Aromaticity
Introduction To Aromaticity
Last updated: June 2nd, 2026 |
What Is “Aromaticity”, Anyway?
In this post we introduce “aromaticity”, a term for describing a collection of three [Note 1] main properties that distinguish benzene from (hypothetical) cyclohexatriene.
- Extremely large resonance energy (36 kcal/mol)
- Delocalized electrons
- Undergoes substitution rather than addition reactions
[This collection of features is not unique to benzene. We’ll describe other aromatic molecules in the next post. See “Rules For Aromaticity“].
In the sections below we go through each of these three features in detail.
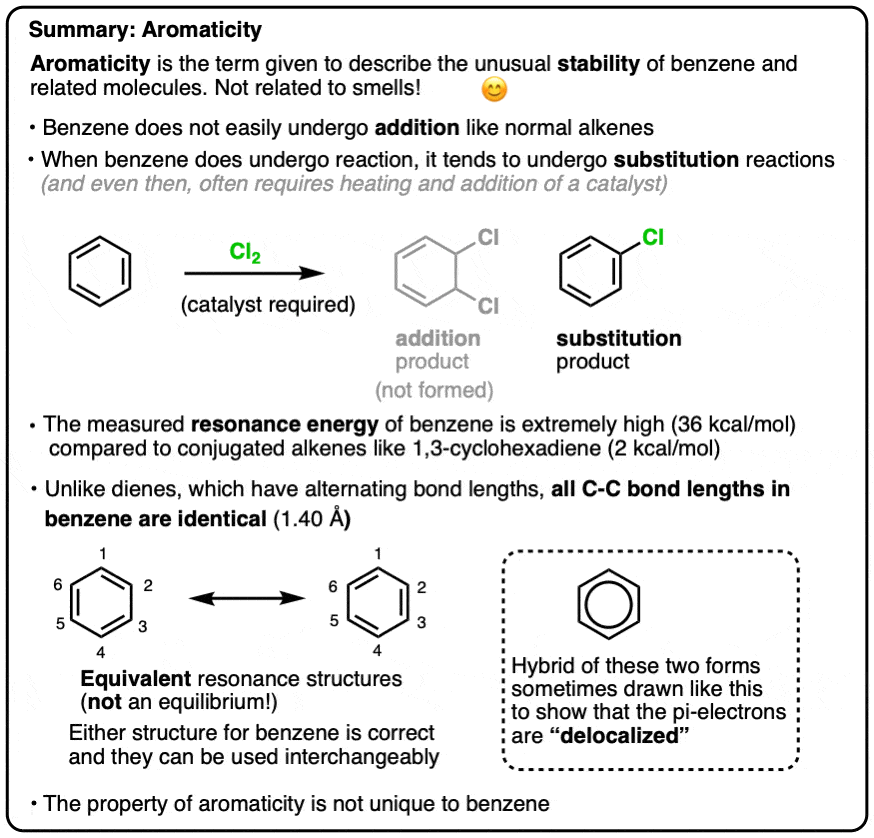
Table of Contents
- The Resonance Energy Of 1,3 Cyclohexadiene Is 2 kcal/mol
- The Resonance Energy Of “Cyclohexatriene” Is 36 kcal/mol (!WOW!)
- The “Emergent Property” Of Aromaticity
- The Structure of Cyclohexatriene Benzene And The “Delocalized” Nature Of Its Pi Bonds
- Benzene Undergoes Substitution Reactions, Not Addition Reactions
- This Collection of Three Special Properties Is Called “Aromaticity”
- Aromaticity Is Not Unique To Benzene
- So What Makes A Molecule Aromatic?
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. The Resonance Energy Of 1,3 Cyclohexadiene Is 2 kcal/mol
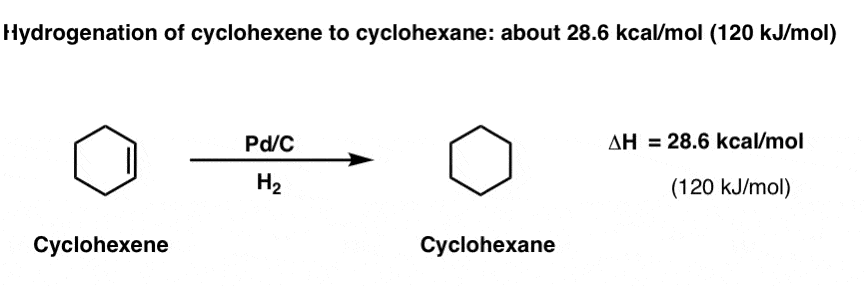
What’s “resonance energy” again? Let’s start with cyclohexene and build our way up.
In case you can’t picture it, cyclohexene is a six membered ring containing a single pi ( π)bond.
If you treat cyclohexene with hydrogen gas (H2) in the presence of a noble metal catalyst such as Pd supported on carbon (Pd/C), you form cyclohexane. The reaction liberates 28.6 kcal/mol (120 kJ/mol) of heat.
If you’re in second semester organic chemistry, there’s no doubt you’ve seen this reaction before.
