Enols and Enolates
Enolates: Formation, Stability, and Simple Reactions
Last updated: May 29th, 2026 |
All about enolates
- Enolates can be formed through removing the proton on carbons adjacent to a carbonyl (i.e. the “alpha-carbon“). The resulting anions are much more stable than typical alkanes since the negative charge can be delocalized to the oxygen atom via resonance.
- Enolates can be formed from aldehydes and ketones, but also from esters, amides, nitriles and many other carbonyl-containing compounds. When an additional electron-withdrawing group is present on the alpha-carbon, enolates become much easier to form.
- Enolates are nucleophiles and have many useful reactions with electrophiles. In this post we will cover the reactions of enolates with H+ , halogens, and simple electrophiles.
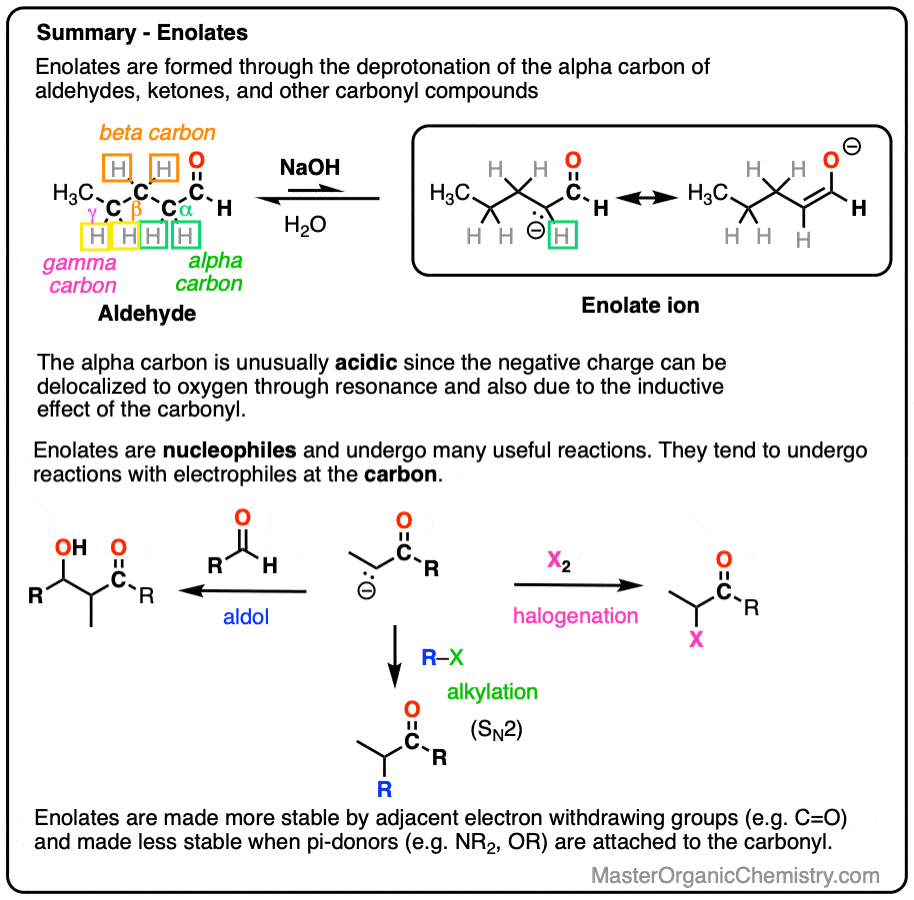
Table of Contents
- What Is An Enolate Ion?
- Keto-Enol Tautomerism via Enolates
- Enolates Tend To React With Electrophiles on the Carbon Atom
- Epimerization (and Deuteration) via Enolates
- Enolates are Good Nucleophiles! Some Simple Examples (Halogenation, Aldol)
- Factors That Affect Enolate Stability
- Enolates of Esters and Amides
- Ketone Enolates
- Conclusion
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. What Is An Enolate Ion?
Aldehydes and ketones will react with strong base [e.g. HO(-) ] to give an enolate ion.
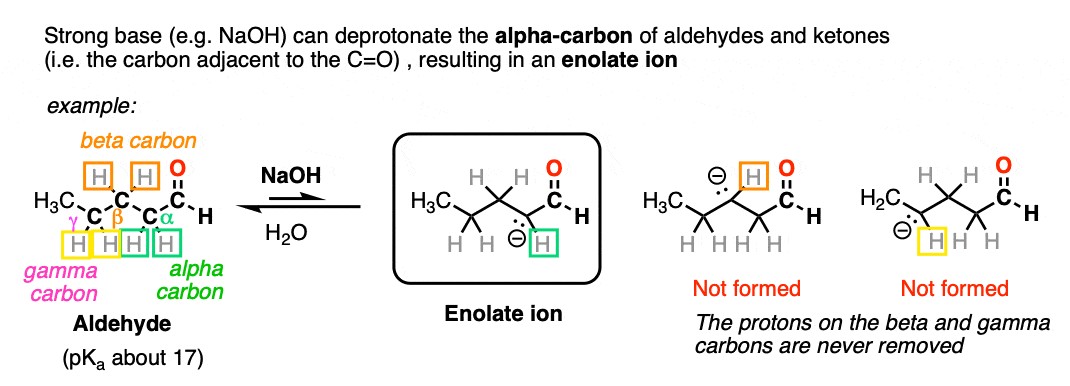
A C-H bond is broken on the carbon adjacent to the carbonyl carbon (i.e. the “alpha-carbon”). The carbons further away from the carbonyl are never deprotonated.
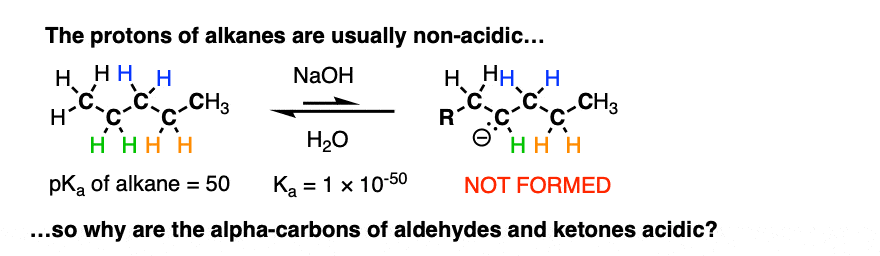
The reaction is reversible and equilibrium favors the starting materials by about 100:1 [Note 1] this is still a pretty surprising result, given all that we know about C-H bonds. After all, alkanes are incredibly resistant to base (pKa = 50) so something here is making the aldehyde or ketone about 30 more pKa units (= 1030 !) more acidic than it usually would be.
Why would this be?
To understand acidity, the first place to look is to examine the stability of the conjugate base. Way back in Org 1 we looked at various factors that affect acidity and noted that the negative charges are stabilized by
- inductive effects (adjacent electron-withdrawing groups)
- resonance delocalization (especially if negative charge can be placed on a more electronegative atom)
- increasing the electronegativity of the atom ( F > O > N > C) bearing the negative charge
(See how all these concepts we learn back in the first quarter of organic chemistry continue to be relevant many chapters later! )
If we compare the conjugate base of an alkane versus the conjugate base of an aldehyde or ketone, two things should stand out.
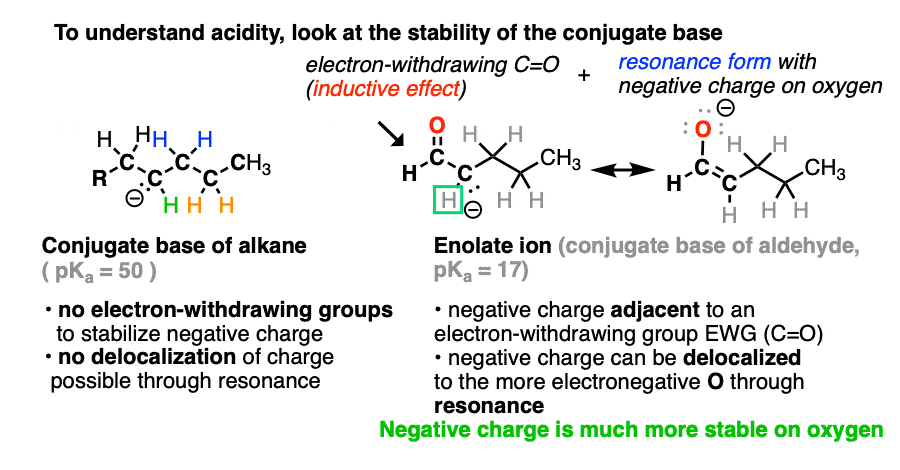
- The enolate ion has an adjacent C=O group to stabilize negative charge through inductive effects.
- If the negative charge is on the alpha carbon, it can be delocalized to oxygen through resonance, which places the negative charge on a more electronegative atom (electronegativity of O is 3.5 versus 2.5 for carbon).
Note that if we form the negative charge on the beta– or gamma– carbon, resonance delocalization isn’t possible.
In order for an enolate to form, there must be a hydrogen on the alpha carbon. If an aldehyde or ketone lacks a proton on the alpha carbon we call it “non-enolizable”.
Also avoid this common mistake: the aldehyde C-H bond is not acidic! [Note 2]
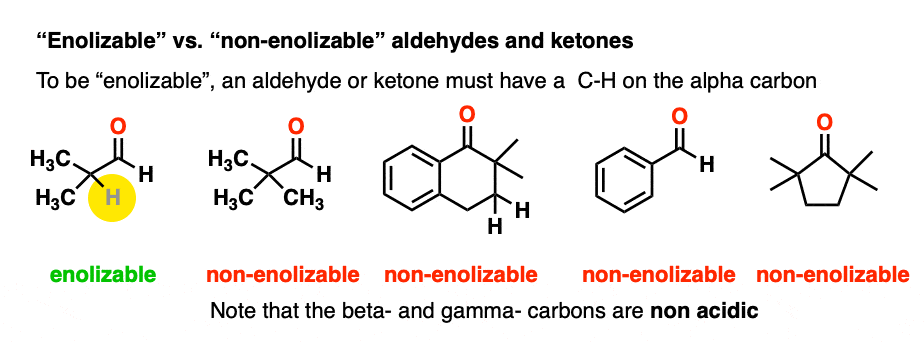
As it turns out, enolate ions are very important intermediates for many reactions and we’ll spend a considerable amount of time getting to know them.
