Acid Base Reactions
Acid Base Reactions Are Fast
Last updated: December 12th, 2024 |
Acid-Base Reactions Are Generally Faster Than Substitution Or Addition Reactions
Here is a very common dilemma in organic chemistry as you move through the latter parts of Org 1 and then into Org 2:
When more than one reaction is possible, how do you know which one will happen?
In a steel cage match between an acid base reaction and other types of reactions, which wins?
In this post we postulate a good rule of thumb to keep in mind: acid-base reactions are fast, relative to other reactions.
Here are three examples – and by the way… these are common trick questions for exams!
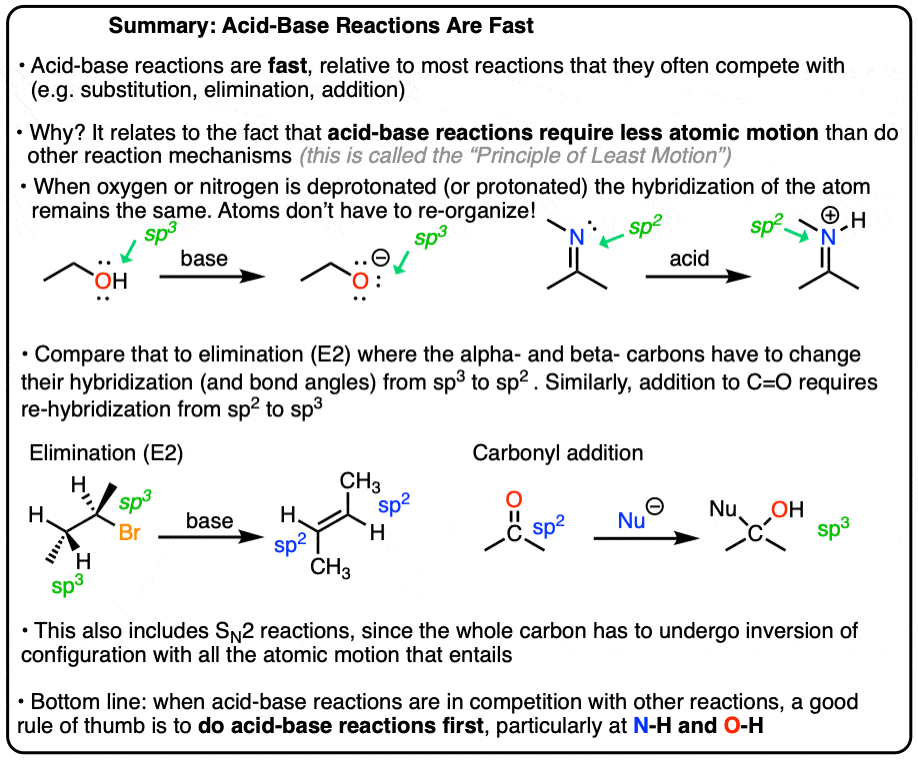
Table Of Contents
- Acid-Base vs Nucleophilic Substitution Reactions (SN2 reactions).
