Organometallics
Reactions of Grignard Reagents
Last updated: May 28th, 2026 |
All About The Reactions of Grignard Reagents
- Grignard reagents are excellent carbon-based nucleophiles as well as strong bases.
- They will add to aldehydes and ketones to form alcohols (after a protonation step)
- They will add twice to esters to give tertiary alcohols.
- They will add to the less-substituted side of epoxides
- Grignard reagents will also react with carbon dioxide (CO2) to give carboxylic acids (after acid workup).
- Grignard reagents will not perform SN2 reactions with alkyl halides. They are also not compatible with carboxylic acids or alcohols.
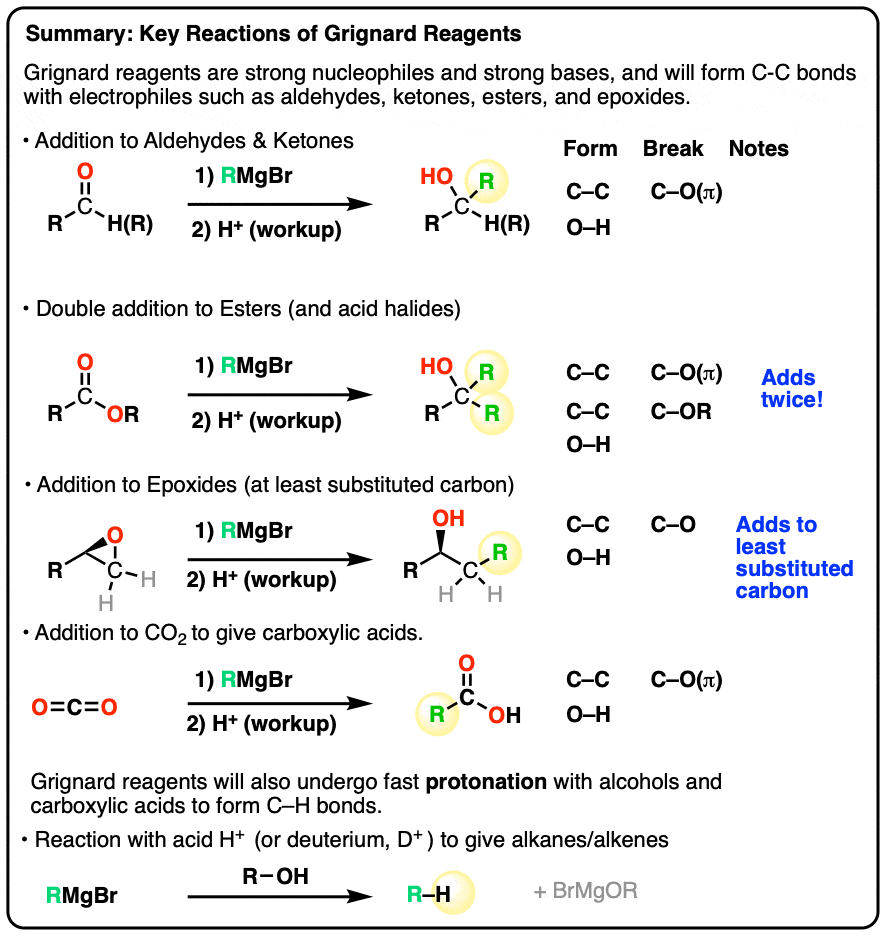
Table of Contents
- Reminder: Grignard Reagents Are Nucleophiles
- Addition of Grignard Reagents To Epoxides
- Reaction of Grignard Reagents With Aldehydes And Ketones
- Reaction of Grignard Reagents With Esters
- Why Do Grignards Add Twice To Esters? The Mechanism
- Summary: Reactions of Grignard Reagents
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. Reminder: Grignard Reagents Are Nucleophiles
So far in this series we’ve introduced organometallic compounds and said that their carbons tend to be nucleophilic. We’ve learned how to make them from alkyl, alkenyl or aryl halides (along with some ways not to make them!) and saw that they are very strong bases.
Most interesting about Grignards is that they are carbon-based nucleophiles and we can thus combine Grignard reagents with various electrophilic carbon species to form new carbon-carbon bonds.
And since carbon-carbon bonds constitute the “backbone” of molecules in organic chemistry, it turns out that this class of reactions is very useful. As a matter of fact, it won its discoverer, Victor Grignard, the Nobel Prize for Chemistry back in 1912.