We’ll start out by covering some of their simple reactions, and then finish up by exploring various factors that affect their stability (and reactivity).
2. Keto-Enol Tautomerism via Enolates
Enolates can be thought of as the conjugate bases of aldehydes and ketones (among others) but they are also the conjugate bases of enols.
Keto-enol tautomerism can occur through addition of either acid or base to an aldehyde or ketone. If you need a review, go back and see this post : Keto-Enol Tautomerism]
Generally speaking the keto tautomer is favored at equilibrium [mostly because the C-O pi bond is about 20 kcal/mol stronger than the C-C pi bond].
When keto-enol tautomerism occurs through the addition of base, it proceeds through the formation of an intermediate enolate before being protonated and converted to the enol form.
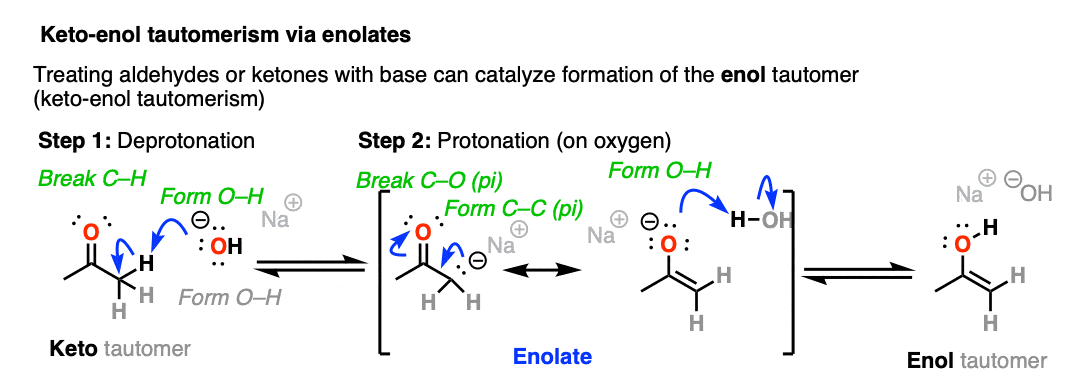
Since the aldehyde / ketone (pKa = 17-19) is a weaker acid than H2O (pKa 15) the equilibrium lies mostly to the left, meaning that the enolate is a minor component of the reaction mixture.
You’ll be forgiven if this doesn’t seem very exciting.
All that’s happening is that base is being used to form a compound (the enol) which is going to be outnumbered at least 100:1 at equilbrium in most cases. Who cares?
The exciting thing is that the enolate itself is a great nucleophile and is very reactive with other electrophiles if they happen to be present, such as:
- aldehydes or ketones (which gives us the Aldol reaction [see post: Aldol reaction]
- halogens (which allows for e.g. bromination of the alpha-carbon)
- alkyl halides (which allows for formation of new C-C bonds at the alpha-carbon via alkylation)
We’ll get to these! But first, some consequences of enolate formation.
3. Enolates Tend To React With Electrophiles On The Carbon Atom
Enolates have two prominent resonance forms and can be drawn with a negative charge on carbon or a negative charge on oxygen.
They are flat, so their most “correct” form is to draw them with their negative charge on oxygen.
However, they tend to react as if they were carbanions, attacking most electrophiles via the carbon atom. There are ways to get the oxygen to be more reactive instead of carbon, but for now we will defer this issue to more advanced courses [Note 3]. For our purposes here we will draw them with the negative charge on carbon.
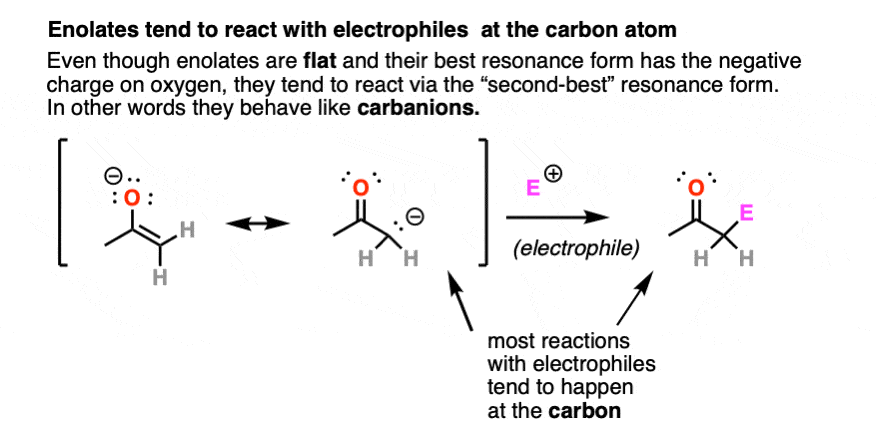
4. Epimerization And Deuteration of Enolates
The flatness of enolates has an important consequence.
If you start with a chiral center on the alpha carbon and treat it with base, the alpha carbon, being flat, will lose its chirality. If the enolate then reacts with another electrophile (e.g. H+) it can do so from either face of the flat enolate, resulting in racemization at that carbon. [To be more specific, we usually call this process epimerization [Note 4] since it won’t result in a racemic mixture of enantiomers if another chiral center is present.
(Chiral centers on the beta- or gamma- carbon are not affected since these C-H bonds are not acidic).
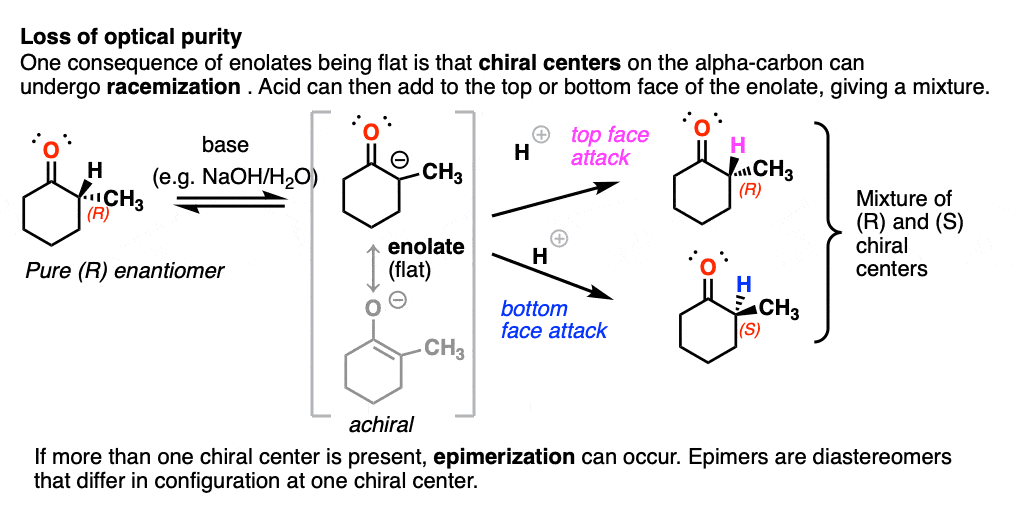
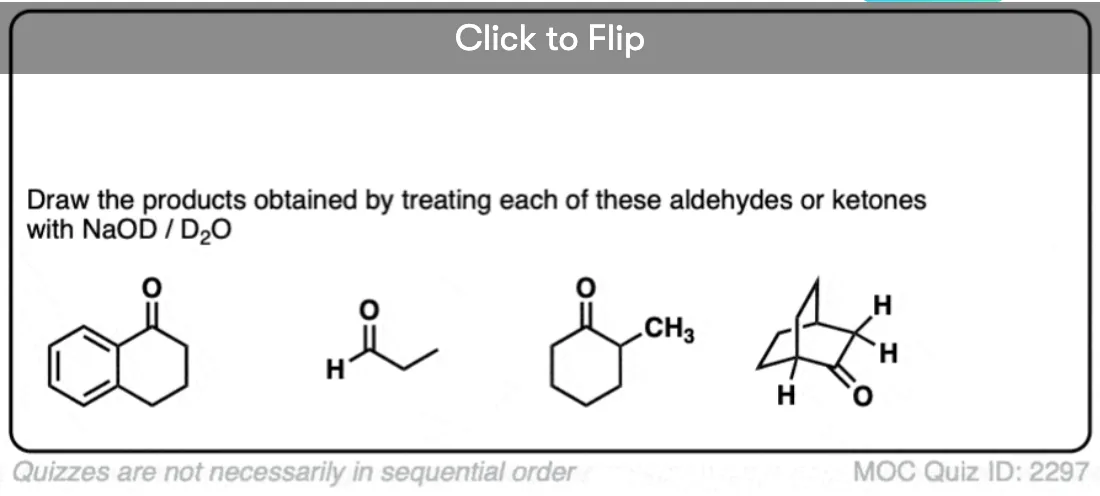
Just for fun, if we use a deuterated solvent like D2O instead of H2O (recall that D is the heavy isotope of hydrogen) then we will replace our C-H bonds with C-D bonds. This can be a useful way of incorporating deuterium.