Now for a slightly more interesting question. What happens to the enthalpy of hydrogenation when we add a second double bond adjacent to the first one: 1,3-cyclohexadiene. (There’s another isomer – 1,4 cyclohexadiene – which we won’t concern ourselves with for this example, because the double bonds are not conjugated).
The enthalpy of hydrogenation in this case is 55.2 kcal/mol (231 kJ/mol) ; almost, but not quite double of that for cyclohexene (2 × 28.6 = 57.2 kcal/mol).
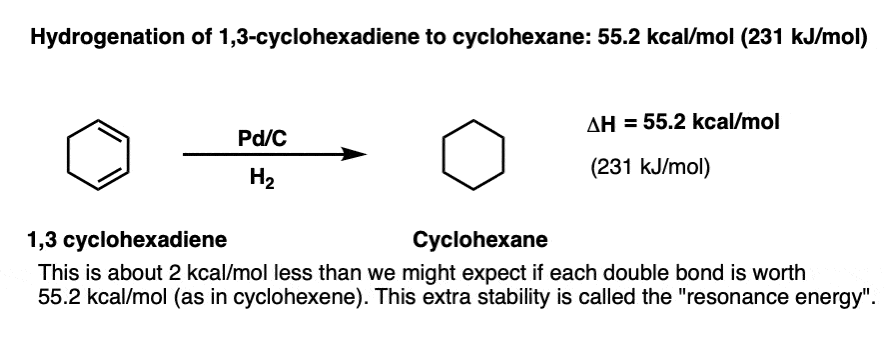
This is still an interesting result, because it tells us that having those two double bonds together results in a little bit of extra stability (2 kcal/mol) versus two isolated pi bonds.
We call this extra stabilization (2 kcal/ mol) “resonance energy” . It’s a consequence of conjugation – extending the p orbital overlap.
2. The Resonance Energy Of “Cyclohexatriene” Is 36 kcal/mol (!WOW!)
Finally, let’s move on to cyclohexatriene. What happens when there are three pi bonds in the ring?
We might naively expect the hydrogenation to liberate about (3 × 28.6 kcal/mol = 85.8 kcal/mol) of heat.
As it turns out, when we try to hydrogenate “cyclohexatriene” under normal conditions (Pd/C, room temperature 1 atm of H2), nothing happens. It’s inert.
Gradually we increase the heat and pressure. Still nothing happens. Cyclohexatriene refuses to hydrogenate.
Something strange is clearly going on here. Clearly, drastic measures are required.
In desperation, we march to the stockroom and break out the heavy artillery.

At 180-220°C and 25-30 atmospheres of hydrogen gas, “cyclohexatriene” finally succumbs to hydrogenation to give us cyclohexane.
The results of the enthalpy measurement are shocking. Recall that we expected the reaction to liberate 85 kcal/mol of heat.
In fact, the reaction liberates 49.8 kcal/mol (208 kJ/mol) of heat. Our guess was off by 36 kcal/mol.
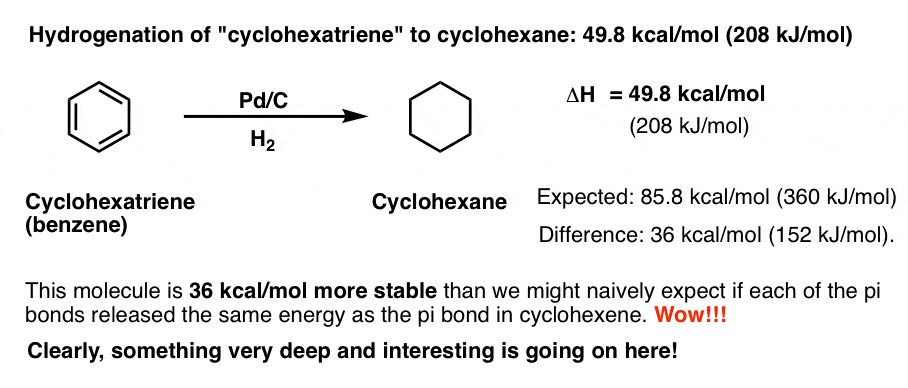
Hot Damn!
3. The “Emergent Property” Of Aromaticity
The heat liberated by hydrogenating cyclohexene and cyclohexadiene grew in roughly linear fashion, but adding that third double bond unlocked an important emergent property.
[We’re going to discuss this special property a lot in the next few posts in this series – it’s called “aromaticity” – but let’s not get too ahead of ourselves.]
