1995 36 (4), 609-612
DOI: 10.1016/0040-4039(94)02315-3
Elemental iodine (I2) can also be used for directly converting alcohols to iodides.Alcohols can be converted to alkyl bromides with PBr3: - Convenient synthesis of labile optically active secondary alkyl bromides from chiral alcohols
Robert O. Hutchins, Divakar. Masilamani, and Cynthia A. Maryanoff
The Journal of Organic Chemistry 1976, 41 (6), 1071-1073
DOI: 1021/jo00868a034 - Synthesis of Optically Active Alkyl Halides
Harry R. Hudson
Synthesis 1969, 112-119
DOI: 1055/s-1969-34195
The main utility of PBr3 is that it allows the conversion of chiral alcohols to bromides with inversion of configuration without rearrangement, as the above two papers demonstrate. They also illustrate the mechanism of the reaction, going through the intermediate alkyl phosphites. - TETRAHYDROFURFURYL BROMIDE
L. H. Smith
Org. Synth. 1943, 23, 88
DOI: 10.15227/orgsyn.023.0088
This procedure from Organic Synthesis, a source of reliable and independently tested experimental organic chemistry procedures, shows how PBr3 is compatible with ethers.
According to the Quick N Dirty guide for Sub. and eli. , in case of 2 degree alcohol when we will treat it with HCL, we get a good leaving group(H2O) and a strong nucleophile(Cl-),so the major pathway should be SN2 but not SN1.How in this case we have to assume SN1?
Sir, may I ask why dry hydrogen halides are used as opposed to aqueous solutions of HX?
H2O will react with hydrogen halides to give H3O+ X-. The product will likely be the alcohol, not the halide.
Sir I have a small confusion. When we use lucas reagent ie ZnCl2 the oxygen of the alcohol coordinates to the zinc center which converts it into a good leaving group. But does using ZnCl2 + HCl in case of primary alcohols promotes Sn1? Cause I have seen a bunch of problems where they have asked to write the product when a primary alcohol is treated with lucas reagent but in all of them they have shown 1,2 shifts which can’t take place unless Sn1 is followed. But apparently as you said and I found in many other sources Sn2 should obviously be taking place.
You can think of the hydride as acting a bit like a nucleophile in an SN2 reaction where the hydride shift happens about the same time as breakage of the C-O bond. That’s probably the best way to look at it.
I am confused how reaction of lucas reagent with primary alcohol with one branching at the beta carbon proceeds (say ch3ch2ch(ch3)ch2oh ). Does it follow Sn1 or Sn2
Hi. In the Lucas reagent synthesis of methyl chloride fr. MeOH + HCl + ZnCl2… Does the excess water in HCl lead to the formation of dimethyl zinc chloride or something else, at all? The answer is two complex for my pea brain, haha
It wouldn’t be dimethyl zinc, since the methyl group in dimethyl zinc is similar in basicity to that of a Grignard reagent (and will be destroyed by water just as easily). Zinc does form strong bonds with oxygen, however…
Why is this resaction considered an nucleophilic substitution and not an electrophilic substitution?
In a substitution reaction, a bond breaks and forms on the same carbon. The reactant which contains that carbon is called the “substrate”.
In nucleophilic substitution, the substrate reacts with a nucleophile (e.g. an alcohol) and a new C-O bond forms and a C-X (carbon-halide) bond breaks.
There is also a reaction called electrophilic substitution. In electrophilic substitution, the substrate (e.g. benzene) reacts with an electrophile (e.g. Br+) resulting in formation of a C-Br bond and breaking of a C-H bond. Still substitution, but since the substrate is reacting with an electrophile, it’s called “electrophilic substitution”.
There is also such a thing as free radical substitution.
Hi,
How would reaction change if we have 2-bromo-1-propanol react with HBr ? instead of just propanol ?
Not the best example, since I’m not sure that’s a very stable compound (it easily snaps shut to form an epoxide) but eventually the alcohol would still be replaced with Br, to obtain 1,2-dibromopropane.
in Case 2 a) – the alkyl shif,
Is CH2 missing????
Yes fixed. Thanks!
