Carbohydrates
Reactions of Sugars: Glycosylation and Protection
Last updated: June 2nd, 2026 |
Finally, Some Reactions Of Sugars: Glycosylation And Protection
In this post we introduce some simple reactions of sugars, especially glycosylation and protection:
- Formation of “glycosides” – just a different name for acetals when they exist at the “anomeric” carbon of sugars
- Hydrolysis of glycosidic bonds (glycosides), which is exactly like hydrolysis of acetals (aqueous acid)
- Reactions on the -OH groups of sugars, including protecting groups
- How to “deprotect” the anomeric (C-1) carbon selectively
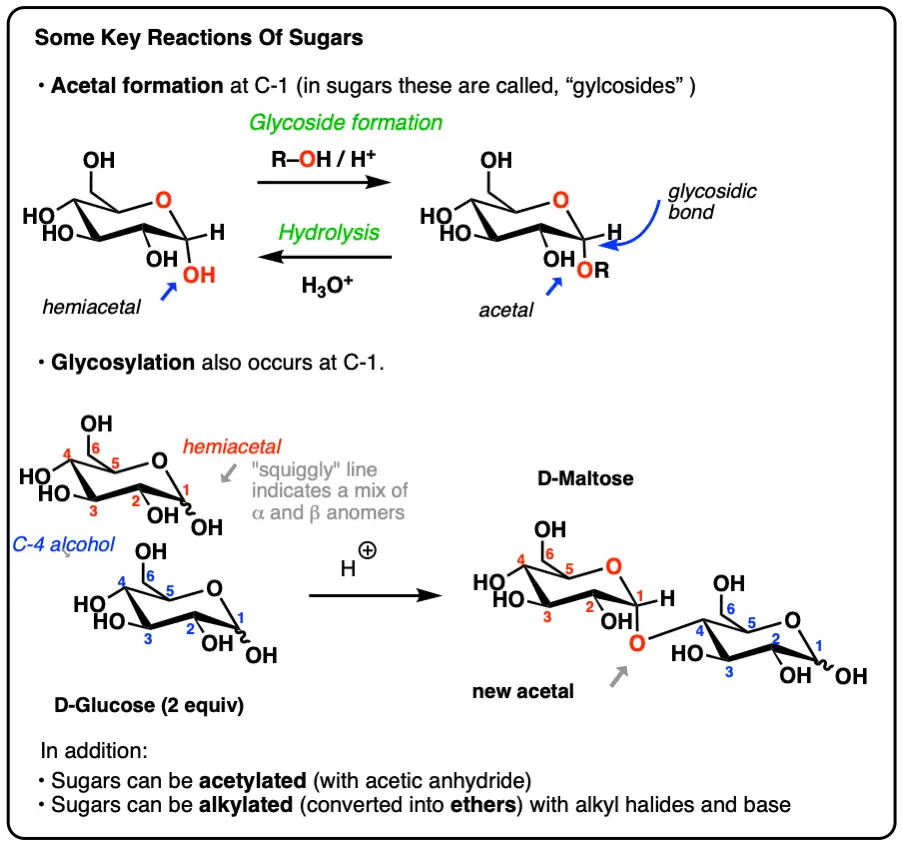
Table of Contents
- Carbohydrates and Sugars: Finally, Some Reactions!
- Reactions At The Hemiacetal (Anomeric) Carbon: Formation And Hydrolysis Of “Glycosides”
- Hydrolysis of Glycosides with Aqueous Acid
- Formation of Disaccharides (In Theory, If Not In Practice)
- Hydrolysis of The Glycosidic Bond Of Disaccharides
- Reactions Of The Alcohol Groups Of Sugars: The Problem Of Selectivity
- A Brute Force Way Around Selectivity: Excess Reagent
- Ethers At C-1 (The Anomeric Carbon) Can Be Cleaved Selectively
- How To Get Around The Selectivity Problem? A Glimpse Into Org 3
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Carbohydrates and Sugars: Finally, Some Reactions!
If reactions are the meat of an organic chemistry course, then nomenclature is the bun – and you’d be forgiven for thinking that the content in this chapter on sugars has been a little, er, “carb-heavy” so far.
A lot of nomenclature and not many reactions!

We’re going to try to fix this deficiency in our chemistry diet today with a discussion of the key reactions of sugars.
The reactions of sugars we will cover really boil down to two main categories.
Part 1: Reactions of the anomeric (hemiacetal) carbon
The anomeric carbon of a sugar can form and break acetals. That’s about it.
- Formation of acetals (“glycosides”), including disaccharides and polysaccharides
- Hydrolysis (cleavage) of acetals (“glycosides”)
Part 2: Reactions of carbohydrate hydroxy groups (alcohols)
The hydroxy groups of sugars can perform all the reactions of alcohols (e.g. ether formation). The trick is getting the right one to react! d
- Ether formation (non-selective, except in special cases with the primary (C6) alcohol)
- Ester (acetate) formation (non-selective) with Ac2O.
We’ll finish up with a few remarks about how reactions of carbohydrates really take us to the limits of what we study in “Org 2” and how it points the way to “Org 3”.
2. Reactions At The Hemiacetal (“Anomeric”) Carbon: Formation Of Glycosides
A few chapters ago, we saw how to convert aldehydes and ketones to acetals, via hemiacetals. In the forward direction, this is accomplished by treating the ketone (or aldehyde) with an excess of alcohol in the presence of acid (such as H2SO4).

Simple sugars (e.g., D-Glucose) have a hemiacetal functional group due to the fact that 5- and 6- membered rings readily form between the carbonyl carbon and the hydroxyl groups, a phenomenon known as “ring-chain tautomerism“.
So, the hemiacetal functional group on sugars can also be converted into a full acetal by treatment with an alcohol in the presence of acid.
In carbohydrate chemistry, these acetals have a special name: “glycosides“.
Here’s an example. Treating D-glucose with ethanol and acid provides a product called Ethyl -D-glucopyranoside.