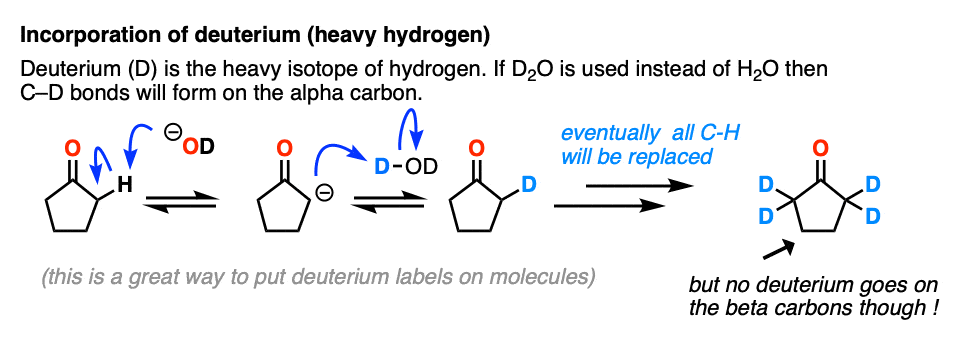
5. Enolates Are Nucleophiles
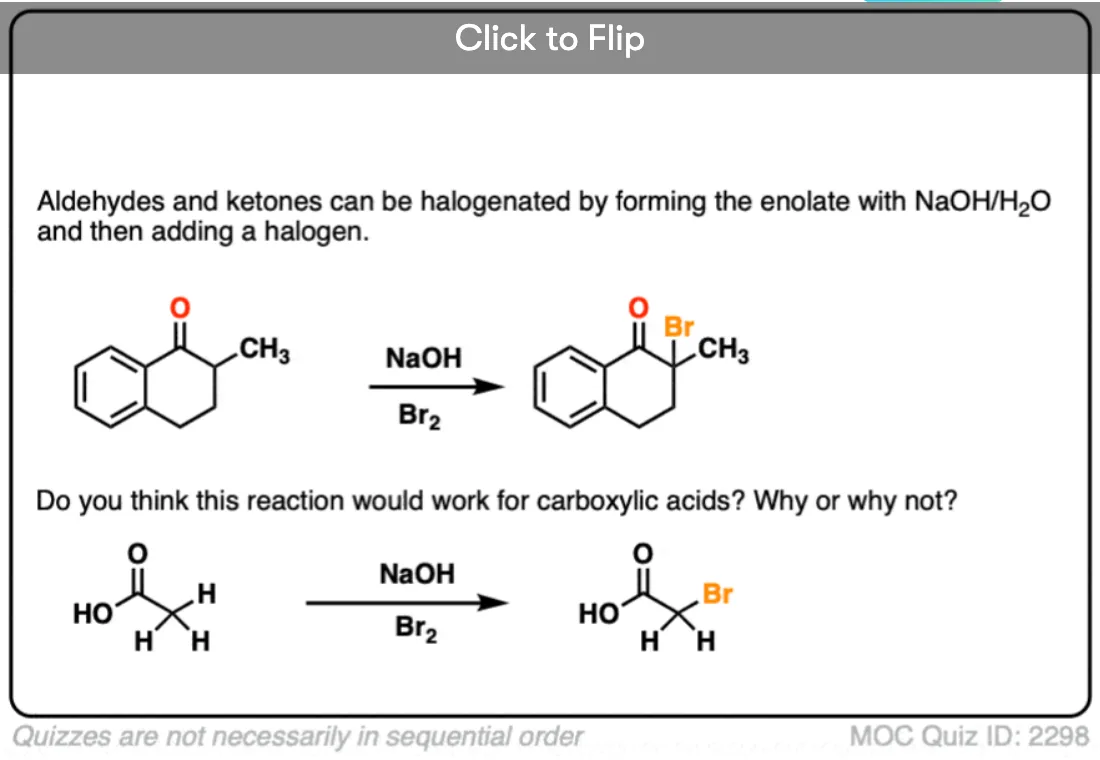
Halogenation
If we make an enolate in the presence of a halogen such as Br2, Cl2, or I2, a new C-halogen bond will form. This is called halogenation of enolates.
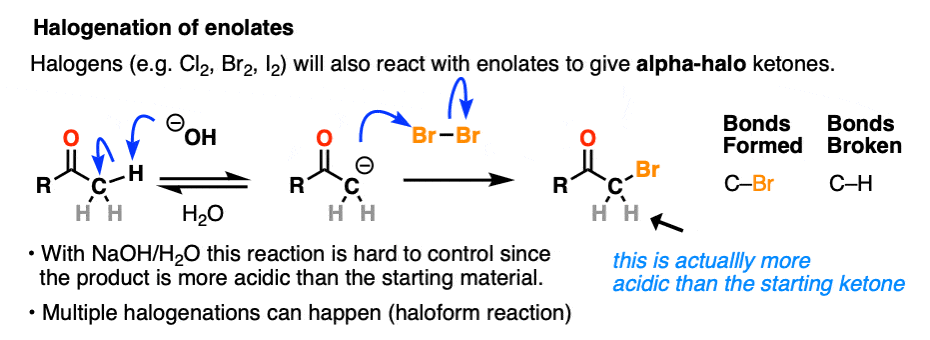
One key thing to watch out for with halogenation under basic conditions is that the reaction has Cookie Monster characteristics (the Cookie Monster never stops at eating just one cookie).
Electron withdrawing groups make adjacent C-H bonds more acidic since they stabilize negative charge. Therefore the resulting product will have a more acidic C-H bond, and be even more likely to be removed by base. It leads to a runaway reaction where multiple halogens can be installed on the same carbon. [see post: The Haloform Reaction]
The better (and more controlled) way to do this is to irreversibly form the enolate with a super strong base like LDA and then add (for example) Br2. No runaway reaction.
Aldol Reaction
Enolates can react with other aldehydes or ketones! This is called the Aldol Reaction (see post: Aldol Reaction)
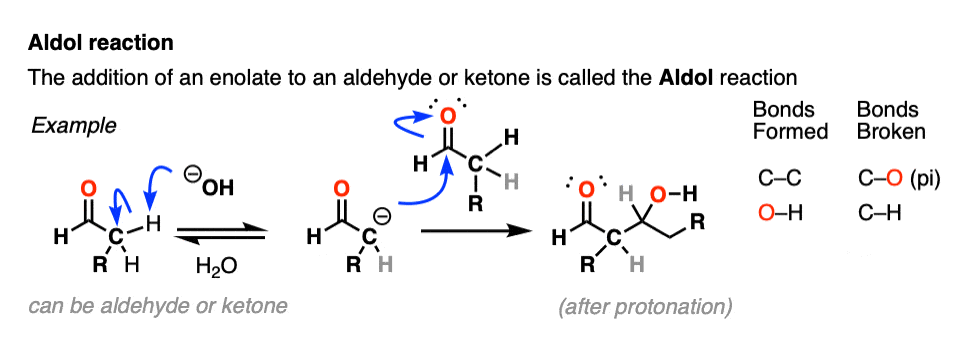
The aldol reaction has many cousins that go by various names like the Claisen condensation, Henry reaction, Knovenagel and whatever. They all boil down to the same essential thing: an enolate nucleophile attacking a carbonyl carbon electrophile.
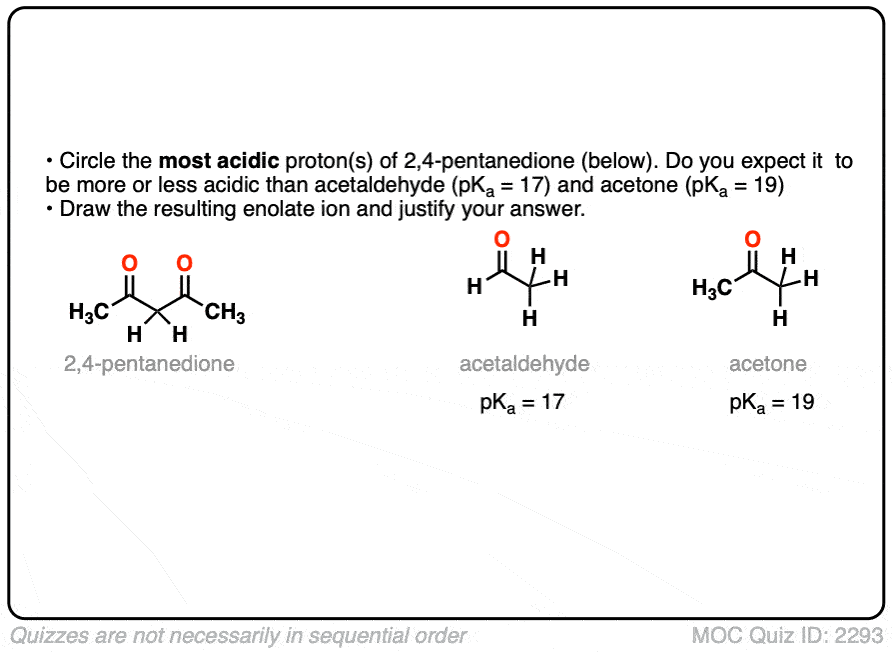
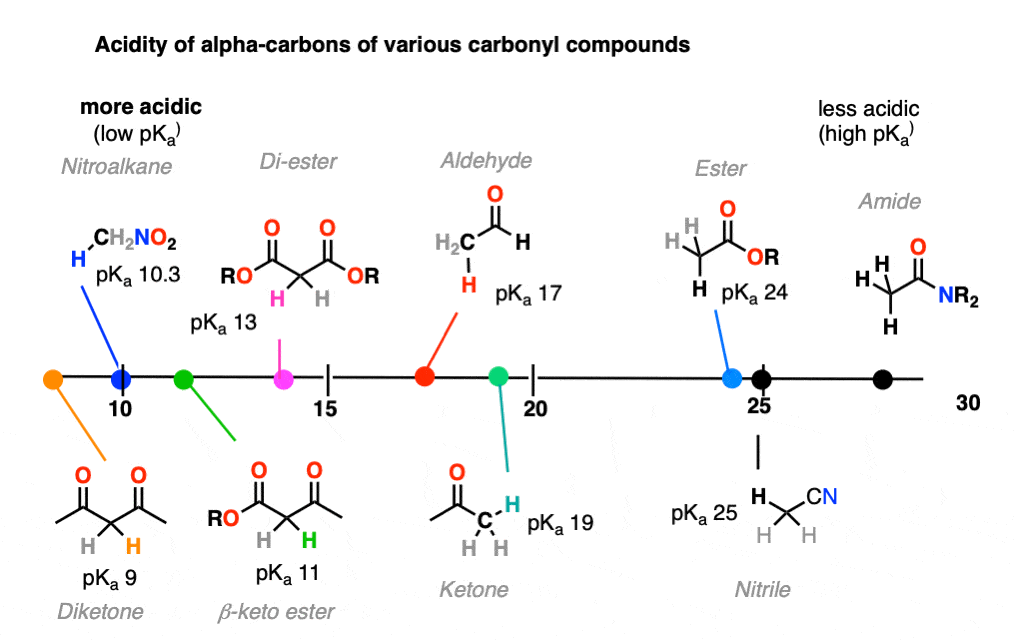
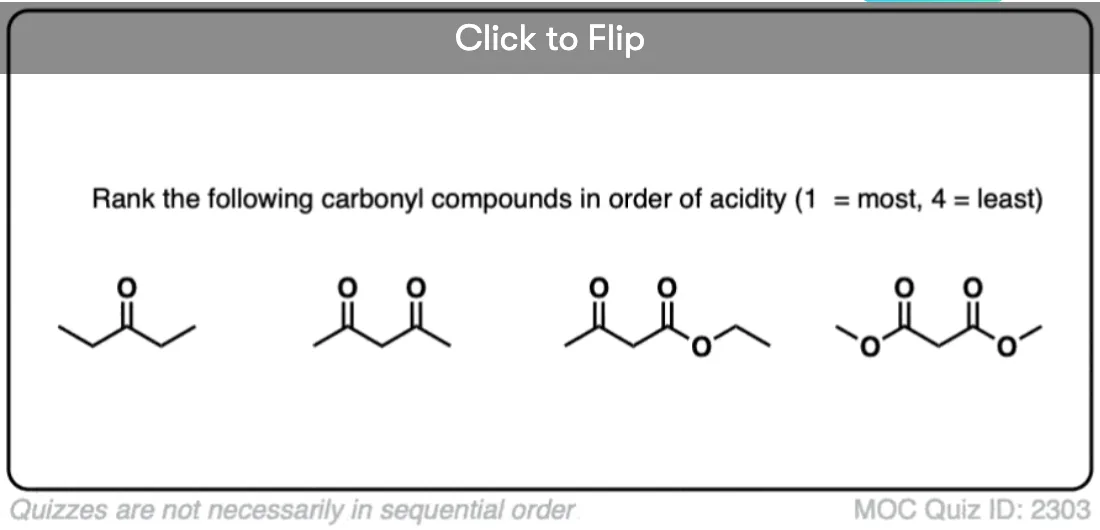
6. Additional Electron-Withdrawing Groups Make Enolates More Stable
So if aldehydes and ketones are reasonably acidic (pKa 17-19) what happens to the acidity of the alpha-carbon when we add an electron withdrawing group to it?