Thank you so much for putting in a lot of efforts and bringing masterpieces to us. Your notes have always helped me and makes Organic Chemistry super fun and interesting as well.
Thanks Sheetal. Glad you find the site useful.
sorry, figure in section (Case 2 a) has an error
Yes, fixed. Thanks for catching it.
I wanna ask why we use nucleophilic substitution in the formation of alkyl halide rather then electrophilic?
The advantage is that substitution is more predictable. You know that the carbon-nucleophile bond will form on the carbon bearing the leaving group. In contrast, when performing electrophilic addition on an alkene, regioselectivity can be a problem. The major product will be determined by Markovnikov’s rule, and even then the selectivity can be bad (4:1). What if you want the anti-Markovnikov product? Or what if you have an alkene where both ends are equally substituted? These problems don’t arise with substitution.
When observing the reaction of butan-2-ol with HCl to produce 2-chlorobutane and water, what can we expect to see? Or will there be no observable reaction? And is it true that when tertiary alcohols react with HCl, the substance becomes more turbid yet with primary alcohols, the substance remains colourless/less turbid than secondary or tertiary reactions? Or does that only pertain to the Lucas Reagent?
Anhydrous Zncl2 is used in the preparation of alkyl chloride from alcohol and conc. HCL. Why?
It’s a good Lewis acid, coordinates to oxygen and assists with oxygen acting as a leaving group.
Conc. HI is not preferred in the preparation of an alkyl halide from an alcohol. Why?
It *will* work on paper, but in the lab, concentrated HI (48%) is nobody’s idea of a fun reagent to work with. There are many other ways. Use them.
Hi, I believe you have an error on the alkyl shift photo. Going from the second to last step to the final you lose a methyl group and in its place is a hi-lighted blue.
Yes, fixed. Thanks!
Is it necessary to use ZnCl2 in the reaction of HCl and Alcohols for preparing Alkyl Halides or can we simply react Alcohols with HCl for preparing Alkyl Halides? Why?
You’re talking about the Lucas test. Conversion of an alcohol to an alkyl halide does not strictly require ZnCl2, although that does speed up the reaction.
I think I’ve spotted a typo. In case 2a), the last structure should have a blue Methyl group rather than a blue H.
Thanks for a great page!!
Yes. Fixed. Thanks!
Possible to do ring expansion on cyclobutane to cyclohexane? Is there a point where a methyl shift is preferred over ring expansion, such as when rings become very large?
Not possible to go from cyclobutane to cyclohexane directly, since the carbocation would have to be adjacent to the 4 membered ring and it always involves a 1,2 shift.
When ring strain is not an issue (carbocations on 5, 6, 7 membered rings) then you can have mixtures of alkyl shift and ring expanded/contracted products.
For making alkyl halide from alcohol, HX is used. This HX is dry and made by reacting NaX with H2SO4 or in case of HI with H3PO4. So why NaX is not used directly to convert alcohol to alkyl halide?
Because OH- is a terrible leaving group compared to its conjugate acid (H2O).
PKa value of HCl is less than for H3O+. So, Cl- remains a weaker base than water. You mentioned in one of the older articles on Sn2 that a weaker base cant displace a stronger base. How can primary alcohols then react with HCl via an Sn2??
Good question.
The “weaker base can’t displace a stronger base” applies, as a rule of thumb, when the difference in pKa is about 8 or greater. When the difference is less, there will be some equilibrium between the two acids, however minor.
As it turns out, chloride ion is actually superior nucleophile to water, reacting with alkyl halides at faster rates. Practically, there will be water as a byproduct of a substitution reaction which might lead to some minor reversibility. This can be removed by adding an excess equivalent of HCl, which will protonate H2O to give H30+ (which of course is a much poorer nucleophile).
Hey James! When is the next article coming out?
I was wondering how to make a anti-Markovnikov halide from an anti-Markovnikov alcohol. Thanks for your help!
Well, this is precisely one situation where you’d want to perform nucleophilic substitution of an alcohol versus formation of the alkyl halide via addition of HX to the alkene.
Great article! Which software do you use for making molecule structures?
ChemDraw. Most educational institutions have a site licence