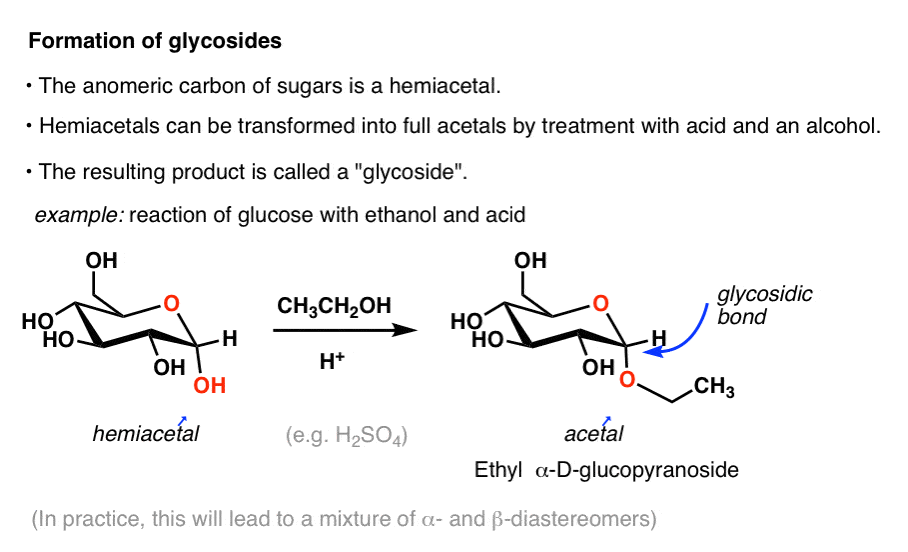
The new C–O bond is called a “glycosidic bond”. In the drawing above, we drew only one anomer (the α), but in practice both will form.
Formation of the glycoside “locks” the ring closed, and it is no longer in equilibrium with the open-chain form, and is therefore no longer a “reducing sugar” [see: Reducing Sugars]. Like all acetals, about the only reaction of significance that it will undergo is hydrolysis back to the starting material with aqueous acid.
3. Hydrolysis of Glycosides With Aqueous Acid
So treatment of a glycoside with water and acid results in the original sugar (as a mixture of anomers, which we can describe with a “squiggly line”).
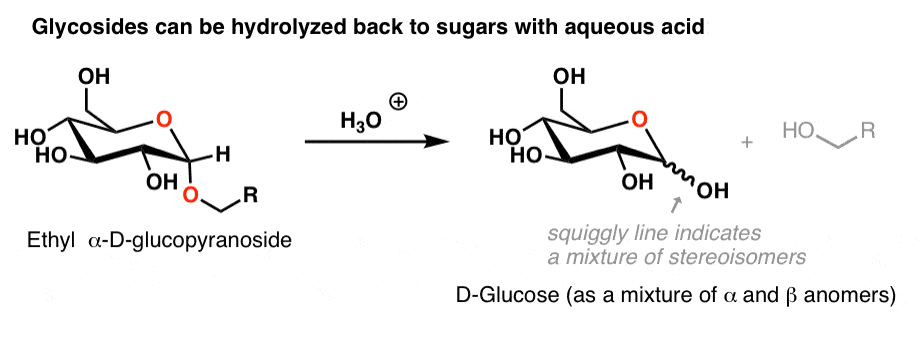
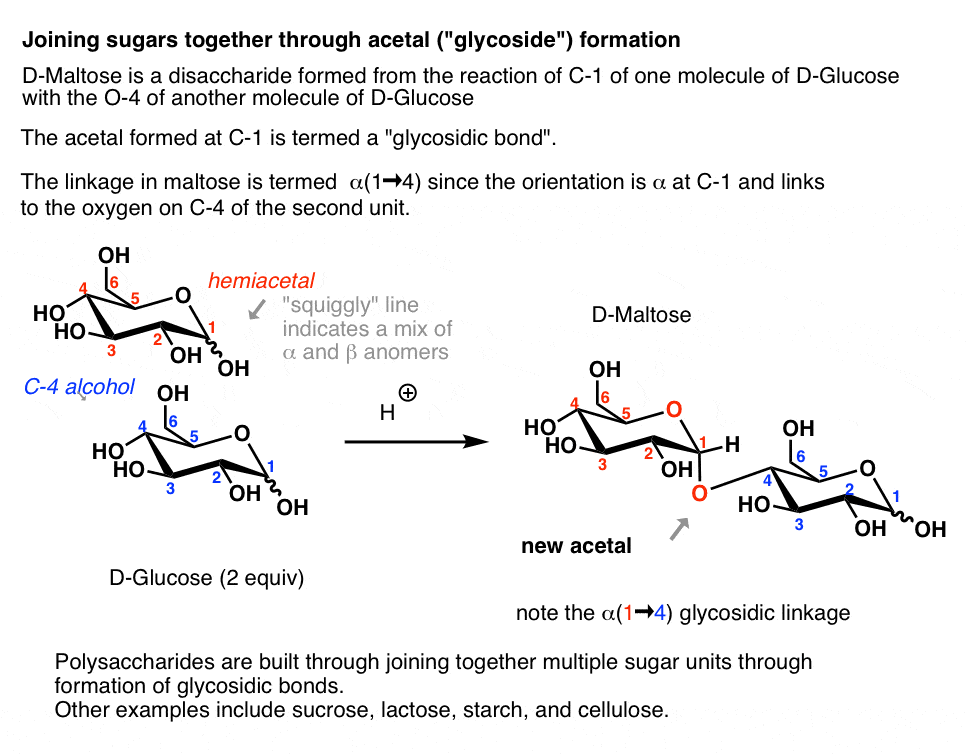
4. Formation Of Disaccharides (In Theory, If Not In Practice)
A particularly important type of glycoside are those formed from the combination of two or more sugars. Maltose, for example, is the acetal formed when C-1 of D-glucose reacts with the C4-OH of another molecule of D-glucose to form an α- glycosidic linkage.