 Click to Flip
Click to Flip
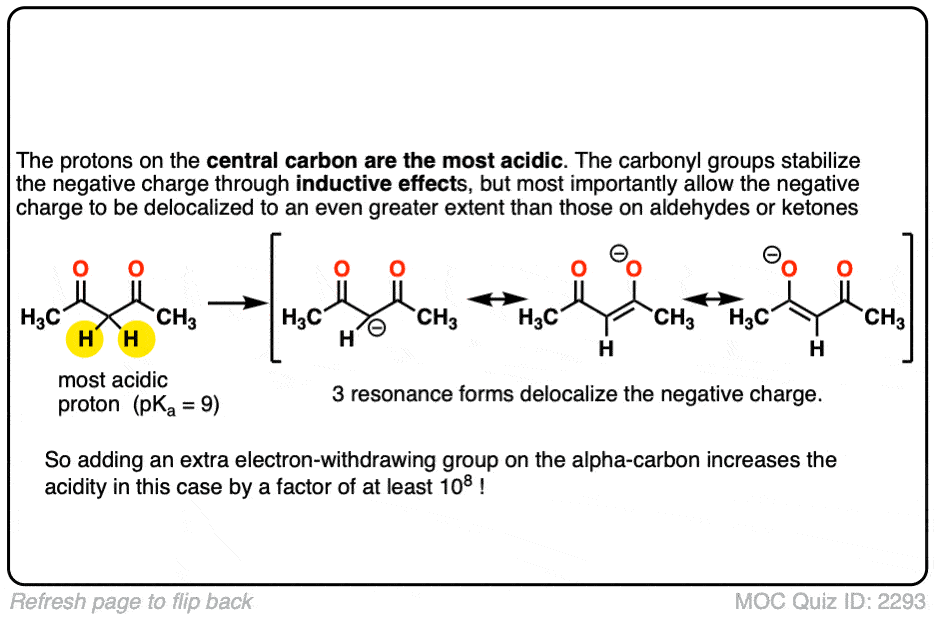
Remember that any factor that stabilizes the conjugate base will increase the acidity. So it should become a much stronger acid since the negative charge of the resulting conjugate base can be spread out over multiple oxygen atoms and also is stabilized by an additional inductive effect.
The bottom line is that enolates that have two adjacent electron withdrawing groups are much easier to form!
For example, beta-keto esters and acetoacetic esters (pKa about 11) can easily be deprotonated with a strong base such as CH3CH2O(-) [Note 5] .
One useful reaction of these enolates is that they will perform SN2 reactions on alkyl halides, forming a new C-C bond. This is called, “alkylation”.
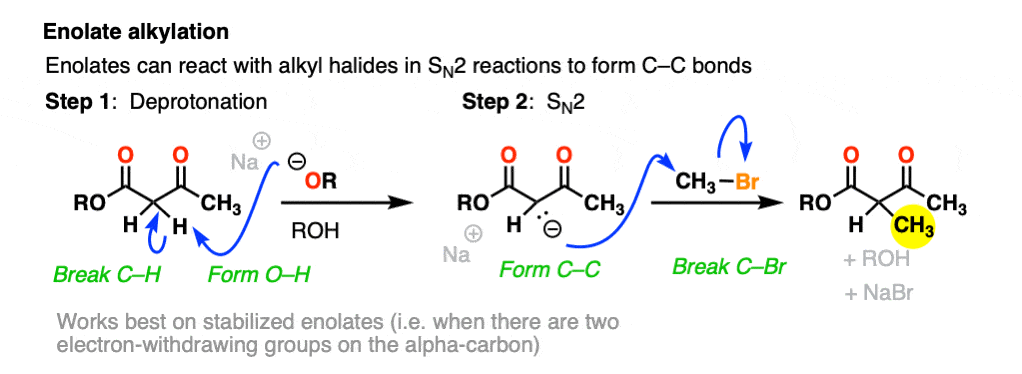
The products can then be made to lose CO2 (decarboxylation) in a process known as the malonic ester synthesis (for di-esters) or acetoacetic ester synthesis (for beta-keto esters). (See post: The Malonic Ester and Acetoacetic Ester Syntheses)
[Note 6 – regarding this reaction with ordinary ketones]
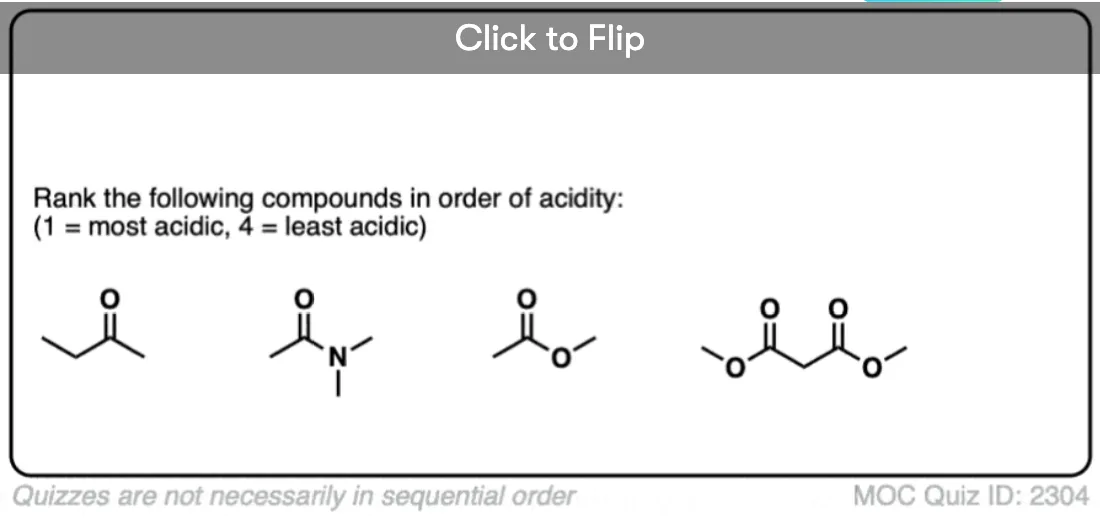
7. Enolates of Esters And Amides
Going in the opposite direction, what happens to the acidity of the alpha-carbon if we switch out a ketone for an ester?
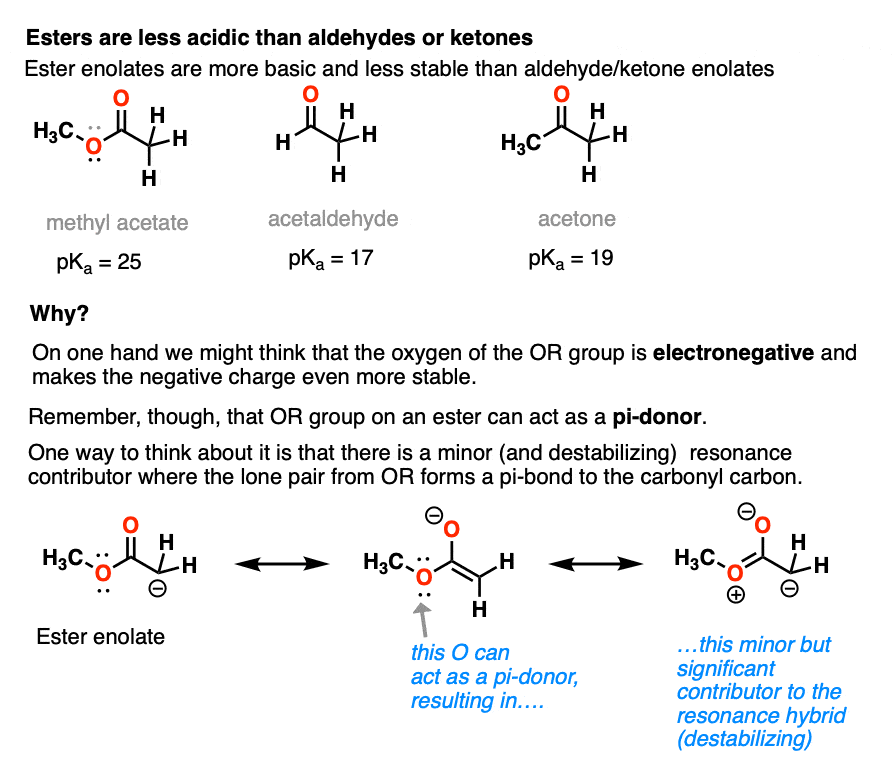
In the case of an ester the alpha-carbon actually becomes less acidic. Therefore ester enolates are less stable than those of aldehydes and ketones.