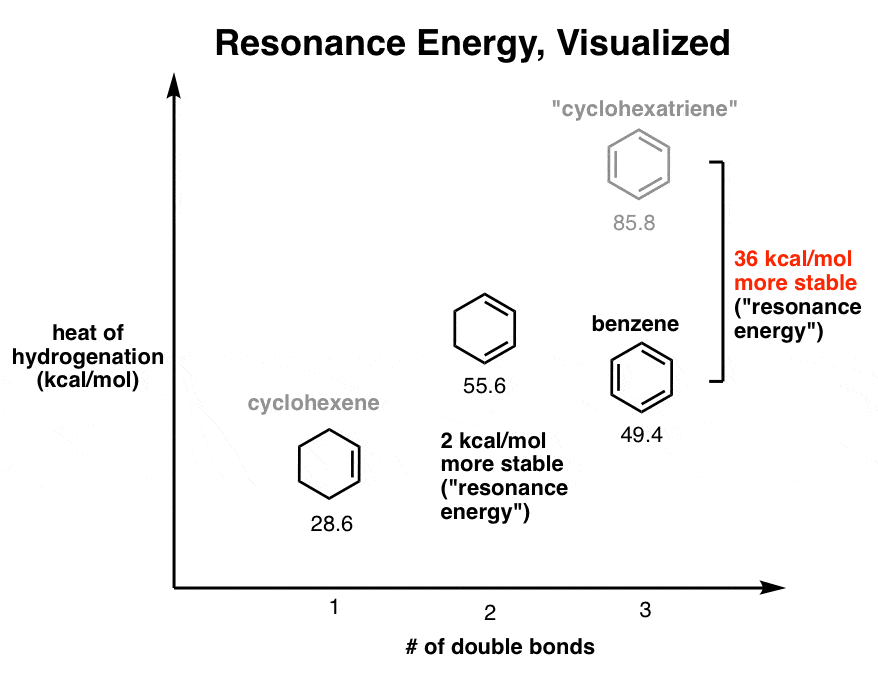
First, it might help to take these energies and visualize them on a graph. Think of it as being a bit like standing on successive rungs of a ladder, with each “rung” representing a new pi bond. The “height above ground” corresponds to the energy released upon hydrogenation of the three successive molecules to the same final product (cyclohexane).
Going from the “second rung” (cyclohexadiene) to the ground state released almost twice as much energy than did going from the “first rung” to the ground state. It was about 3% less (2 kcal/mol) due to resonance energy.
We expected the “third rung” of our imaginary ladder to be another 26 kcal/mol or so higher up. In fact, the third rung is about 6 kcal/mol lower than the second rung.
Have you ever climbed a set of stairs with your eyes closed and, reaching the top, stumbled after your foot expected to land on a step that’s not there? It’s a lot like that.
4. The Structure of Cyclohexatriene Benzene And The “Delocalized” Nature Of Its Pi Bonds
As you probably know by now, our “cyclohexatriene” actually goes by a different name: benzene.
Benzene has been known since at least the Middle Ages. Michael Faraday isolated it and originally determined its composition as C12H6 (later revised to C6H6 when the atomic mass of carbon was corrected) through burning a sample and measuring the samples of CO2 and H2O given off. Here is Faradays sample of benzene, which he named “bicarburet of hydrogen”.
{kind=link}
By the 1860s it was known that the molecular formula of benzene was C6H6, all of the hydrogens were equivalent, and that valence bond theory required that there be 4 bonds to carbon.
Many structures were proposed for this molecule. It fell to August Kekulé in 1865 to propose the correct, cyclic structure of benzene, which may or may not have been inspired by a dream about a serpent eating its own tail.
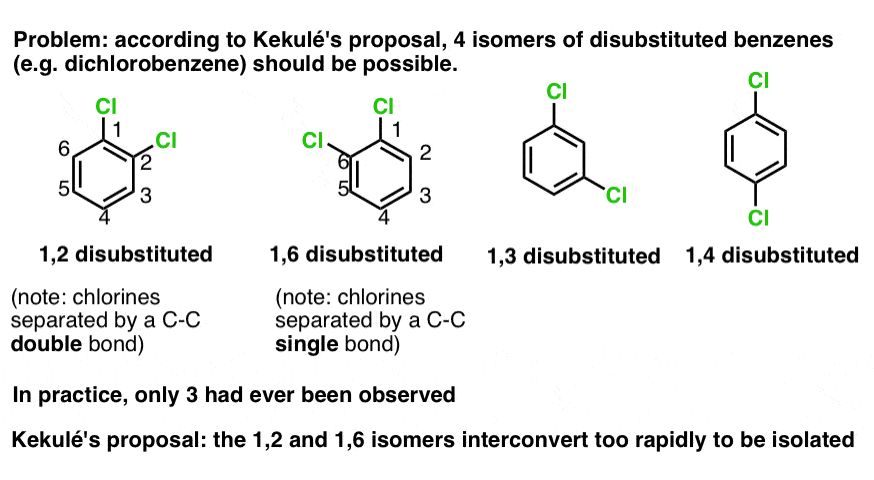
Something still bothered Kekule about this structure, however: disubstituted benzenes always seemed to be missing an isomer.
Take dichlorobenzene for example. From this structure, you’d expect 4 isomers: 1,2, 1,3, 1,4, and 1,6 (the difference between 1,2 and 1,6 is subtle, but they are different nonetheless).
The problem is that only 3 isomers of dichlorobenzene had ever been isolated and characterized.