A quick way of describing this linkage is (α-1→4), where 1 and 4 indicate the numbers of the carbons flanking the glycosidic bond, and “α” indicates the stereochemistry at the anomeric carbon. [quick review of alpha and beta]
In theory, it’s possible to carry out this reaction by treating D-glucose with acid:
[To see the mechanism hover here for a pop-up image or click on link for image.
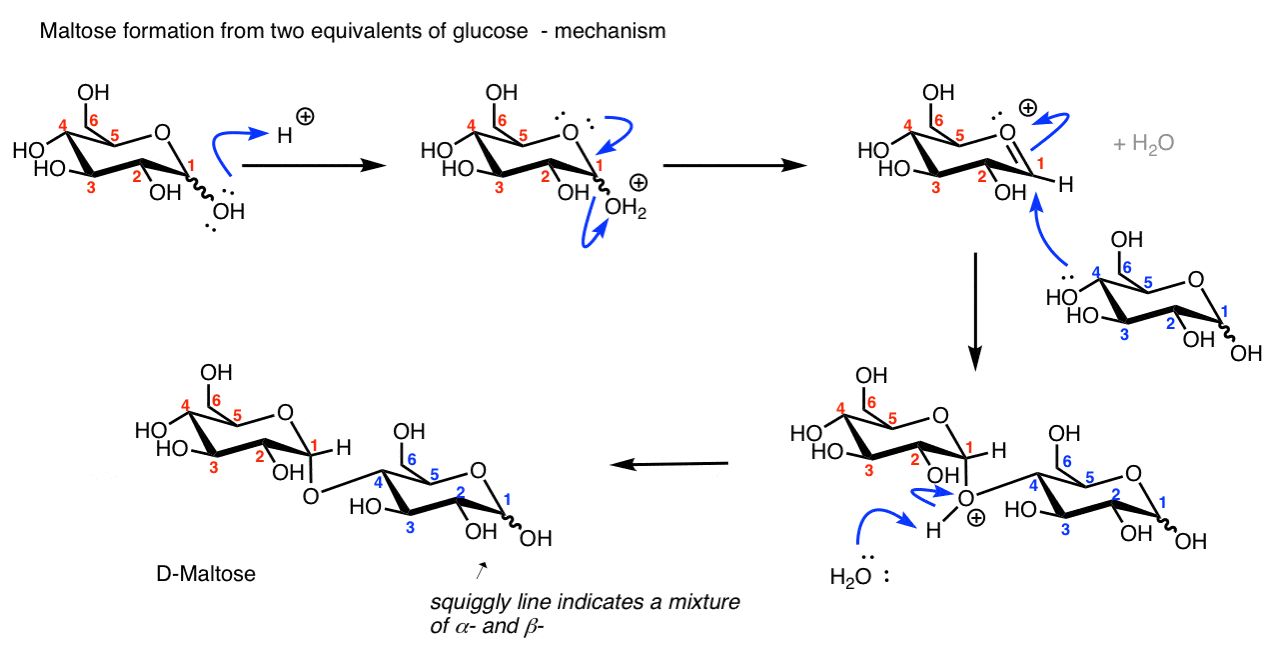{kind=link}
In practice? Well, the drawing above is a bit misleading.
In the lab, adding acid to glucose might get you some maltose, but it will also deliver a ton of other side-products from the reaction of other hydroxy groups (C1-OH, C2-OH, etc.) with the C-1 carbon. (it’s the kind of reaction my friend Jeff would write in his lab notebook as “BFM”, where “M” stands for “mess”).
In nature, very specific enzymes have evolved that combine sugars together with exquisite site-selectivity (“regioselectivity” for C4-OH in this case, versus the other possible alcohols as nucleophiles) and stereoselectivity (α- for maltose).
[For example, it’s very important that the orientation at the C-1 acetal is drawn alpha (α) for maltose. The beta (β) stereoisomer is another disaccharide entirely (cellobiose).]
Similarly:
- sucrose (table sugar) is a disaccharide formed through acetal formation between C-1 of glucose and C-2 of fructose (α-1→β-2 linkage)
- lactose is a disaccharide formed between C-1 of galactose and C4-OH of glucose (β-1→4 linkage).
- Amylose, one of the two components of starch, is a polysaccharide of glucose linked through (α-1→4) glycosidic bonds.
All are built through formation of glycosidic bonds between sugars – in other words, good-old acetal formation.
5. Hydrolysis of The Glycosidic Bond of Disaccharides
Just as with ethyl D-glucopyranoside, above, adding aqueous acid to a polysaccharide will hydrolyze it back into the constitutent sugars.
D-lactose, for instance, can be hydrolyzed back into D-Galactose and D-Glucose with aqueous acid:
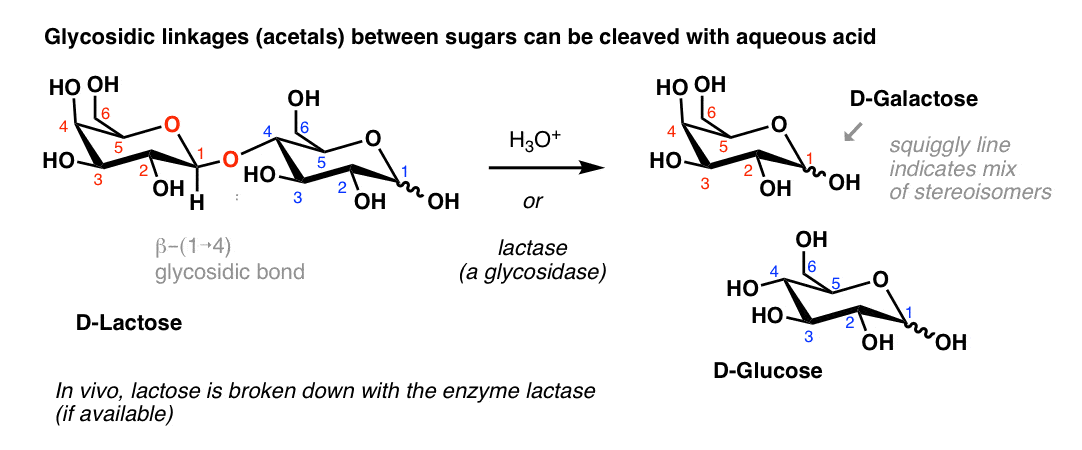
In organisms, enzymes perform the hydrolysis of glycosides – these are called glycosidases . The familiar condition of “lactose intolerance” is a result of the body lacking the necessary enzyme (lactase) to break down the glycosidic bond of lactose.