This might seem surprising. Why are ester enolates less stable? After all, wouldn’t we expect that it should be *better*, since it has that electron-withdrawing OR group?
The answer is related to the reason why OH and OR are activating groups on aromatic rings instead of deactivating groups.
Yes, oxygen is electronegative, but the lone pairs mean that it is also a pi-donor, capable of forming a C-O pi bond with the carbonyl carbon. The pi-donation outweighs the electronegativity factor.
In a sense the C=O of an ester is less able to stabilize the negative charge of the enolate because the enolate anion is “competing” with the lone pairs from the -OR group.
In addition to esters, other groups capable of acting as pi-acceptors such as NO2, CN, and amides will also form enolates.
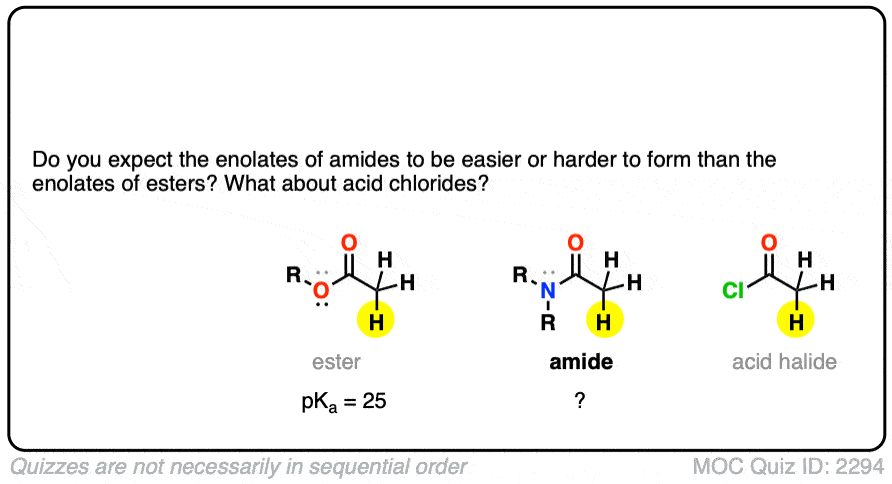
Knowing what you know now, do you think the enolates of amides (NR2) would be more or less stable than those of an ester (hint: think back to activating groups on aromatic rings)
 Click to Flip
Click to Flip
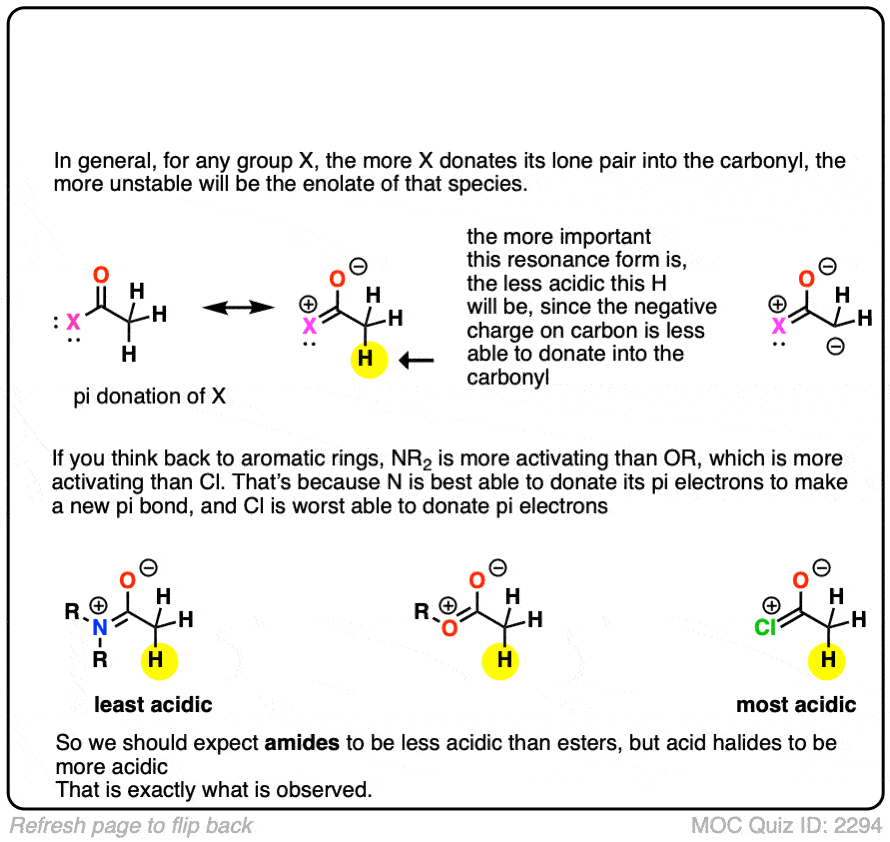
One simple way to think about it is to think of the C=O as a bucket for accepting an electron pair from the lone pair on carbon. When there is already a pi-electron donor like an OR or an NR2 adjacent to the C=O, there’s going to be less room in the bucket for the pair of electrons on the carbon, and the enolate will be less stable.
It might be helpful to put these trends together on a figure like this:
8. Ketone Enolates
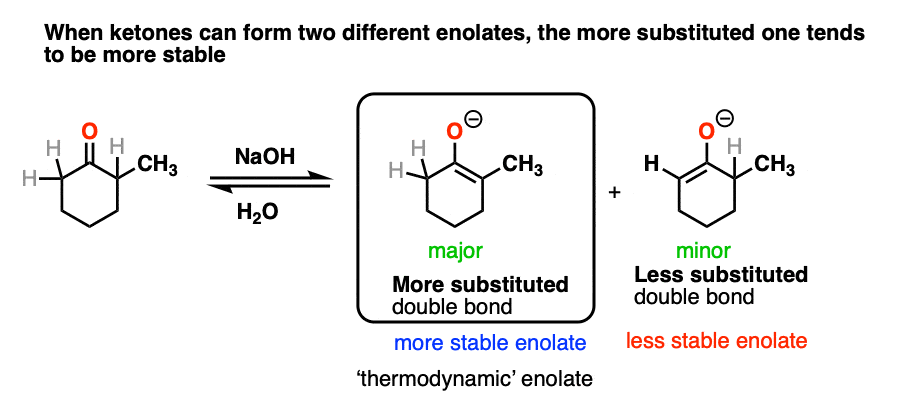
This article would not be complete without briefly mentioning some of the complications that come with ketone enolates.
In many cases treating a ketone with base could result in two different enolates.
So how do we know which one “wins?”.
We treat it the same way we do with double bonds, i.e. Zaitsev’s rule. Just as with alkenes, the more substituted enolate will be the most stable.
[Note 7]
For this reason the more substituted enolate is called the “thermodynamic enolate” – it is thermodynamically more stable.
However, this tendency to form the more substituted enolate can interfere with our carefully-laid plans! There are many times when we would like to form an enolate on the less substituted side of a ketone, regardless of what it “thermodynamically” might want.
One way to overcome this tendency is to use a strong, but very bulky base. The base lithium di-isopropyl amide (LDA) fits the bill nicely. It is like a big hammerhead shark that has a hard time sticking its nose into tight corners. So when presented with a ketone bearing two different alpha carbons, it will overwhelmingly remove the one attached to the less substituted carbon atom.
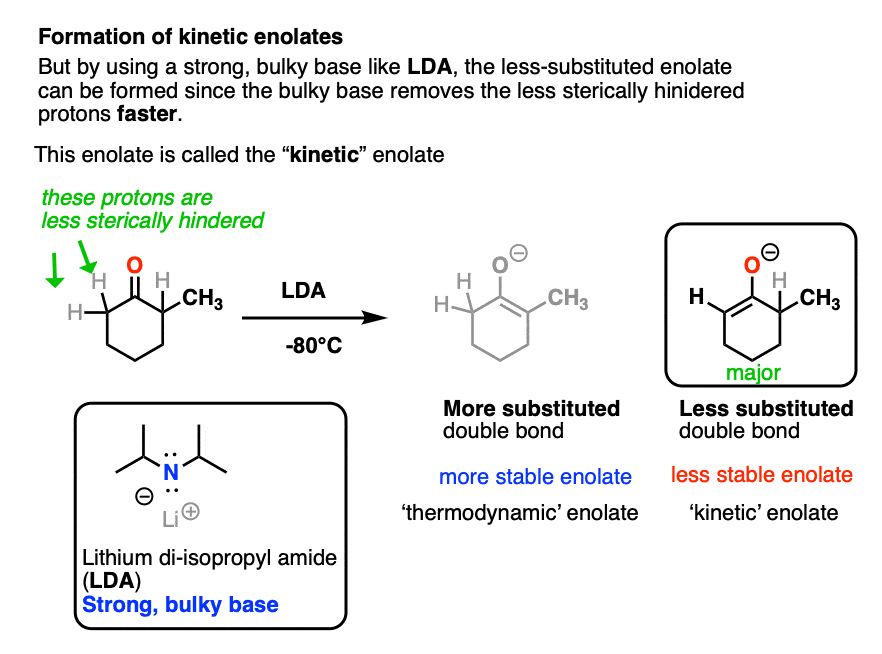
LDA is also useful for making the enolates of esters, amides, and nitriles, but we’ll have more to say about that in a future article.
9. Summary
What have we learned?