Kekule’s solution to this problem was to propose that 1,2 dichlorobenzene and 1,6 dichlorobenzene interconvert too rapidly to allow for either to be separated.
It wasn’t until X-ray crystallography was developed that this picture was shown to be incorrect. In 1928 Kathleen Lonsdale showed that all six C-C bond lengths in hexamethylbenzene were equivalent. She later determined the structure of benzene itself, which established the length of each C-C bond as 1.40 Å (140 picometers).
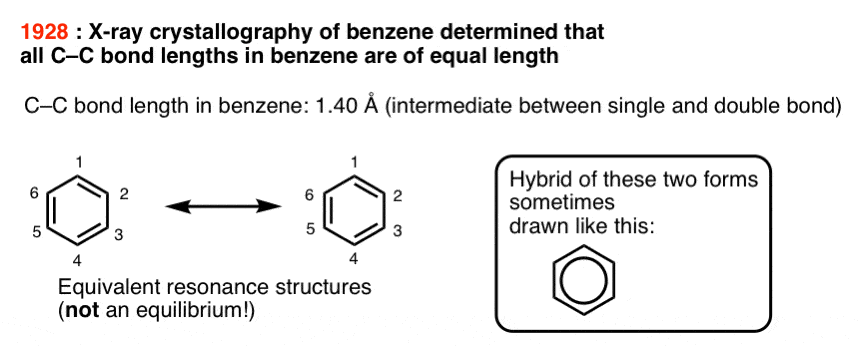
Equal bond lengths is inconsistent with two interconverting molecules.
Rather, it suggests that “1,2” and “1,6” dichlorobenzene are actually resonance forms of the same molecule and the “true” structure of benzene is a hybrid of these two structures.
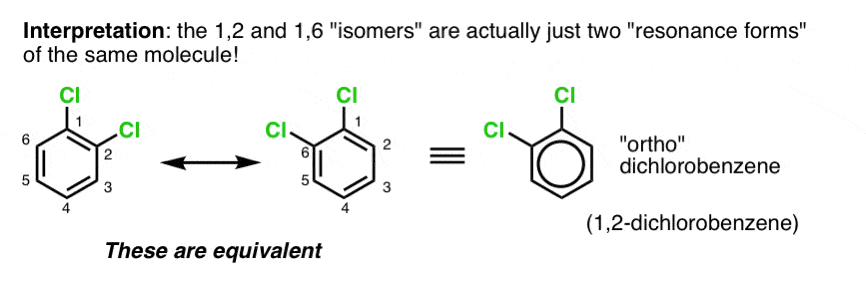
One way of describing this is that the pi electrons in benzene are delocalized throughout the perimeter of the molecule, forming a kind of circle, or torus.

This is sometimes conveyed by drawing benzene as a hexagon with a circle in the middle.
5. Benzene Undergoes Substitution Reactions, Not Addition Reactions
Let’s go back to reactions.
We saw that benzene is a lot less reactive in hydrogenation reactions than what we’d expect from an alkene. In order for hydrogenation to occur, you have to subject it to tremendous heat and pressure.
{kind=link}
You might reasonably ask: does benzene differ in its reactivity from alkenes in other ways as well?
You bet it does.
Here’s one of the most prominent differences.
As we’ve seen, alkenes typically react with electrophiles to give “addition” products (break C-C pi, form two new adjacent single bonds to carbon). Case in point: chlorination (see below).
Benzene is much less reactive toward chlorine than a typical alkene – it generally requires a catalyst in order for chlorination to occur (FeCl3 is a popular choice here). When it does react, instead of giving addition products, we instead see a substitution product (break C-H, form C-Cl).
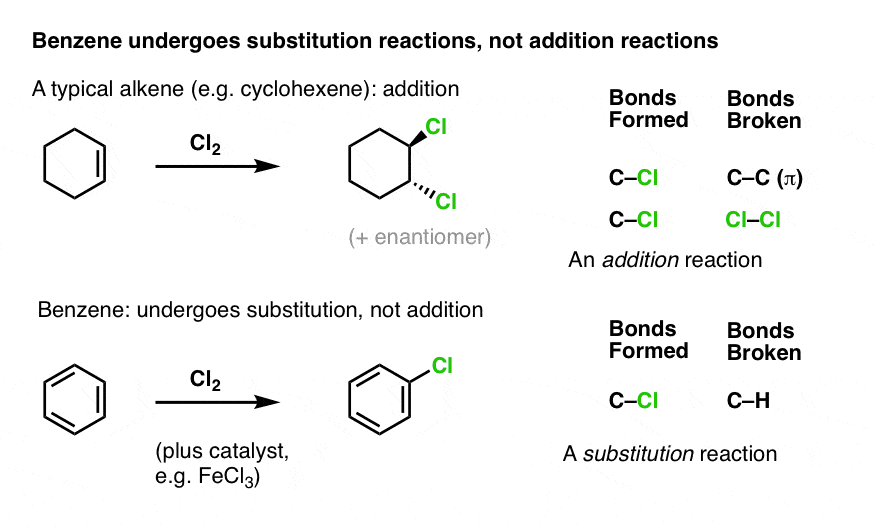
This turns out to be a very common class of reaction with benzene and its derivatives, a reaction we call “Electrophilic Aromatic Substitution”.