Enzymes can be exquisitely sensitive to stereochemistry. Starch, a polymer of glucose with (α-1→4) glycosidic bonds, is easily broken down in our bodies to units of D-Glucose, but cellulose [with (β-1→4) glucose linkages] is not. [Fun fact: Grass-eating mammals like cows rely on microorganisms in their gut to convert cellulose to glucose].
6. Reactions Of The Alcohol Groups Of Sugars: The Problem Of Selectivity
What kind of reactions do the hydroxy groups of sugars undergo? They’re essentially just alcohols, right?
We’ve explored many important reactions of alcohols previously. So let’s say we wanted to do a meat-and-potatoes reaction of alcohols – the Williamson Ether synthesis – on the C3-OH of glucose.
You may recall that to perform a Williamson, all we need to do is add base and an alkyl halide, and voila – an ether forms.
What could possibly go wrong?
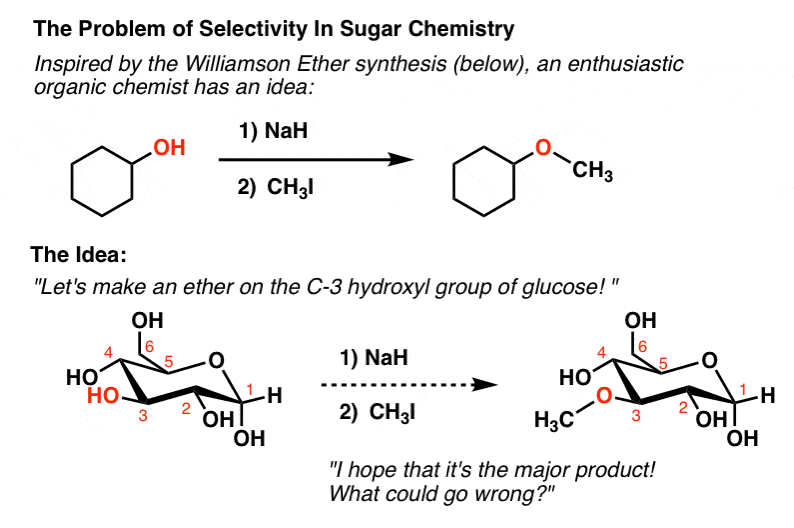
Well, at least four things could go wrong, actually. The C-3 OH isn’t significantly more selective toward CH3I than any of the other four hydroxyl groups.
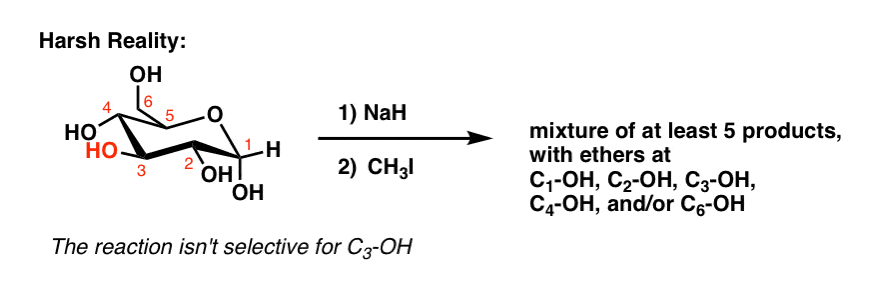
The result is a mixture of products which requires a tedious separation.
Even with all the advances of modern organic chemistry, there’s no known way to get the Williamson ether synthesis to occur selectively with, say, the C3-OH of glucose in good yields without also forming ethers at the other hydroxyl groups.
This might seem hard to believe but it is true. The more we learn about organic chemistry, the more we learn to appreciate just how incredible Nature is in devising extremely selective catalysts for reactions on sugars at specific sites without the use of protecting groups.
There’s still lots to discover in organic chemistry!