For our purposes, the key carbon-based electrophiles that Grignard reagents react with are epoxides, aldehydes, ketones, and esters. Let’s go through them in turn.
2. Addition of Grignard Reagents To Epoxides
Epoxides (“oxiranes” if you are an IUPAC stickler) are 3-membered cyclic ethers which possess considerable ring strain. As we’ve seen, this ring strain makes them somewhat “spring loaded” toward attack by nucleophiles, which will result in formation of a new bond to carbon and opening of the ring.
Negatively charged nucleophiles (such as Grignards) tend to react with epoxides in a manner similar to the SN2 reaction: attack occurs at the least substituted carbon of the epoxide. Here’s an example:
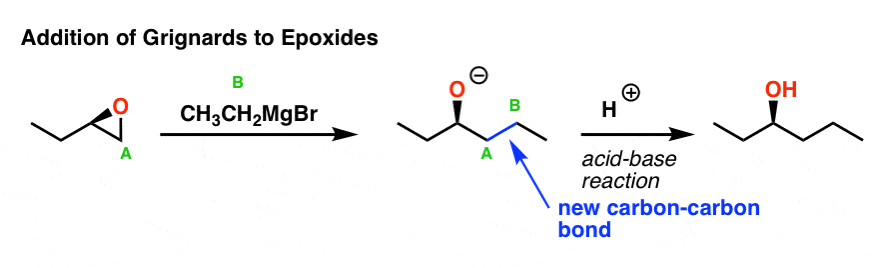
Note the bonds that formed and broke here: we formed a new C-C bond (between carbons A and B), and broke a C-O bond (between carbon A and the oxygen). This resulted in a negatively charged oxygen (alkoxide): to produce final alcohol product, we typically quench the reaction with a source of acid, forming O–H.
Here’s how the reaction works. The hard thing is to recognize that the nucleophile is the pair of electrons in the C-Mg bond: remember from previous posts that carbon is strongly δ- (nucleophilic) because of its greater electronegativity as compared to magnesium.
It might be helpful to imagine the Grignard reagent below as CH3CH2– . Other than that the reaction is fairly straightforward if you’ve seen an SN2 reaction before: we simultaneously form C-C and break C-O.
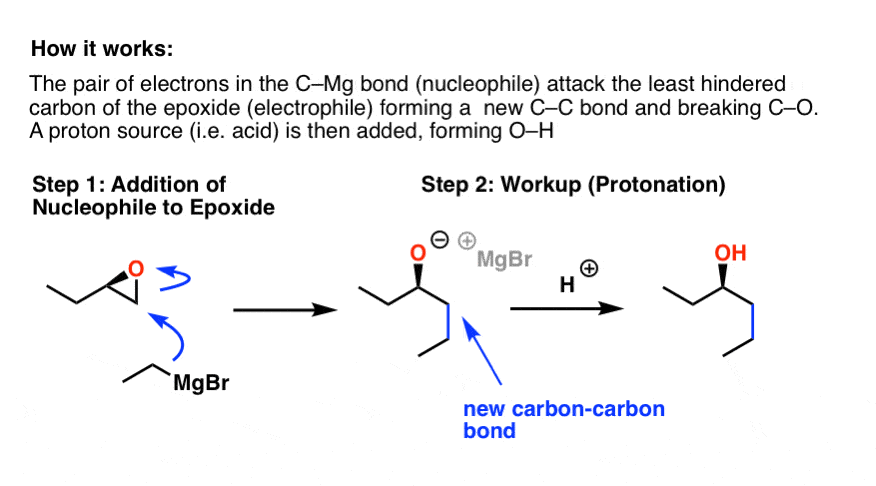
Note that this reaction also forms an “alkoxide”. In order to obtain our neutral alcohol product at the end, we must perform second step: a “workup” (“quench”) with a source of acid. This is written a variety of ways – H+, H3O+, H2O, or just “acid workup”. This step occurs after our key Grignard reaction, for what should be obvious reasons – being strong bases, Grignard reagents are destroyed by acid.