- Enolates result from the removal of a proton on the carbon adjacent to a carbonyl (the “alpha carbon”). They are stable due to inductive effects, but more importantly due to the delocalization (via resonance) of the lone pair on carbon to more electronegative oxygen.
- Aldehydes/ketones without a C-H bond on the alpha carbon are called, “non-enolizable” since they lack an acidic proton.
- Enolates are flat. If an enantiomerically pure ketone with a chiral alpha carbon is treated with base, watch out for a loss of optical purity due to deprotonation/protonation.
- Base-catalyzed keto-enol tautomerism of aldehydes and ketones goes through an enolate.
- Enolates will react with halogens (e.g. Br2, Cl2, I2) aldehydes/ketones, and alkyl halides.
- Enolates with two electron withdrawing groups attached to the alpha carbon are particularly easy to form.
- Enolates of esters and amides are more difficult to form than enolates of aldehydes and ketones.
- In many cases, treating a ketone with base could result in two different enolates. With a strong base such as NaOH or NaOR the tendency is to form the more substituted enolate because alkene stability increases with substitution.
- This tendency can be overcome by using a strong sterically hindered base like lithium diisopropyl amide (LDA) which forms the “kinetic” enolate.
Notes
Note 1. We can calculate how favored this reaction is by looking at the four components of our acid base reaction (acid, base, conjugate acid, conjugate base) comparing the pKas of the acid and the conjugate acid. The acid is the aldehyde (pKa = 17) and since our acid is HO(-), the conjugate acid is H2O (pKa =15.7). The equilibrium constant K would therefore be roughly (16-17 = -1 ) or 10-1 in the direction of product. So very little enolate would be present at any given time.
Note 2. The aldehyde C-H is not acidic, even though it seems like it should be!
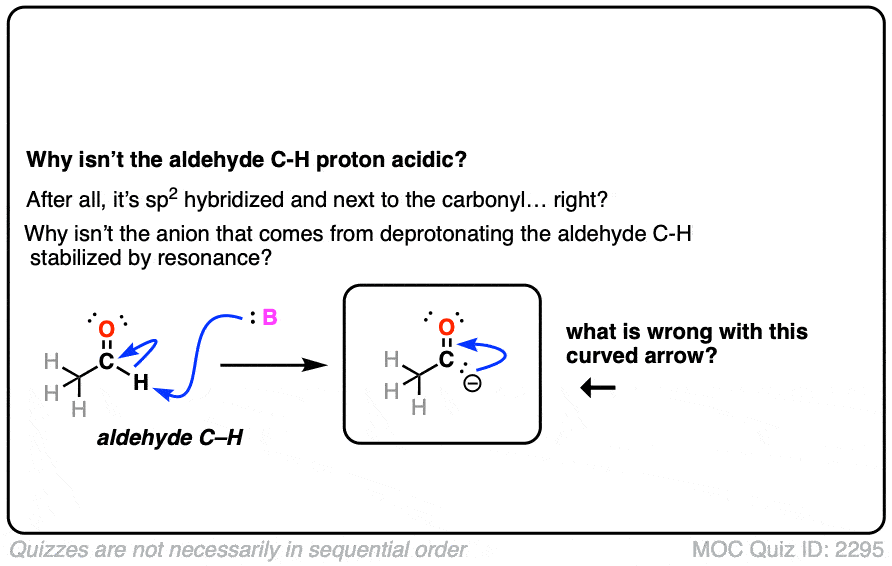 Click to Flip
Click to Flip
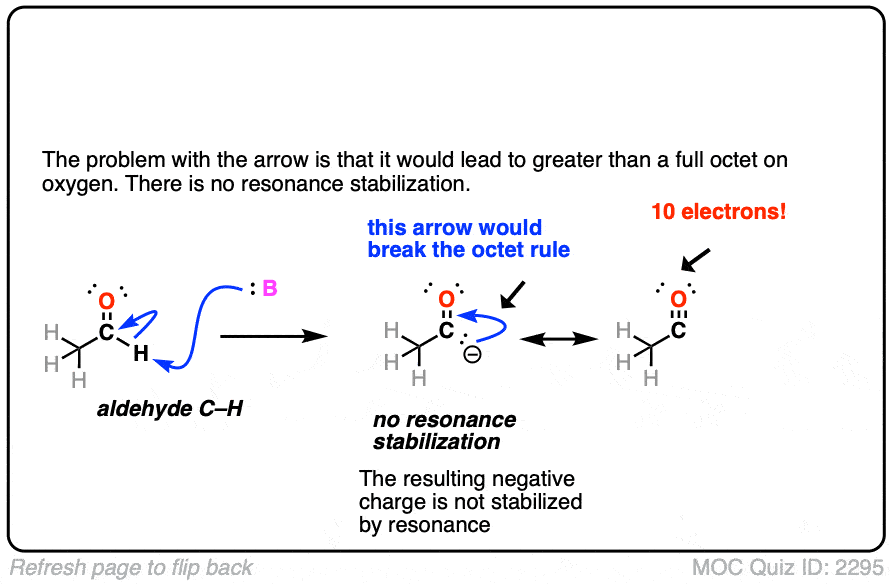
Note 3. Enolates tend to react at carbon rather than oxygen. One reason is that the oxygen tends to be tightly bound to the counter-ion of whichever base is used (e.g. Li+ or Na+). One way to get the oxygen to be more reactive is to use alkali metal salts that are bigger and form a weaker ionic bond with oxygen (e.g. potassium, K+).
Note 4. When only one chiral center is present, and it’s on the alpha-carbon, the result will be racemization. When there are multiple chiral centers, and one of the is on the alpha carbon, only the chiral center on the alpha-carbon will be affected.
This can lead to the formation of epimers. Epimer is a special name we give to diastereomers that differ in their configurations at only one chiral center.
For example, treating this optically pure (2S, 3S)-dimethylcyclohexanone with base will result in a mixture of (2S, 3S)-dimethylcyclohexanone and (2R,3S)-dimethylcyclohexanone. They aren’t enantiomers since they have the same configuration at C-3.
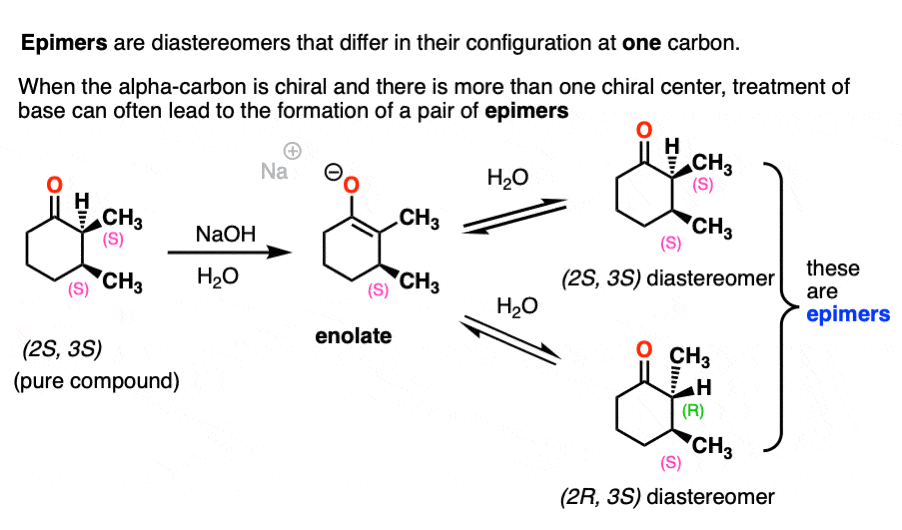
Note 5. We have to be careful with the base we use here. If we use NaOH, we will end up performing saponification of the ester (basic hydrolysis). If we use a strong base with a different R group we could end up performing transesterification.
Note 6. Alkylation of ordinary ketones using NaOR as base and alkyl halides such as CH3I is a mixed bag. It can often lead to multiple alkylations happening. The best way to perform these reactions is to irreversibly form the enolate using the strong base LDA and then use an alkyl halide.
The classic study on this was by House and Kramar. Highly recommend.
Note 7. A typical ratio of thermodynamic:kinetic enolates under “thermodynamic conditions” (NaOR/ROH) is about 4:1 but it can vary widely. Page 2 of this article provides a great overview on forming thermodynamic enolates.
Note 8. One question I get asked a lot is why Grignard reagents are able to add to ketones without deprotonating them first. After all the pKa of an aldehyde or ketone is about 16-18 and the pKa of an alkane is around 50, so why wouldn’t you expect an acid-base reaction?
The answer is that alpha protons are acidic but they are only acidic if the C-H bonds are aligned with the p-orbitals of the C-O pi bond. That’s because resonance delocalization is only possible when the p-orbitals can overlap.
This makes deprotonation at carbon quite slow, relative to addition to the carbonyl and also relative to deprotonation of O-H which doesn’t require orbital overlap.
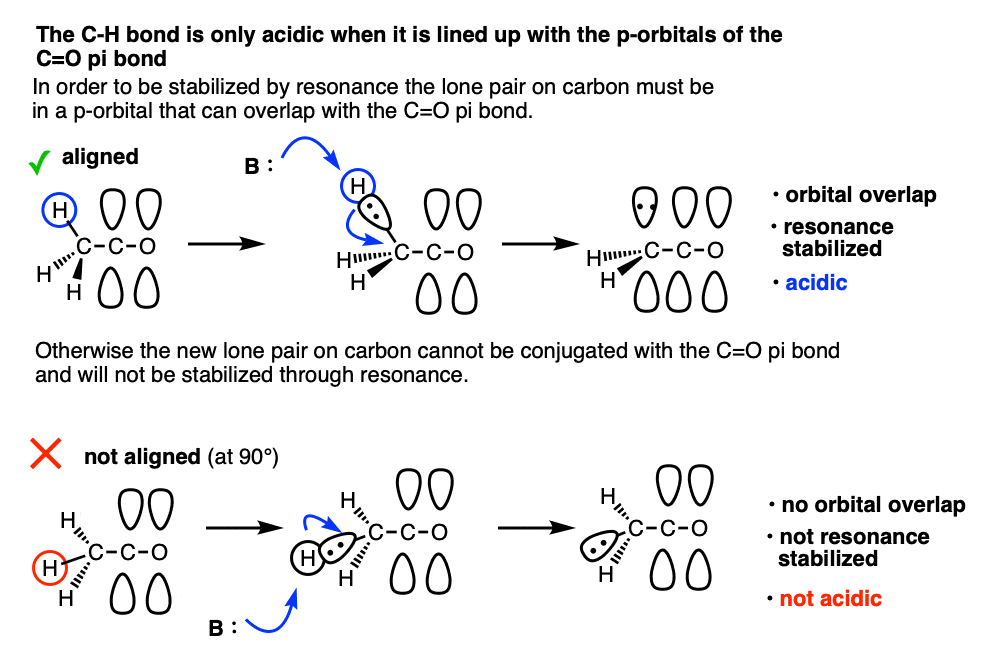
The classic example here is the bridgehead C-H bond of bicyclic ketones.