[There is one reliable way to put a single ether group on a sugar like glucose. The C6-OH is a primary alcohol, and therefore less sterically hindered than the other alcohol groups. It is possible to use a very bulky alkyl halide like trityl chloride and have it react selectively there. See Note 1 ]
No matter what reaction you try – oxidation, acetylation, halogenation, or silylation – you’ll almost always run into this problem with unprotected sugars. A workaround is in order. [Note 2]
7. A Brute Force Way Around Selectivity: Use Excess Reagent
If we abandon all hopes of selectivity, good yields can be obtained by just treating the sugar with a vast excess of a given reagent. For example if we treat glucose with excess methyl iodide in the presence of base, we get the penta-ether in high yield. [We sometimes call this, “exhaustive methylation”]. [Note 3]
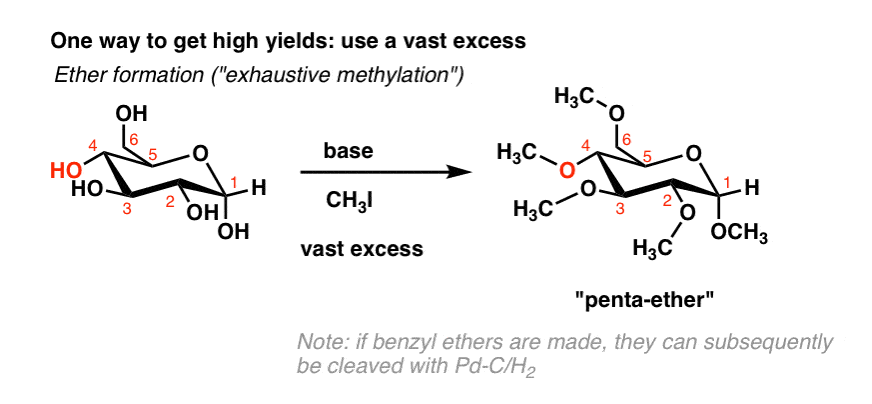
Using an excess of acetic anhydride, one can also form the “penta-acetate”:
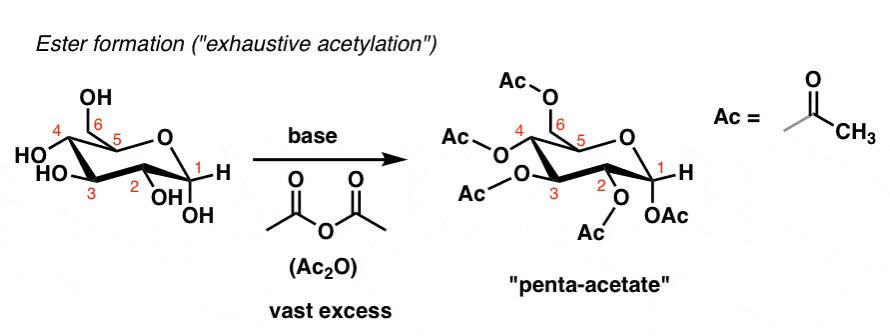
Some very familiar plastics from modern life are the result of treating the polysaccharide cellulose with various reagents under “exhaustive” conditions:
- Cellulose acetate film (“safety film”, from treating cellulose with excess acetic anhydride)
- Celluloid (the first plastic, originally used as film stock, obtained through nitration of cellulose)
- Rayon (where cellulose is treated with excess carbon disulfide to form xanthates)
8. Ethers At C-1 (The Anomeric Carbon) Can Be Cleaved Selectively
One note. It’s possible to free up the C-1 hydroxyl group on the penta-ether through acidic hydrolysis, because the anomeric carbon (C-1) is part of an acetal.
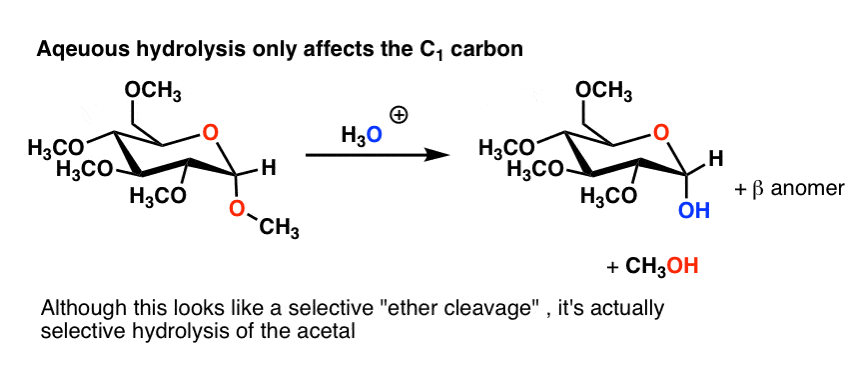
This might look like an ether cleavage (which usually requires a very harsh acid like H-I) but is actually just hydrolysis of an acetal. (A good “trick” exam question!).
9. How To Get Around The Selectivity Problem?
Until we develop catalysts that rival enzymes in their ability to react selectively with the hydroxyl groups of sugars, we’re left with protecting group strategies.
We briefly covered protecting groups in the chapter on alcohols. [see: “Protecting Groups For Alcohols“] With sugars, protecting group strategies are taken to a whole new level. It’s a big topic – one we don’t have time for in this post today. [Here is a start, though.]
A Glimpse Into The Land Beyond (Org 3)
I’d argue that protecting group strategies of sugars is really a topic for what you might call “Org 3”.
What does that mean?
Org 1 and Org 2 are courses that introduce the properties of the various functional groups and their major reactions. The functional groups are largely treated in isolation.
It’s a bit like chess. Org 1 and Org 2 is like learning the rules of how the game is played, and how the pieces (functional groups) move.
In Org 3, life gets more complicated. We learn how to devise strategies to deal with the many real-life situations where a molecule has multiple reactive functional groups (like glucose!) , and we need to selectively form a bond at just one of them.
Or, in chess terms, it’s where you start to learn how to coordinate all your pieces together in an overall strategy.
In the meantime, I hope this post has been more beef and less bun.
Notes
The trityl group (triphenylmethyl) is so frickin’ massive that it tends only to fit on primary alcohols, so it will react selectively with the C-6 hydroxyl group:
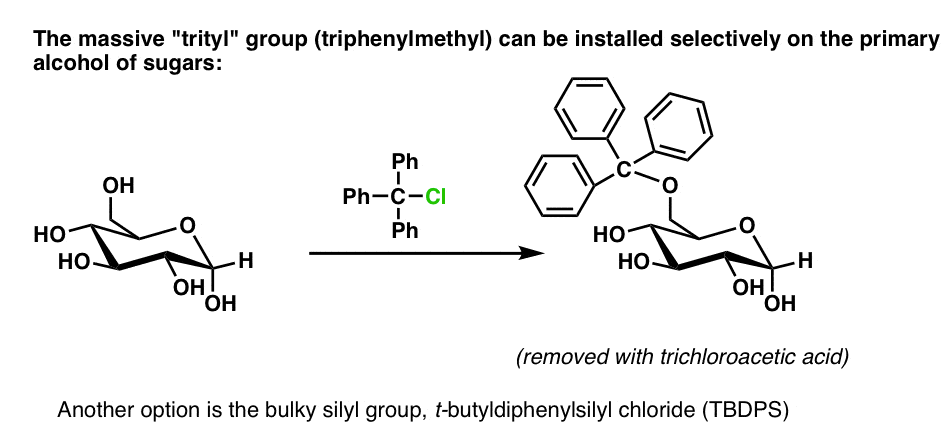
I can say from experience that it also feels really good after you put it on, since the mass of your product skyrockets (“started with 400 mgs, and now I almost have a gram. YESSS!”). This feeling disappears, however, when it’s removed with trichloroacetic acid. Another option is the bulky silyl group t-butyldiphenylsilyl chloride (TBDPS).