We’re not going to get into why this happens just yet. For now, lets just satisfy ourselves with the what.
More on that in a few posts.
6. This Collection of Three Special Properties Is Called “Aromaticity”
Let’s review the three* [Note 2 ] important properties we’ve noticed about benzene so far.
- Extremely large resonance energy (36 kcal/mol)
- Delocalized electrons
- Undergoes substitution rather than addition reactions
This collection of special properties has been known since the late 1800’s, and was given a special name that we are now stuck with: aromaticity. It actually has nothing to do with their aroma, although it was noticed that a number of sweet-smelling molecules all had these properties in common.
You might reasonably ask: is aromaticity unique to benzene and its derivatives?
7. Aromaticity Is Not Unique To Benzene
The answer is no – it turns out that aromaticity is such a fundamental and important property that it deserves its own chapter(s) in introductory textbooks, which is why we will be writing a whole series on this phenomenon.
Aromaticity is a property shared by a large number of molecules, including
- molecules with multiple rings (e.g. naphthalene, anthracene, indole)
- molecules with ring sizes other than 6
- molecules with rings containing atoms other than carbon (furan, indole, pyridine) [aromatic molecules containing an atom other than carbon are often called “heterocycles”].
- certain molecules bearing a positive charge (the “tropylium ion”)
- certain molecules bearing a negative charge (the “cyclopentadienyl anion”)
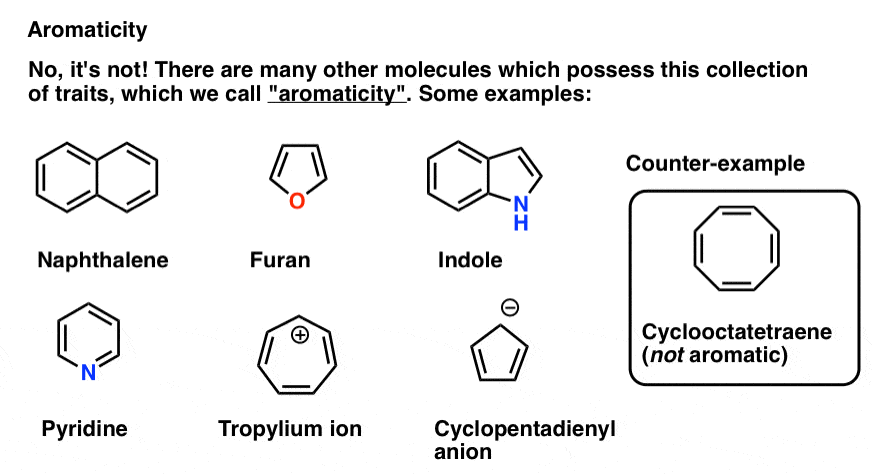
There’s even molecules entirely lacking in carbon which have some properties of aromaticity (borazine). The bases of DNA (adenine, cytosine, guanine, thymine) have aromatic character.
However, molecules are not aromatic simply by virtue of being cyclic and having double bonds at the perimeter. Cyclooctatetraene (above right) is not aromatic, and behaves like a normal conjugated alkene.
It’s enough to make you scratch your head.
8. So What Makes A Molecule Aromatic?
So why is benzene aromatic, but cyclooctatetraene is not? How can we tell if a molecule is aromatic? What are the rules?
Great, great question. But we’ve run out of time to answer that today. More on that in the next post. See next: Rules For Aromaticity
Thanks to Matt Knowe for assistance with writing this post.
Notes
Note 1. 36 kcal/mol is a huge deal in energy terms. A C-C pi bond is worth about 60 kcal/mol, so the energy stabilization of aromaticity is worth about half that number.
Even 3 kcal/mol difference between two compounds in equilibrium will result in about a 99:1 distribution towards the more stable component (remember cyclohexane chair A-values?)
Note 2. There’s a fourth diagnostic property of aromaticity – ring currents – which is observed in an applied magnetic field, such as when obtaining nuclear magnetic resonance (NMR) spectra. There’s no need to get into this right now, so it will get no further comment
Quiz Yourself!