Note 9. Two other ways of making enolates that we encounter in introductory organic chemistry bear mention.
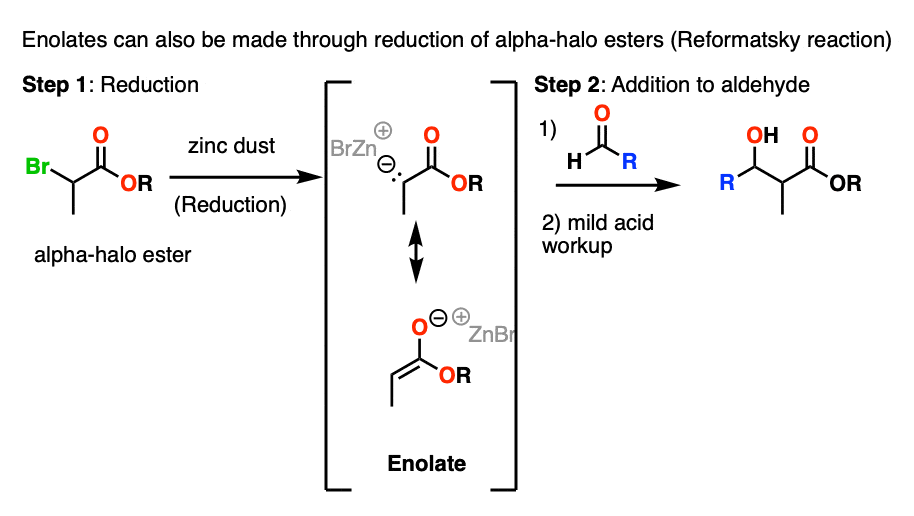
The first one is that alpha-halo esters can be treated with Zn to give enolates in a process called reductive formation of enolates . This is much like Grignard reagent formation.
The resulting enolates can add to aldehydes and ketones in a reaction known as the Reformatsky reaction.
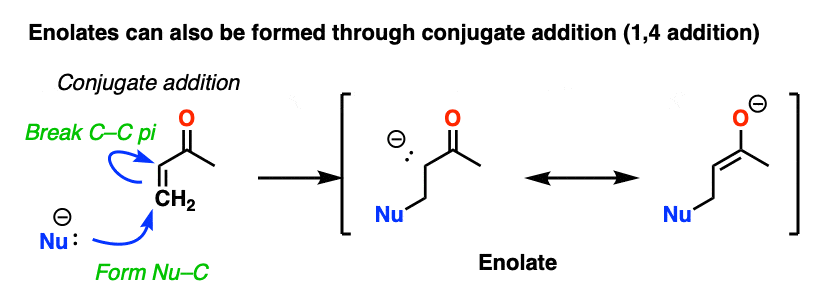
Another way of making enolates is through conjugate addition. Addition of a nucleophile to an enone gives a new enolate.
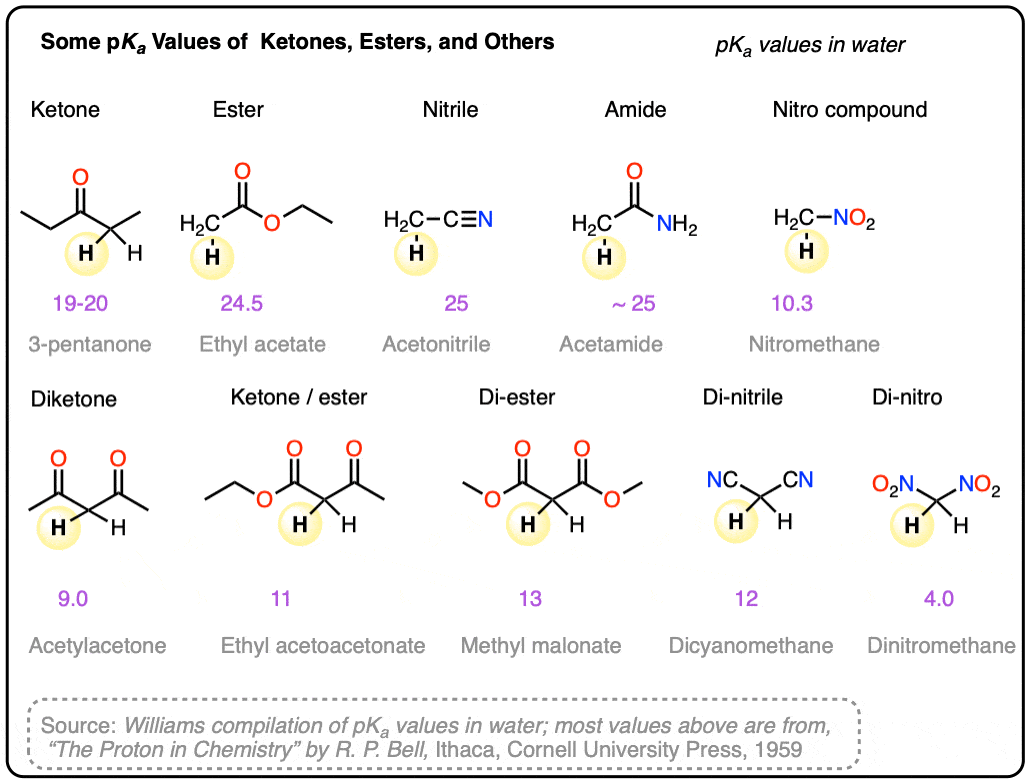
Note 10. Some pKa values in water. Main source.
Quiz Yourself!
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
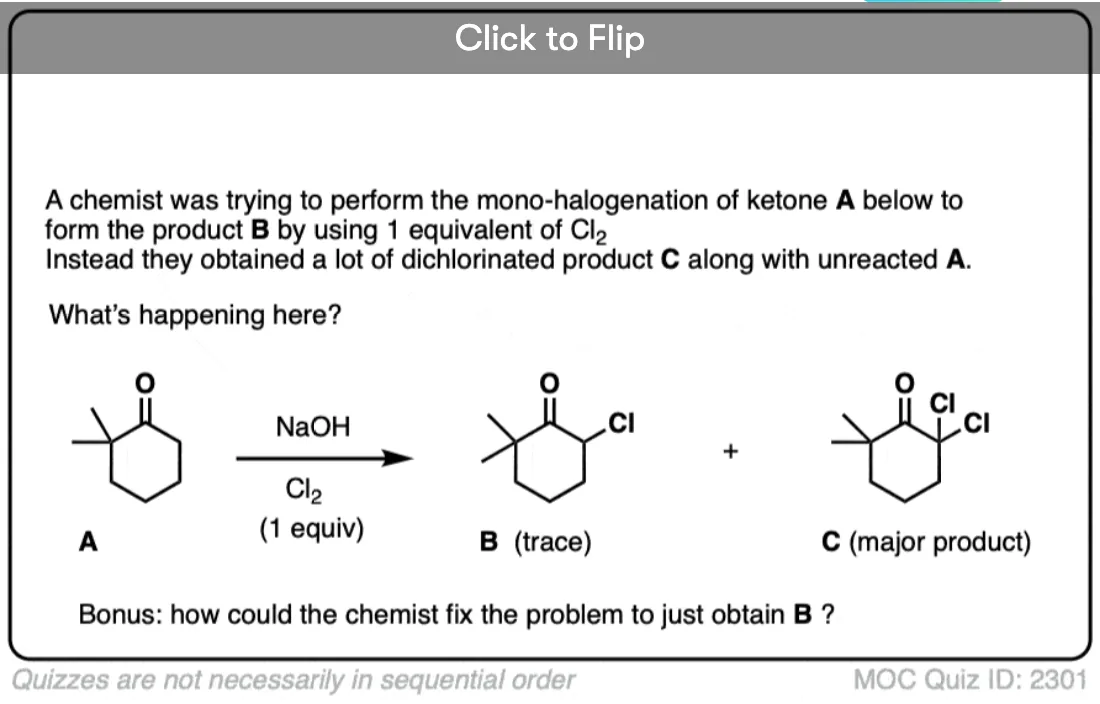
Become a MOC member to see the clickable quiz with answers on the back.