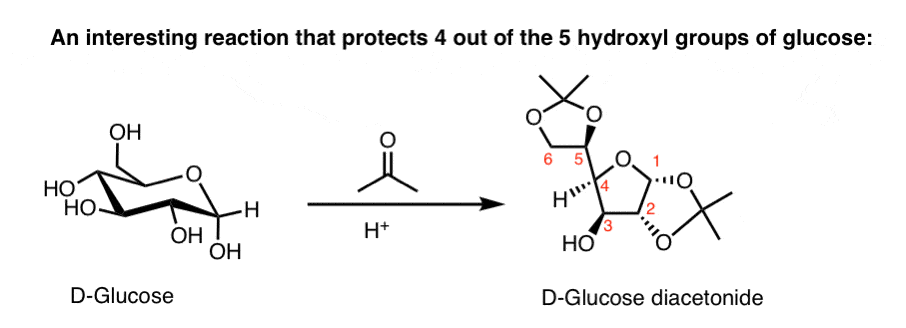
Note 2. This is way above Org 2, but here’s a very useful protection sequence that allows for selective functionalization of C-3 of D-glucose. Treatment with acetone and acid (one can also use a Lewis acid, like CuSO4) ties up 4 of the 5 hydroxyl groups as “acetonides” (acetals of acetone). This leaves the C-3 OH, which can then be selectively methylated, if desired. [This was used in the Nicolaou synthesis of Leucomycin A to put a methyl ether on C-3 of glucose. See “Adventures In Carbohydrate Chemistry” for many more wonderful examples. ]
Note 3. A more reversible variant of this reaction forms benzyl ethers instead of methyl ethers (which can be notoriously difficult to cleave). The benzyl ethers can be removed by treatment with Pd/C and hydrogen gas.
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
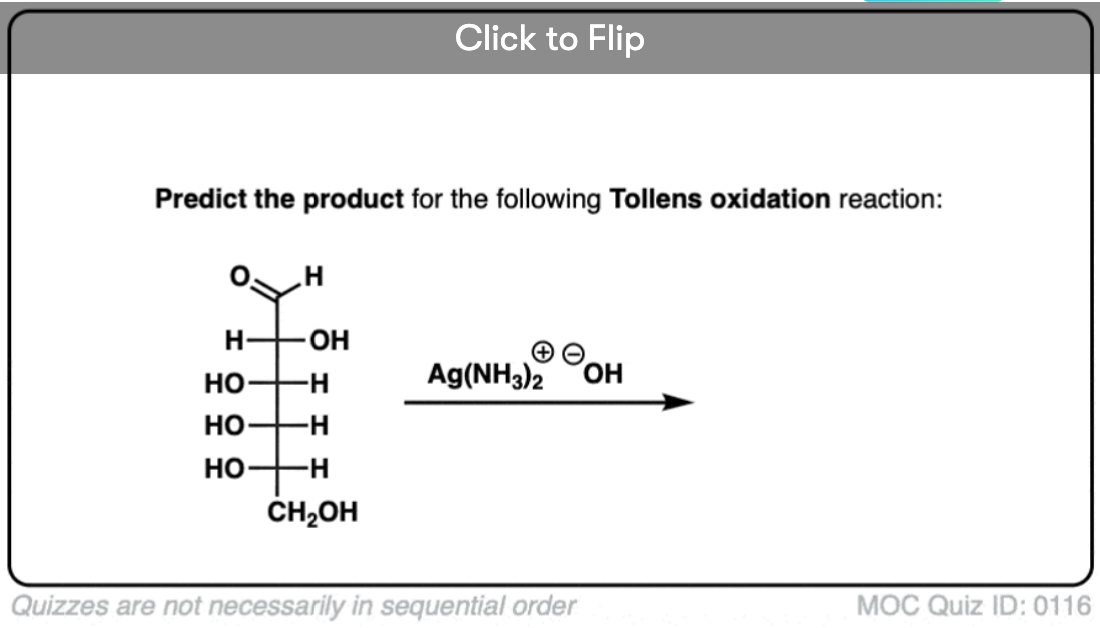
Become a MOC member to see the clickable quiz with answers on the back.