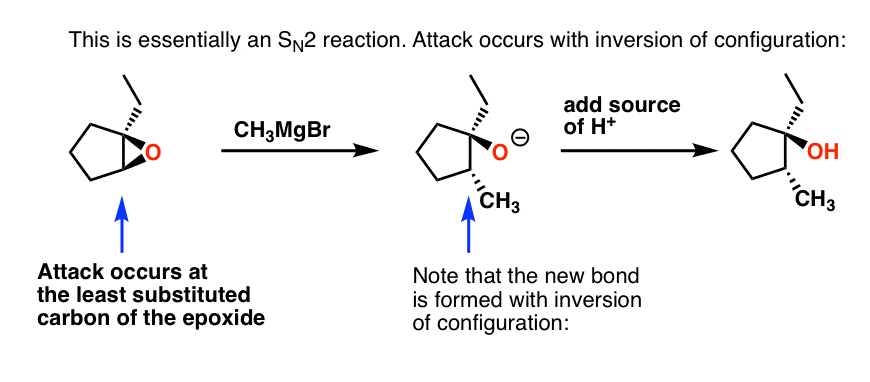
Another thing to keep in mind is stereochemistry of the epoxide.Consistent with an SN2 reaction, if the reaction occurs at a secondary carbon, we will observe inversion of configuration:
3. Reaction of Grignards With Aldehydes and Ketones
A second class of important electrophiles that react with Grignards (and arguably THE most important class of electrophiles) is aldehydes and ketones. If you haven’t covered the reactions of these functional groups yet, a short summary would be this: the carbonyl carbon is an electrophile, and when nucleophiles react at this carbon, it’s accompanied by cleavage of the C-O pi bond (π bond). (For more on the addition mechanism to carbonyls, see post: Nucleophilic Addition)
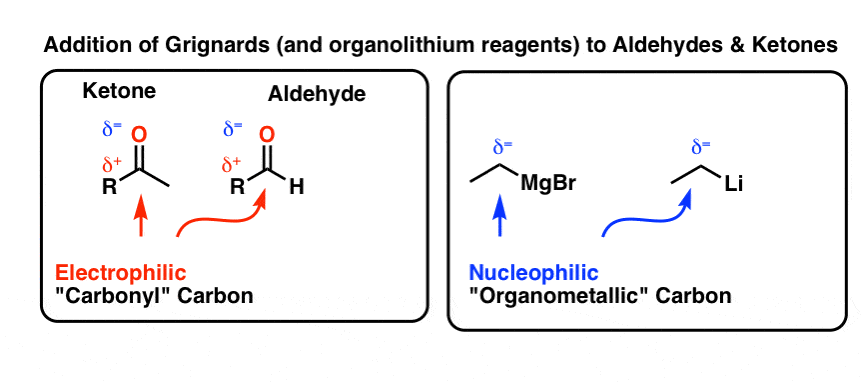
Here are some examples of reactions of Grignards with aldehydes and ketones. Note that in each case we are forming a new bond between the carbonyl carbon (labelled A) and the carbon bound to magnesium (labelled B), and we are breaking the C-O pi bond in the process.
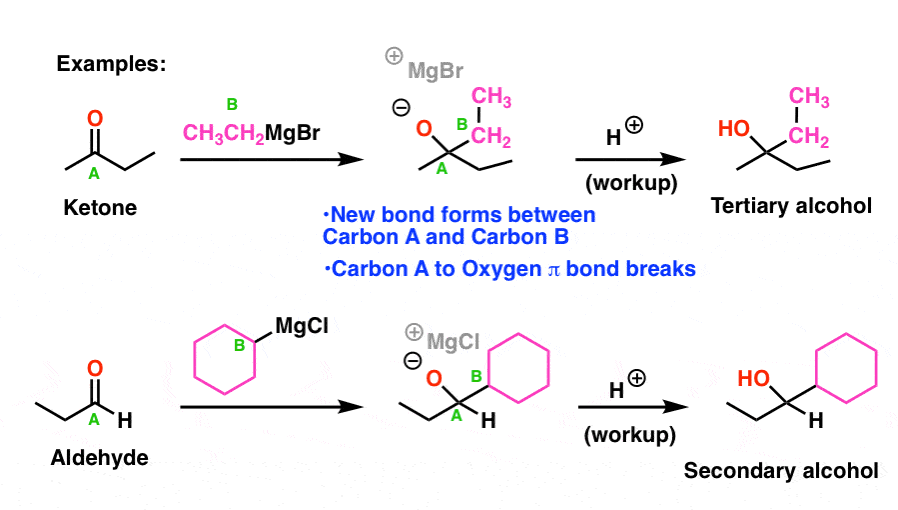
So how does this reaction work?