- Acid-Base Reaction vs Addition To A Carboxylic Acid
- Grignard Reaction To A Molecule With A Hydroxyl Functional Group
- Why Acid-Base Reactions Are Fast: The Principle of Least Motion
- Notes
- (Advanced) References and Further Reading
1. Acid-Base vs Nucleophilic Substitution Reactions (SN2 reactions).
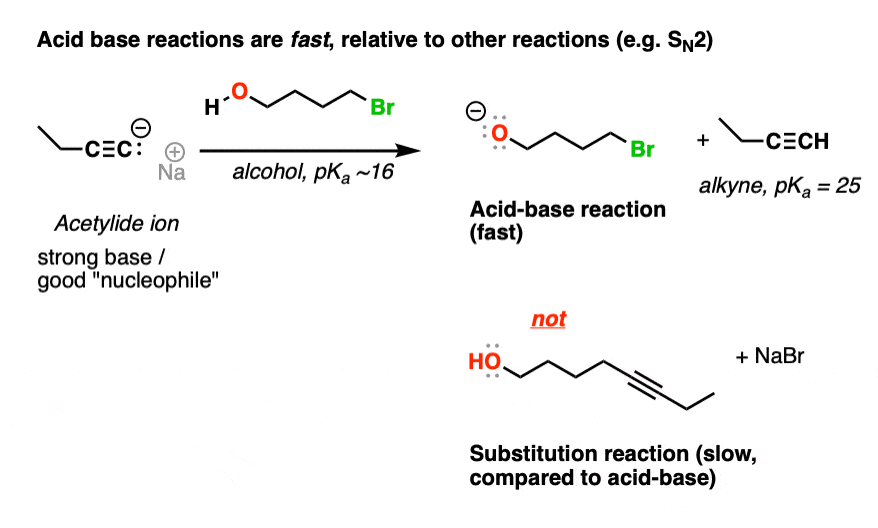
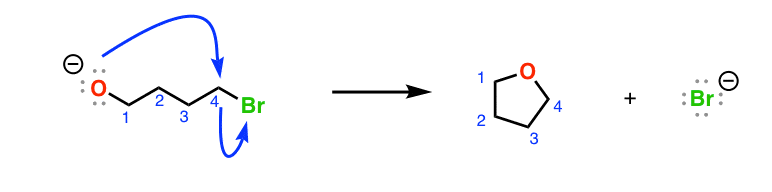
Here, note that our nucleophile (the conjugate base of an alkyne, pKa 25) can remove the proton of an alcohol (pKa ~15) or perform an SN2 reaction on the primary alkyl halide.
With a difference of 10 pKa units between the alkyne and the alcohol, the acid-base reaction between the deprotonated alkyne (“acetylide”, stronger base) to produce a deprotonated alcohol (“alkoxide”, weaker base) is extremely favorable.
And since acid-base reactions are fast, relative to other reactions, the preferred first reaction here is deprotonation of the alcohol to give the conjugate base (“alkoxide“)
Bonus question: what would be the final product of this reaction, after the deprotonation? Answer below [Note 2]
2. Acid-Base Reaction vs Addition To A Carboxylic Acid
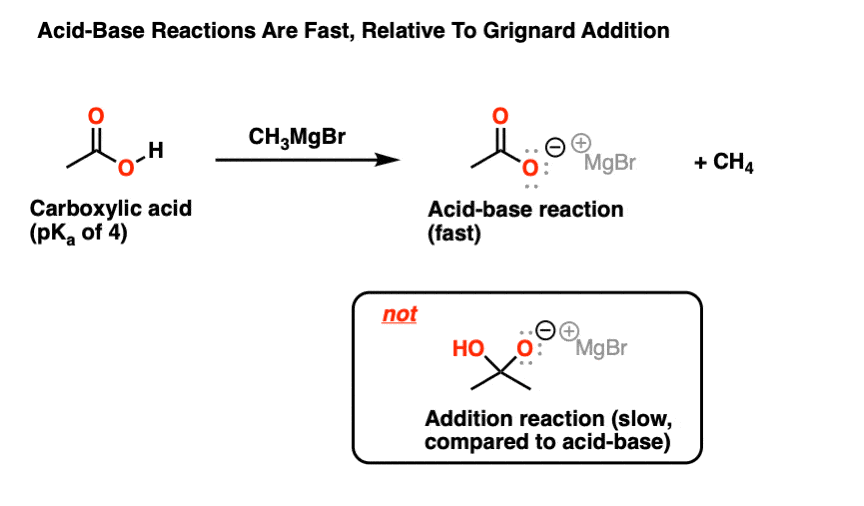
Grignard reagents are very good nucleophiles – reacting with carbonyl compounds such as ketones, aldehydes, and esters. But as the conjugate bases of alkanes (pKa ~ 50) they are also extremely strong bases.