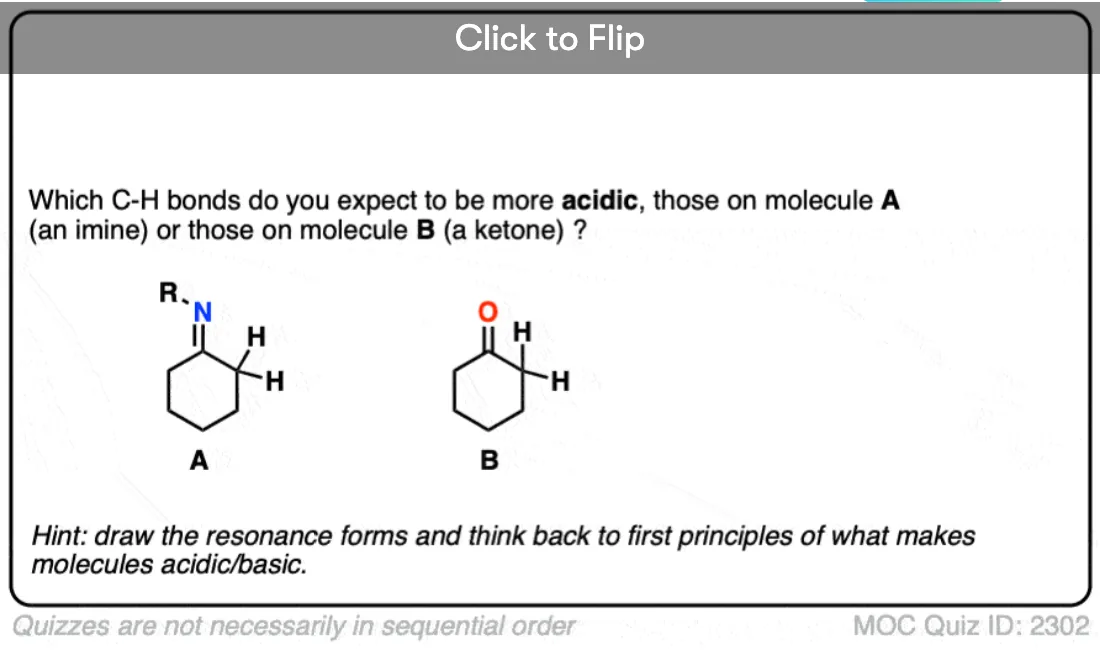
Become a MOC member to see the clickable quiz with answers on the back.
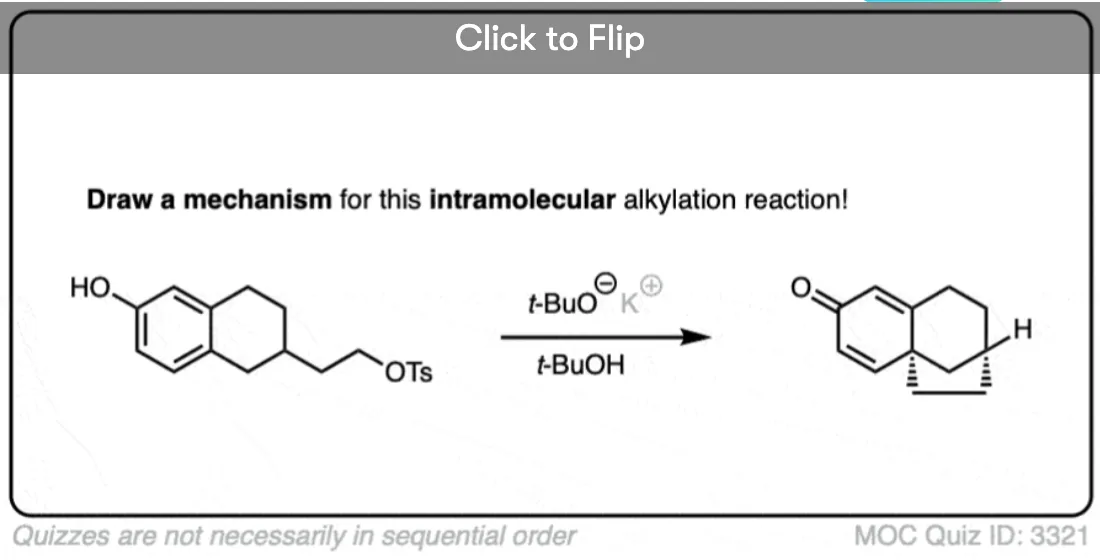
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
References
- Optical Activity in Relation to Tautomeric Change. Part VI. Comparison of the Rates of Racemization and Bromination of a Ketone.
Shing Kong Hsu and Christopher L. Wilson
J. Chem. Soc. 1936,623-625
DOI: doi.org/10.1039/JR9360000623
Early study on enolates showing that the rate of racemization was equal to the rate of halogenation, implying the reaction proceeds through an enolate intermediate. - Tetrahedron report number 25: Ketone enolates: regiospecific preparation and synthetic uses
Jean d’Angelo
Tetrahedron 1976, 32 (24), 2979-2990
DOI: 10.1016/0040-4020(76)80156-1
This review covers various methods for enolate formation, and has data on the composition of various ketone-enolate mixtures formed under kinetic and thermodynamic conditions.Prof. H. O. House (MIT, then Georgia Tech) published a series of papers on carbanion and enolate chemistry, studying kinetic and thermodynamic enolate formation in detail. A selection of these papers is below: - The Chemistry of Carbanions. V. The Enolates Derived from Unsymmetrical Ketones
Herbert O. House and Vera Kramar
The Journal of Organic Chemistry 1963, 28 (12), 3362-3379
DOI: 10.1021/jo01047a022 - The Chemistry of Carbanions. IX. The Potassium and Lithium Enolates Derived from Cyclic Ketones
Herbert O. House and Barry M. Trost
The Journal of Organic Chemistry 1965 30 (5), 1341-1348
DOI: 10.1021/jo01016a001 - Chemistry of carbanions. XV. Stereochemistry of alkylation of 4-tert-butylcyclohexanone
Herbert O. House, Ben A. Tefertiller, and Hugh D. Olmstead
The Journal of Organic Chemistry 1968, 33 (3), 935-942
DOI: 10.1021/jo01267a002 - Thermodynamic and Kinetic Controlled Enolates: A Project for a Problem-Oriented Laboratory Course
Augustine Silveira Jr., Michael A. Knopp, and Jhong Kim
Journal of Chemical Education 1998, 75 (1), 78
DOI: 1021/ed075p78
A paper from J. Chem. Ed. that covers how to demonstrate the concepts of kinetic and thermodynamic enolates in an undergraduate laboratory session. - Chemistry of carbanions. XVI. Stereochemistry of alkylation of the 1-acetyl and 1-cyano derivatives of 4-tert-butylcyclohexane
Herbert O. House and Thomas M. Bare
The Journal of Organic Chemistry 1968, 33 (3), 943-949
DOI: 1021/jo01267a003
In ketone and ester enolates that are exocyclic to a conformationally biased cyclohexane ring there is a small preference for the electrophile to approach from the equatorial direction. If the axial face is further hindered by addition of a substituent, the selectivity is increased. - A stereoselective total synthesis of (±)-gymnomitrol
Steven C. Welch and Suthep Chayabunjonglerd
Journal of the American Chemical Society 1979, 101 (22), 6768-6769
DOI: 1021/ja00516a057
The first step in this total synthesis is the alkylation of the Li enolate of 2-methylcyclohexanone, which is made by deprotonation by LDA at -78 °C, followed by equilibration to the thermodynamically more stable substituted enolate anion at room temperature for 4-5 hours. - THE FORMATION AND ALKYLATION OF SPECIFIC ENOLATE ANIONS FROM AN UNSYMMETRICAL KETONE: 2-BENZYL-2-METHYLCYCLOHEXANONE AND 2-BENZYL-6-METHYLCYCLOHEXANONE
Martin Gall and Herbert O. House
Org. Synth. 1972, 52, 39
DOI: 10.15227/orgsyn.052.0039
This procedure from Organic Syntheses has detailed experimental conditions for regioselective alkylation of an unsymmetrical ketone through kinetic and thermodynamic enolate formation conditions. - Thermodynamic and Kinetic Controlled Enolates: A Project for a Problem-Oriented Laboratory Course
Augustine Silveira Jr., Michael A. Knopp, and Jhong Kim
Journal of Chemical Education 1998, 75 (1), 78
DOI: 1021/ed075p78
A paper from J. Chem. Ed. that covers how to demonstrate the concepts of kinetic and thermodynamic enolates in an undergraduate laboratory session. - Kinetic Enolate Formation by Lithium Arylamide: Effects of Basicity on Selectivity
Linfeng Xie, Keith Vanlandeghem, Kurt M. Isenberger, and Carolyn Bernier
The Journal of Organic Chemistry 2003, 68 (2), 641-643
DOI: 10.1021/jo0263465
This paper is on the topic of enolate formation and examines how the stereochemistry of the enolate formed varies depending on the nature of the Li-amide base.Some papers on advanced topics related to enolate chemistry: - Pseudoephedrine as a Practical Chiral Auxiliary for the Synthesis of Highly Enantiomerically Enriched Carboxylic Acids, Alcohols, Aldehydes, and Ketones
Andrew G. Myers, Bryant H. Yang, Hou Chen, Lydia McKinstry, David J. Kopecky, and James L. Gleason
Journal of the American Chemical Society 1997, 119 (28), 6496-6511
DOI: 1021/ja970402f
The enolates derived from N-acylation of pseudoephedrine can be selectively facially alkylated. It is easier to induce diastereoselectivity in a reaction than enantioselectivity, and one way to do that is to use a chiral auxiliary – a chiral scaffold that you attach to the substrate and then remove after you’ve alkylated it. This is a paper that demonstrates practical chemistry – at the time, pseudoephedrine was easier to obtain than it is now, and Prof. Myers (now at Harvard) has extensively documented the reaction and the procedures in order to make it reproducible, robust, and reliable. - Asymmetric alkylation reactions of chiral imide enolates. A practical approach to the enantioselective synthesis of .alpha.-substituted carboxylic acid derivatives
D. A. Evans, M. D. Ennis, and D. J. Mathre
Journal of the American Chemical Society 1982, 104 (6), 1737-1739
DOI: 10.1021/ja00370a050
This is a landmark paper, in which Prof. Evans pioneered the use of chiral auxiliaries and showed how they could be used for asymmetric aldol reactions. The auxiliary used here is a chiral oxazolidinone, and this chemistry is known as the “Evans Asymmetric Aldol” or the “Evans chiral auxiliary”. - Acyclic stereoselection. 7. Stereoselective synthesis of 2-alkyl-3-hydroxy carbonyl compounds by aldol condensation
Clayton H. Heathcock, Charles T. Buse, William A. Kleschick, Michael C. Pirrung, John E. Sohn, and John Lampe
The Journal of Organic Chemistry 1980 45 (6), 1066-1081
DOI: 10.1021/jo01294a030
This paper introduces the concepts of stereoselectivity in aldol reactions when an enolate is adding to another carbonyl compound. Transition-state models are discussed, and the role of Li ions in chelation is also illustrated.
Good work
Is it always true that we use second best resonance form as in while drawing the mechanisms?