Let’s get familiar with a VERY important mechanism called “addition” (sometimes called, “1,2-addition”). This is by far the most important reaction of the carbonyl group, and if you give yourself a chicken for every time you will see variations of it in Org 2, you will have a lot of eggs in your room by the end of the semester.
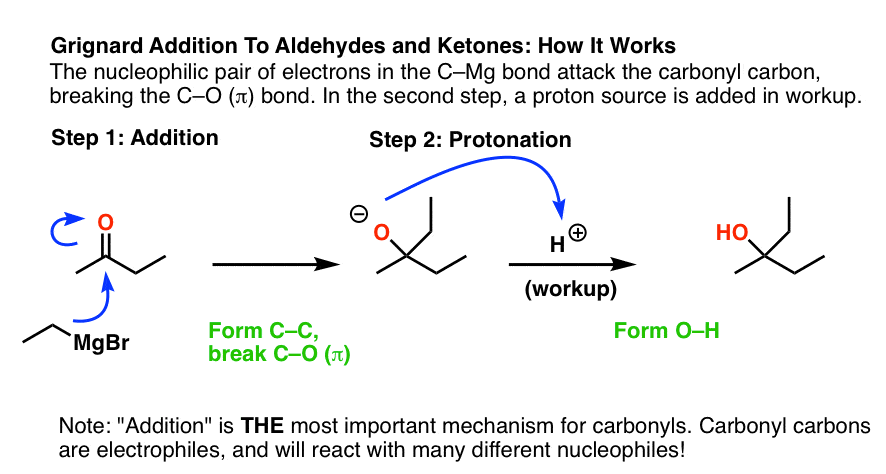
Note that this reaction also forms an “alkoxide”. In order to obtain our neutral alcohol product at the end, we must perform a “workup” (“quench”) with a source of acid, forming O-H.
4. Reaction of Grignard Reagents With Esters
Esters are close relatives of aldehydes and ketones: they consist of a carbonyl group directly attached to an OR group. As you might expect, they react with Grignards in a similar fashion to aldehydes and ketones: with formation of a new C-C bond and breakage of a C-O (pi bond).
However, there’s a twist with the reaction of esters that isn’t present with aldehydes and ketones. Look carefully: what’s different?
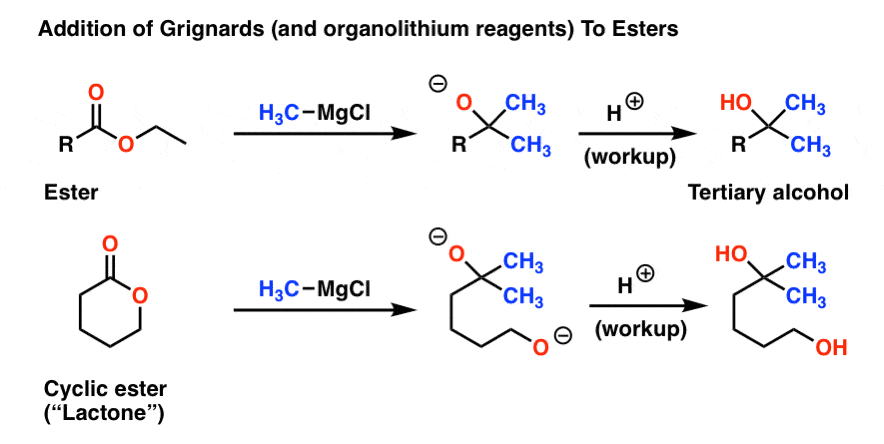
Note that in both cases we added two equivalents of our Grignard reagent to the ester, forming a tertiary alcohol.
Wait a minute – how did this happen?!
5. Why Is There “Double Addition”With Esters? The Mechanism
This reaction incorporates the second most important mechanism of carbonyls (next to “addition”), namely, “elimination“. In fact “elimination” is the exact reverse of “addition” ! Let’s walk through it. There are 4 steps
- In the first step, the Grignard performs an addition reaction on the ester, forming C-C and breaking C-O (pi), giving us an intermediate with a negatively charged oxygen. We’ve seen this type of reaction before in the addition of Grignards to aldehydes and ketones.
- Now comes the new step: elimination (sometimes, “1,2 elimination”). This intermediate has a reasonably good leaving group (OCH2CH3 in the case below). What happens next is reformation of the C-O pi bond with expulsion of the leaving group (CH3CH2O– in the case below). In other words, we form C–O π and break a C–O single bond. The new product is a ketone.