When combined with a carboxylic acid (pKa ~4 or 5) the result is not an addition to the carbonyl, but an acid base reaction (45 pKa units makes for a pretty favorable reaction!).
It’s always helpful to remember that carboxylic acids… are acids!
3. Grignard Reaction To A Molecule With A Hydroxyl Functional Group
The same applies for reactions of Grignard reagents with molecules that have hydroxyl groups in addition to aldehydes or ketones. If merely one equivalent is added, the first thing to happen will be deprotonation of the alcohol, which is faster than addition to the ketone carbonyl carbon.
It’s only after addition of a second equivalent of Grignard that addition to the ketone will occur.
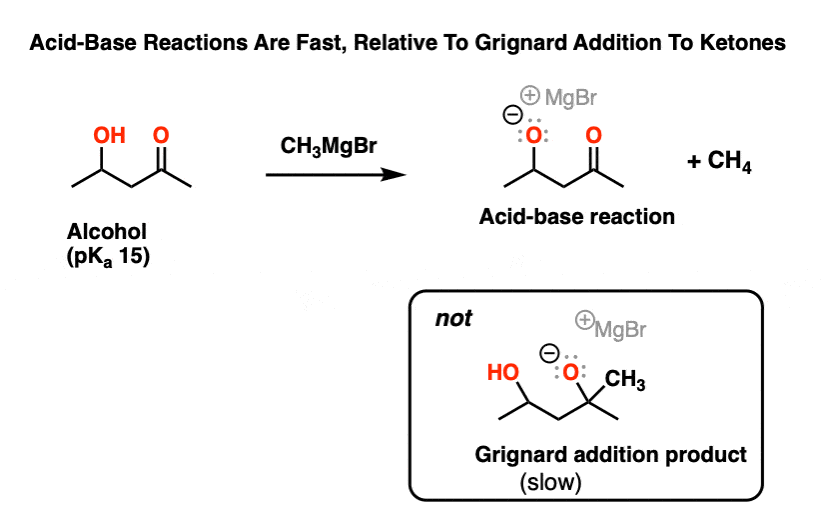
So what’ s going on?
4. Why Acid-Base Reactions Are Fast: The Principle of Least Motion
What’s going on here is an application of a handy principle in chemistry called the Principle of Least Motion.
Simply stated, it’s this.
Acid-base reactions on “heteroatoms” (that means atoms other than carbon, such as O, N, and S) generally require very little reorganization of the nuclei in the structure after loss of a proton.
Therefore these reactions are fast, relative to reactions where the nuclei have to move or be reorganized.
Think about removing a proton from an O-H.
After loss of hydrogen, the oxygen gains a new lone pair. But its hybridization doesn’t change – it started as sp3, and it’s still sp3. So the nuclei (other than the H, of course) don’t significantly change positions in these reactions. No extra atomic motion, in other words.
However when bonds are formed or broken at carbon – such as in the SN2 reaction or in additions to carbonyl carbons – a lot of atomic furniture has to get rearranged.
For instance, the SN2 proceeds through a backside attack, which means that the geometry of the molecule changes from tetrahedral to trigonal bipyramidal (that’s the 5-coordinate transition state) and then back to tetrahedral.
In addition reactions to carbonyl compounds, we’re changing the hybridization of carbon from sp2 to sp3. That requires a shift from trigonal planar to tetrahedral geometry.
Extra atomic motion means it will be a slower reaction, relative to an acid-base reaction.
Bottom line: acid base reactions on oxygen, sulfur, or nitrogen are fast. So long as the acid base equilibrium is reasonable [How to use a pKa table, a handy rule of thumb for acid-base reactions] do them first.
Next Post: Acid-Base Reactions – What’s The Point?
Notes
Note 1. It’s interesting that Grignard reagents (the conjugate bases of alkanes, pKa ~50) don’t usually deprotonate the alpha-carbon of aldehydes (pKa ~18) or ketones (pKa ~20).