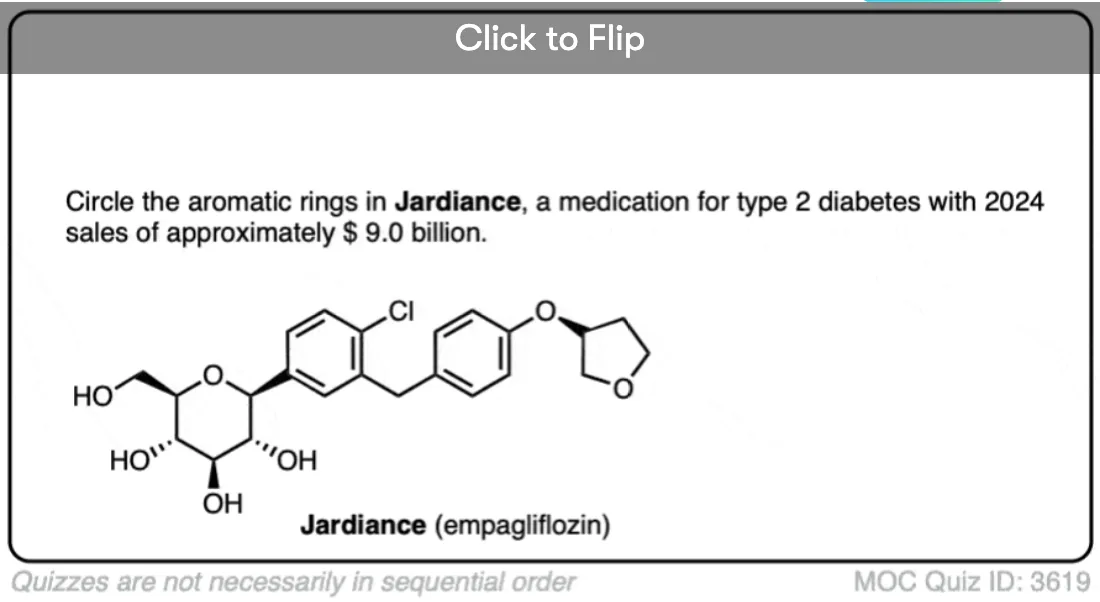
Become a MOC member to see the clickable quiz with answers on the back.
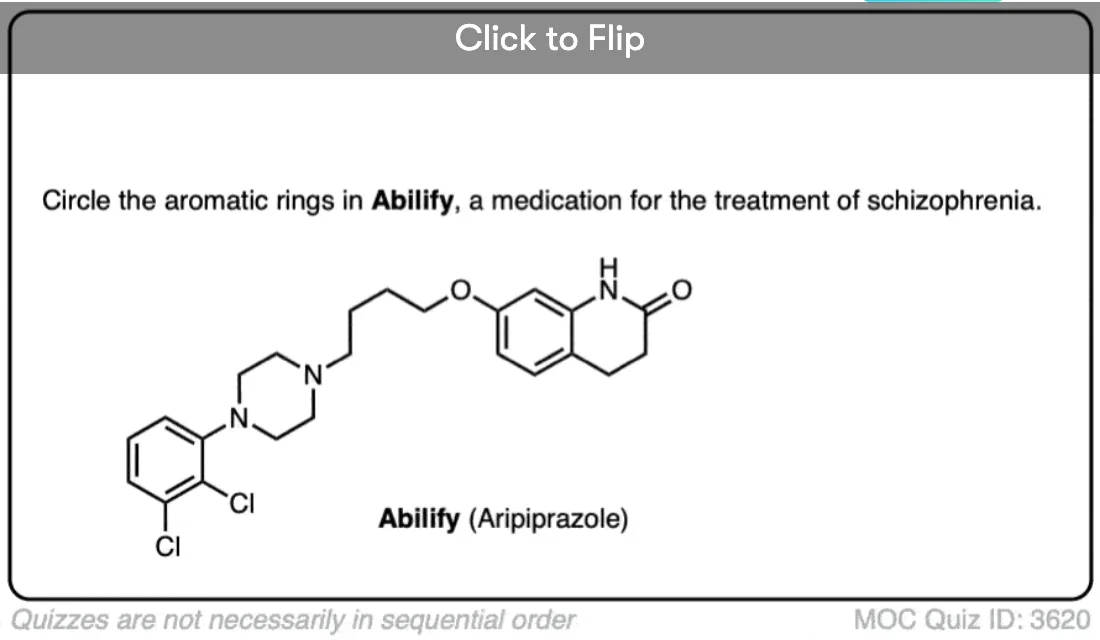
Become a MOC member to see the clickable quiz with answers on the back.