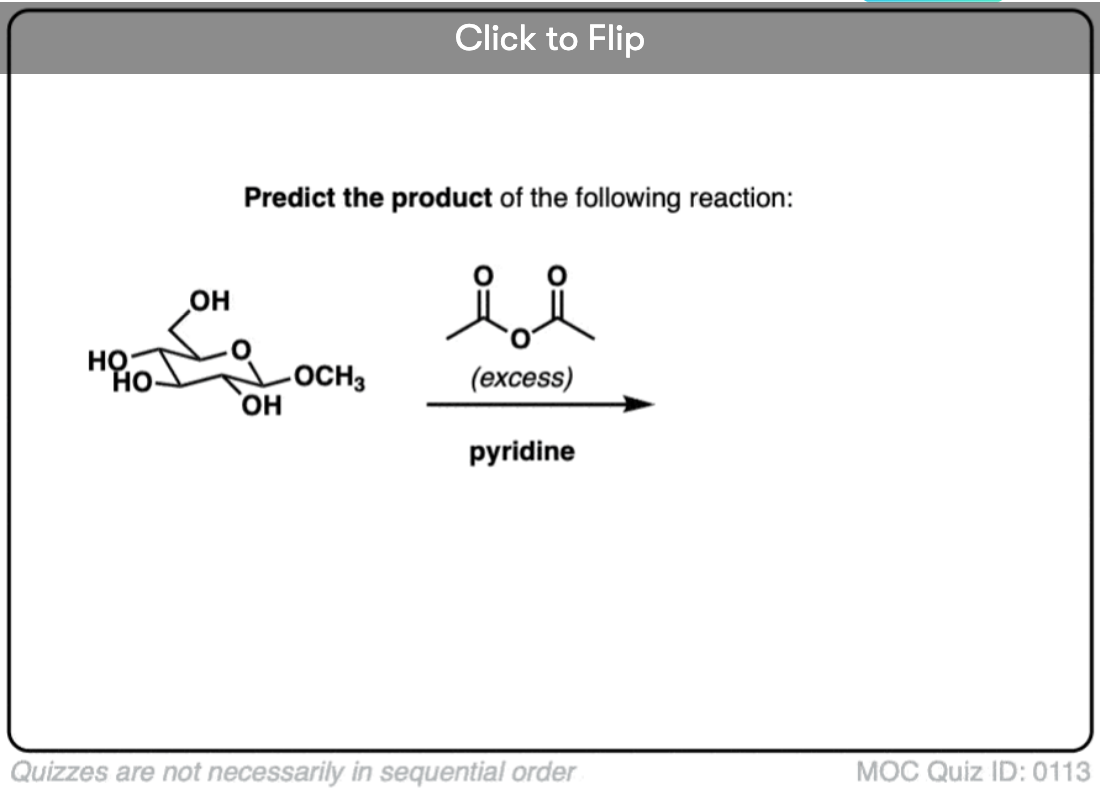
Become a MOC member to see the clickable quiz with answers on the back.
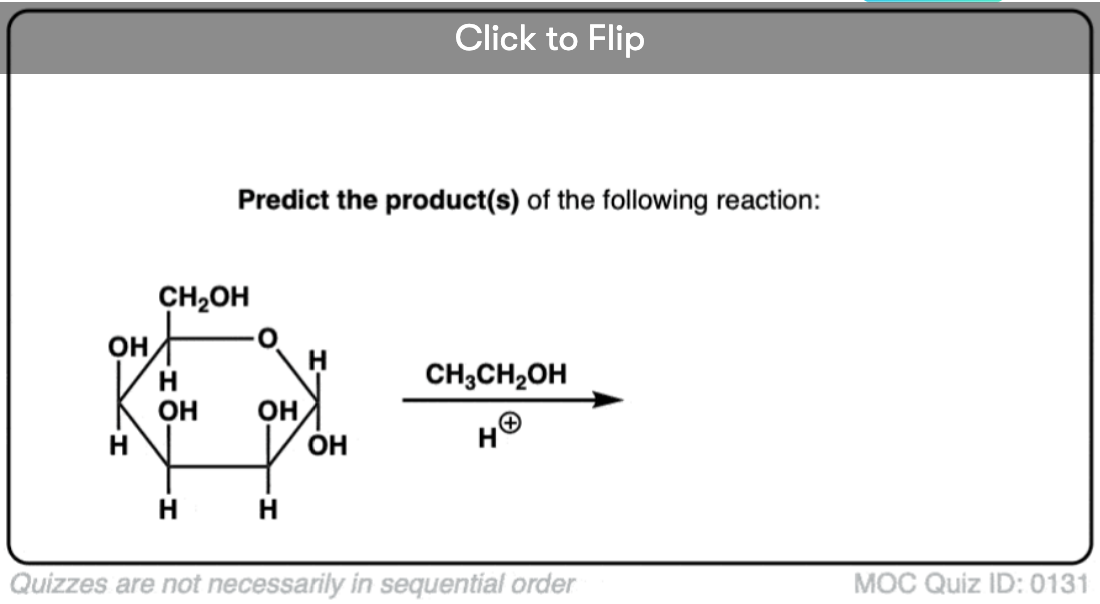
Become a MOC member to see the clickable quiz with answers on the back.
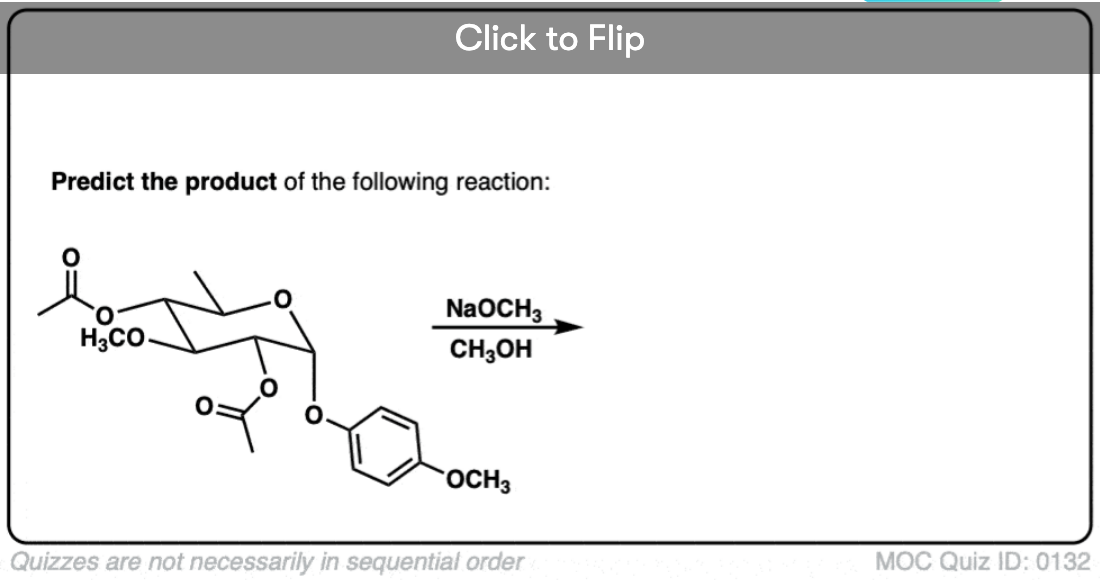
Become a MOC member to see the clickable quiz with answers on the back.