That’s another application of this principle. Removing a proton from an aldehyde or ketone requires breaking a C-H bond, and the resulting base (called an “enolate”) will undergo a change in hybridization from sp3 to sp2. Therefore, it’s slow.
Note 2. After the first acid-base reaction, the deprotonated alcohol can then do an SN2 reaction on the primary alkyl bromide.
(Advanced) References And Further Reading
- The Principle of Least Nuclear Motion.
Hine, J.
Advances in Physical Organic Chemistry, Academic Press, Volume 15, 1977, Pages 1-61. ISSN 0065-3160.
DOI: 10.1016/S0065-3160(08)60117-3
In this book chapter, Prof. Hine of The Ohio State University reviews the principle of least ncuelar motion. Particularly notable is the graph on page 40, which shows that HCN has the lowest barrier to deprotonation of any carbon-based acid, consistent with a small change in internal geometry.
The essence of Richard Feynman’s path integral formulation is that every possible trajectory contributes to the particle’s behaviour with a certain probability amplitude. Only when the contributions of paths interfere with each other in the classical limit lead to principle of least action? How can the principle of least action be applied to atomic motion if they are at the quantum scale?
That’s principle of least action. This is principle of least nuclear motion. Different thing.
https://www.sciencedirect.com/science/article/abs/pii/S0065316008601173?via%3Dihub
In many acid-base reactions, the structure of the transition state greatly resembles that of the reactants; very little atomic reorganization is required.
In nucleophilic substitution (and other) reactions, there is re-organization of the atoms in the transition state versus the starting materials. That re-organization requires more energy.
That said, I should probably re-write this to clarify.
Why in Aldol Condensation OH- being a weaker base than Grignard Reagent able to deprotonate alpha H and Grignard Reagent Isn’t
Grignard reagents add to the carbonyl – irreversibly. Hydroxide can add to the carbonyl to form a hemiacetal but it is reversible, and eventually deprotonation occurs to give the enolate.
With Grignards, deprotonation at the alpha position only tends to occur when there is a lot of steric bulk around the carbonyl carbon. The C-H bond has to be oriented at 90° in order for deprotonation to occur, otherwise the resulting carbanion won’t be in resonance with the carbonyl pi bond
Thank you so much for all your work on the website. I got orgo 1 summary sheet and finding it very helpful.
Just a question. Why breakage of C-H bond results in rehybridazation om aldehyde and ketone?
Wouldn’t it be still sp3 with the lone pair after deprotonation?
There is no
Thank you again.
Hi, if you deprotonate C-H adjacent to a ketone, the carbon will not be sp3 hybridized for long. It is energetically favourable for the carbon to re-hybridize to sp2, which puts the lone pair in a p orbital and can then be conjugated with the p orbitals in the C=O group.
It’s this atomic motion which makes deprotonation of C-H “slow”.
Thank you James for your reply!
I purchased the summary, and realized I wasn’t ready for summary yet, and going through the posts now, and it’s really helpful. I just thought it will be really good if you can put together these post contents into printable pdf. I would definitely pay a reasonable price to have this contents easier to read, carry, and make notes..!
Thanks so much for summarizing!
I’ve always had trouble understanding why certain actions are preferred over others. It’s an important point that you clarified so well!
Thank you!
Forgot to add that yes, I realize the shifts can be used in SN1 / SN2 reactions…maybe it’s best if I keep an eye out for any potential carbocation rearrangements that could make a carbon center more stable.
I have a question that’s been bothering me for a while now–mostly because my organic chem prof doesn’t explain concepts fully–when is the best time an alkyl or hydride shift can be done on a reaction mechanism problem? I know alkyl / halide shifts can be used in a problem where you have an alcohol and you’re reacting it with some type of haloalkane like HBr, but are there any other times the shifts can be used?
Any time you form a carbocation, watch out for rearrangements.