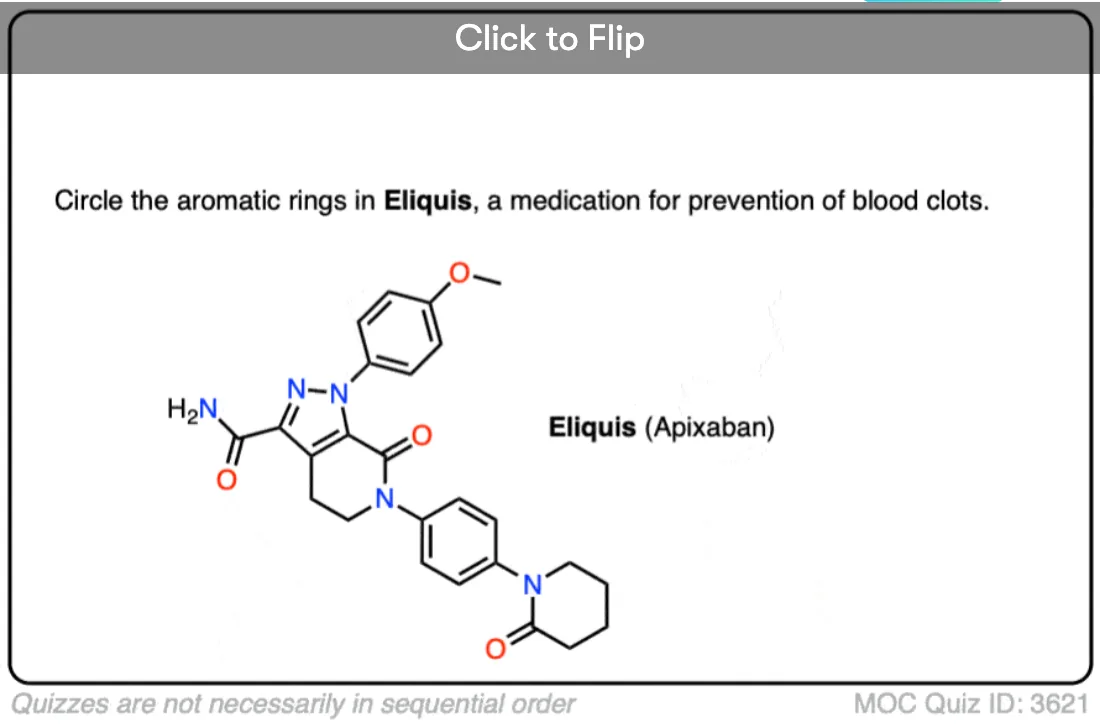
Become a MOC member to see the clickable quiz with answers on the back.
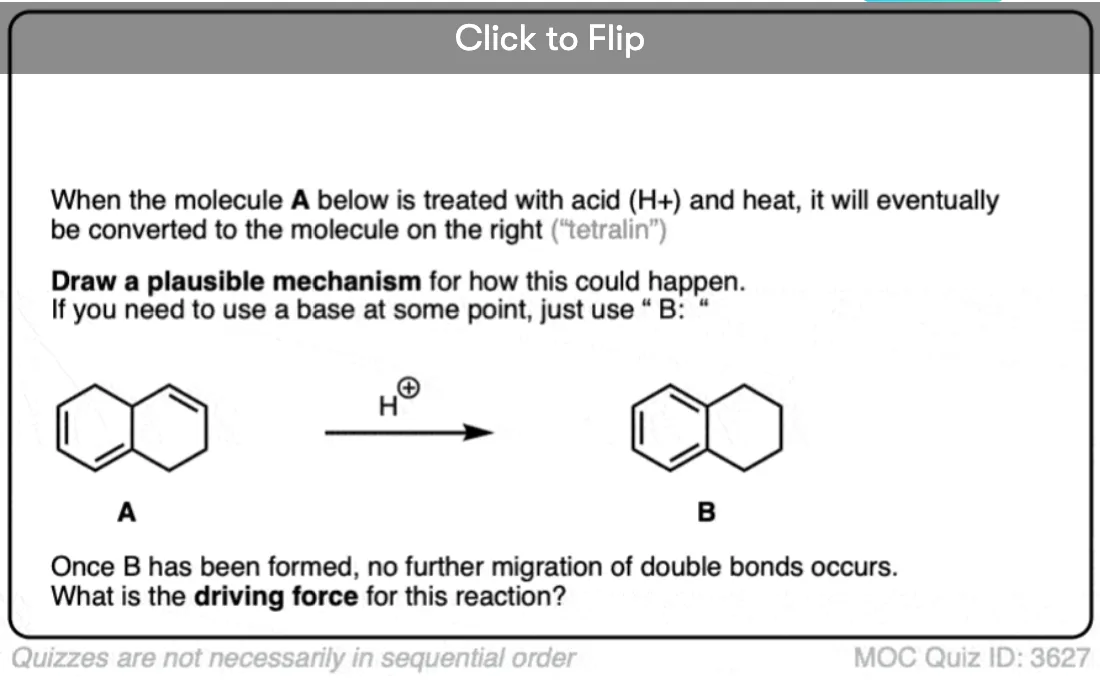
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- Sur la constitution des substances aromatiques
A. Kekulé
Bull. Soc. Chim. Fr. 1865, 3, 98–110
LINK
Kekulé’s famous paper on the structure of benzene and aromatic compounds, in French. - Zur Constitution des Benzols
Ladenburg
Just. Lieb. Ann. Chem. 1874, 172 (3), 331-356
DOI: 10.1002/jlac.18741720314
Ladenburg proved all 6 positions of benzene were equivalent (this paper is in German). - Ueber die Constitution des Benzols. Ueber die Hexahydroisophtalsäure
Adolf Baeyer, Victor Villiger
Lieb. Ann. Chem. 1893, 276 (3), 255-265
DOI: 10.1002/jlac.18932760302
This paper is by the same Adolf von Baeyer and Victor Villiger of Baeyer-Villiger reaction fame. Here, they describe the reduction of various benzene derivatives to cyclohexadiene, cyclohexene or cyclohexane compounds and determined the constitutions by standard methods. They also disproved a prism structure for benzene (this paper is in German). - Zur Kenntniss der ungesättigten Verbindungen. Theorie der ungesättigten und aromatischen Verbindungen
Johannes Thiele
Lieb. Ann. Chem. 1899, 306 (1-2), 87-142
DOI: 10.1002/jlac.18993060107
Thiele was the first person to discuss “conjugation” between double bonds, and potentially might have been the first to draw benzene with a circle in the middle (this is in German). - The structure of the benzene ring in C6(CH3)6
Kathleen Lonsdale
Proc. Royal Soc. A 1929, 123 (792), 494-515
DOI: 10.1098/rspa.1929.0081
Paper that describes the X-ray structure of hexamethylbenzene, showing that all the aromatic C-C bonds are the same length, 1.42 Å. - The Nature of the Chemical Bond. VI. The Calculation from Thermochemical Data of the Energy of Resonance of Molecules Among Several Electronic Structures
Linus Pauling and J. Sherman
J. Chem. Phys. 1933, 1, 606
DOI: 10.1063/1.1749335
This paper is the origin of the term ‘resonance energy’. - Heats of Organic Reactions. IV. Hydrogenation of Some Dienes and of Benzene
B. Kistiakowsky, John R. Ruhoff, Hilton A. Smith, and W. E. Vaughan
Journal of the American Chemical Society 1936, 58 (1), 146-153
DOI: 10.1021/ja01292a043
The commonly cited value of 49.8 kcal/mol for the complete hydrogenation of benzene is from this paper. - The use of 90°-1,3-butadiene as a reference structure for the evaluation of stabilization energies for benzene and other conjugated cyclic hydrocarbons
Philip George, Mendel Trachtman, Charles W. Bock, Alistair M. Brett
Tetrahedron 1976, 32 (12), 1357-1362
DOI: 1016/0040-4020(76)85010-7
The isodesmic reaction approach has been applied to the calculation of the resonance stabilization of benzene. This approach can be taken using either experimental thermochemical data or energies obtained by MO calculations. If the resonance energy of butadiene is assigned as zero, then the resonance energy of benzene would be 21.2 kcal/mol. If butadiene is considered to have a delocalization energy, the calculation must be modified to reflect that fact. Using 7.2 kcal/mol as the butadiene delocalization energy gives a value of 42.8 kcal/mol as the benzene resonance energy. - Quantentheoretische Beiträge zum Benzolproblem