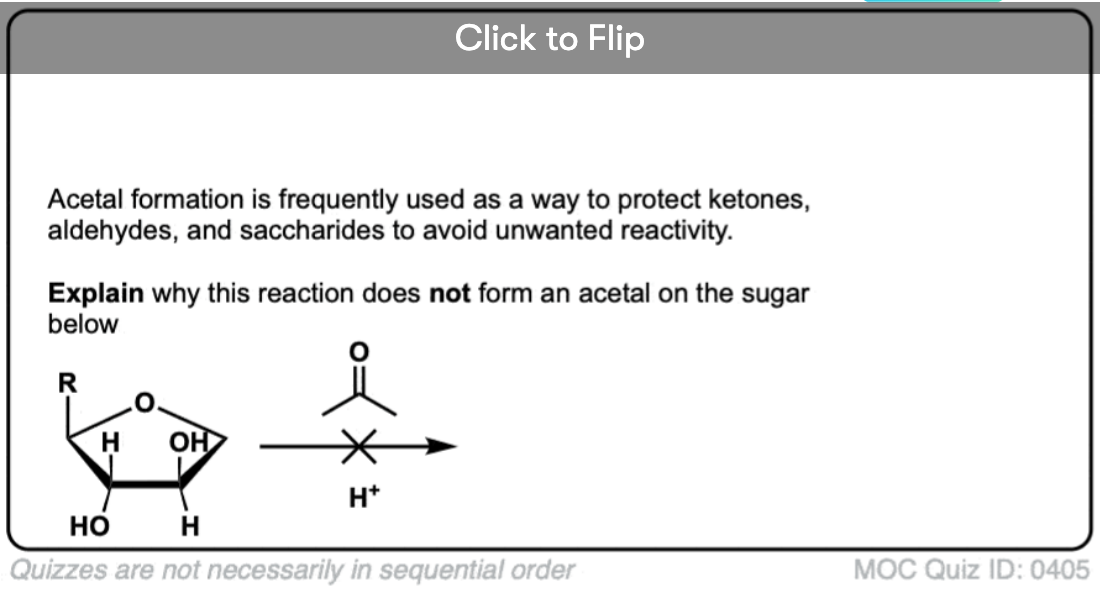
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- Esterification of the primary alcoholic groups of carbohydrates with acetic acid: a general reaction
R. B. Duff
J. Chem. Soc., 1957, 4730-4734
DOI: 10.1039/JR9570004730
An early paper showing that selective monoacetylation on the primary hydroxyl is possible, since that is significantly more reactive. - Exhaustive Methylation of Glucosamine
Norbert J. Wojciechowski, Ralph Daniels, and Bernard Ecanow
Pharm. Sci. 1961, 50 (10), 888-889
DOI: 10.1002/jps.2600501024
The abstract states: “A reported synthesis of a glucosamine quaternary derivative was investigated by chemical and infrared spectra methods. Under the conditions reported the glucosamine was found to become degraded and tetramethylammonium iodide was formed” - Applications of Tin-Containing Intermediates to Carbohydrate Chemistry
Bruce Grindley
Adv. Carbohydrate Chem. and Biochem. 1998, 53, 17-142
DOI: 10.1016/S0065-2318(08)60043-8
The regioselective acylation and alkylation of tin acetals is very useful for the selective protection of a number of polyol containing systems, including protection of equatorial over axial hydroxyl groups and for differentiation between two equatorial hydroxyl groups with different steric environments. - Adventures in Carbohydrate Chemistry: New Synthetic Technologies, Chemical Synthesis, Molecular Design, and Chemical Biology
K. C. Nicolaou Prof. Dr. Helen J. Mitchell Dr.
Angew. Chem. Int. Ed. 2001, 40 (9), 1576-1624
DOI: 10.1002/1521-3773(20010504)40:9
A tour de force review by Prof. K. C. Nicolaou (now at Rice U.) that exhaustively covers carbohydrate chemistry and its applications in natural product total synthesis.
when glucose reacts with hno3 , then (considering Fischer structure of glucose) only the aldehyde group and lowermost alcohol group in the structure gets oxidized to produce saccharic acid. so my question is , why won’t other alcohol groups in the structure of glucose get oxidized
Hello James, awesome topic!! My question is about Mesylation/Tosylation or even using Tf2O. Are they still selective on OH-6 like trityl group?
No, you really need a very bulky protecting group like trityl or even tri-isopropylsilyl(TIPS) will also do the job. Tosyl and mesyl and triflyl are not selective enough; they’ll hit everything. I don’t recommend using Tf, as OTf is a *great* leaving group and you’ll likely get a mess.
In the attack of bulky substituent how u took the primary alcohol of C6 as C1 carbon is also having primary oh group as the oh attached to C1 is further jst attached to C2 i.e one alkyl so it’s primary na????
The C1 carbon is not primary; it’s a hemiacetal (a masked aldehyde)
The OH on C-6 is significantly farther away from the rest of the molecule than any of the other OH groups, including the hemiacetal on C1, making it considerably more reactive towards a bulky protecting group like the trityl anion
What if you an acetal coming off the sugar other than the actual acetal involved in a glycosidic linkage, then can you hydrolyze that acetal?
Dear James, thanks for your efforts. I would like to ask you if we react vanillin with D glucose. What should happen?
In the absence of any acid or other reagent, the most exciting thing that can happen is that you form a hemiacetal between one of the glucose hydroxyls and the aldehyde of vanillin. Not very exciting.
Hey James
love your site
What would happen if you reacted d-glucose with R-SH, so a thiol, in acid? d-glucose with two S-R where the carbonyl oxygen was?
James, you’re the god of chemistry man!
I most certainly am not, but if you found this article useful, I’m glad.