Die Elektronenkonfiguration des Benzols und verwandter Verbindungen
Erich Hückel
Zeitschrift für Physik 1931, 70, 204–286
DOI: 10.1007/BF01339530
Erich Hückel achieved recognition by elaborating, together with Peter Debye, the theory of strong electrolytes in 1923 and later by applying a simplified version of quantum theory to p-electrons in conjugated molecules, which became known as Hückel molecular orbital (HMO) theory. Although he never explicitly formulated a “4n + 2 rule”, this was obvious from his work. Hückel showed that monocyclic systems with continuous conjugation having 6, 10, 14, etc. p-electrons benefited from extra stabilization and were aromatic. But it is more accurate to refer to the “Hückel 4n + 2 p-electron rule,” rather than to “Hückel’s rule.” - Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe
Paul von Ragué Schleyer, Christoph Maerker, Alk Dransfeld, Haijun Jiao, and Nicolaas J. R. van Eikema Hommes
Journal of the American Chemical Society 1996, 118 (26), 6317-6318
DOI: 10.1021/ja960582d
This paper is an advanced topic but worth including here, as it is one of Prof. Schleyer’s most highly cited papers. Aromaticity is a difficult concept to accurately define, but one way to empirically measure it is to use computational methods. Here, Prof. Schleyer describes the “NICS effect” as a method of measuring aromaticity, based on magnetic susceptibility exaltation. Aromatic compounds have a ‘ring current’ due to the conjugation of the p orbitals and the presence of delocalized p electrons, and are therefore diamagnetic. This can be measured experimentally or probed computationally. - Aromaticity Today: Energetic and Structural Criteria
Mikhail Glukhovtsev
Journal of Chemical Education 1997, 74 (1), 132
DOI: 1021/ed074p132
This paper discusses two of the criteria for establishing aromaticity – planarity and a positive stabilization energy. The latter can be verified by computational methods, as the article demonstrates.
Excellent notes I am wayy behind in advanced organic chemistry class and i feel a lot better reading this along with my lecture notes. I am understanding a lot of simple concepts essential to understanding harder things.
That’s the goal. Thanks for stopping by Davion!
Here i coupling 2-flouru-5-hydroxy benzaldehyde and 4-flouro- benzonitrile presence of k2co3 in dmf as solvent at 95-100 degree. Reaction is not going upto 90% just 70% going then remain 30% dimer impurity forming, here what i need no need to formation of dimer is there any suggestions please
For such a specialized questions you may want to access the collective wisdom of many different people. I suggest asking this question on a forum such as this one:
http://www.chemicalforums.com/index.php?board=35.0
That’s a forum for graduate students, and others, seeking help with synthesis.