Together, these two steps are often referred to as Nucleophilic Acyl Substitution (See post: Nucleophilic Acyl Substitution)
Elimination does not occur in addition to aldehydes and ketones because the leaving group would have to be the extremely strong bases H(-) or R(-). It is reasonably favorable for esters because the leaving group RO(-) is of comparable basicity to the negatively charged oxygen of the tetrahedral intermediate. [Note 1]
- But wait! There’s more! After Step 2, we have a new ketone. As we’ve seen before, Grignards will react quickly with ketones in yet another addition reaction [Step 3]. Here, as in Step 1, we form C–C and break C–O (pi). The result is a tertiary alkoxide (the conjugate base of a tertiary alcohol).
[Wait, you might ask. If we just use one equivalent of Grignard reagent, is it possible to get the reaction to stop at the ketone stage? The short answer is “no”. [See Note 3 for the long answer]]
- Finally, protonation of this tertiary alkoxide yields the tertiary alcohol (Step 4).
Here’s the graphical walkthrough:
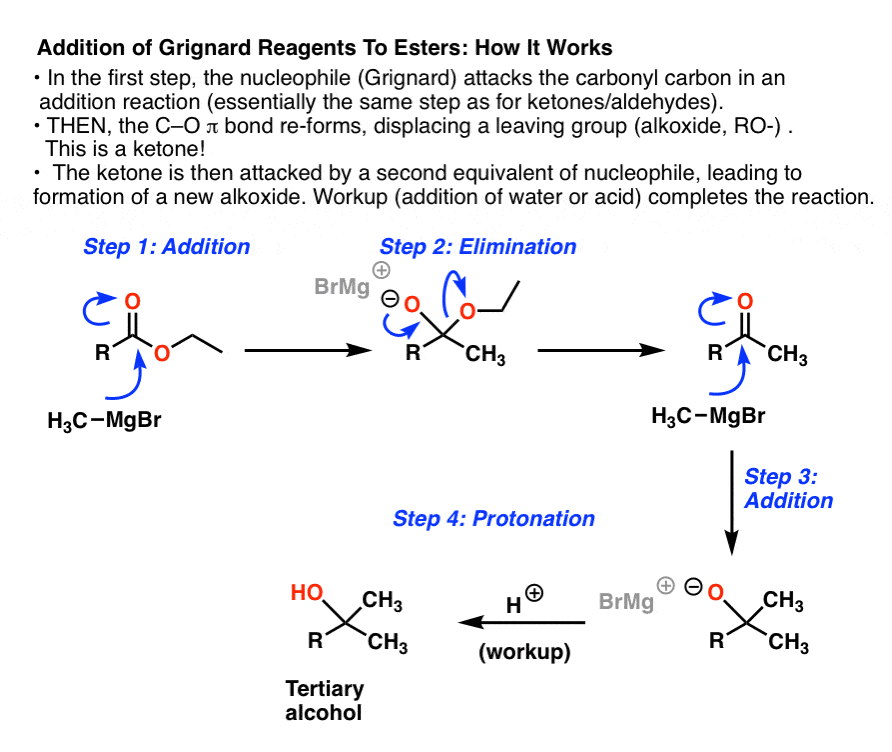
6. Summary: Reactions of Grignard Reagents
That does it for the key reactions of Grignard reagents you’ll see in most Org 1 and Org 2 courses.
In the next post we’ll talk about yet another way to screw up formation of Grignard reagents, and it involves the reactions in this post.
Next Post: Protecting Groups In Grignard Reactions
Notes
Note 1: Although alkoxides (RO–, the conjugate base of alcohols, pKa 16-18) are not on anyone’s list of Great Leaving Groups, they are some 25 orders of magnitude better leaving groups than hydrides (H–, the conjugate base of hydrogen, pKa 40) and more than 30 orders of magnitude better than alkyl groups (R- , the conjugate base of alkanes, pKa 50). Thus, when the alkoxide intermediate is formed in Step 1, there is not any deep energetic penalty for the C-O pi bond to reform and for RO- to be expelled: after all, we are simply replacing a strong base (the O- ) with one of comparable basicity.
Note 2. Why are ketones more reactive towards Grignard reagents than esters? This requires understanding the phenomenon of pi donation. The lone pair on oxygen donates electron density into the carbonyl carbon. This is worthy of a separate post, but here’s the bottom line:
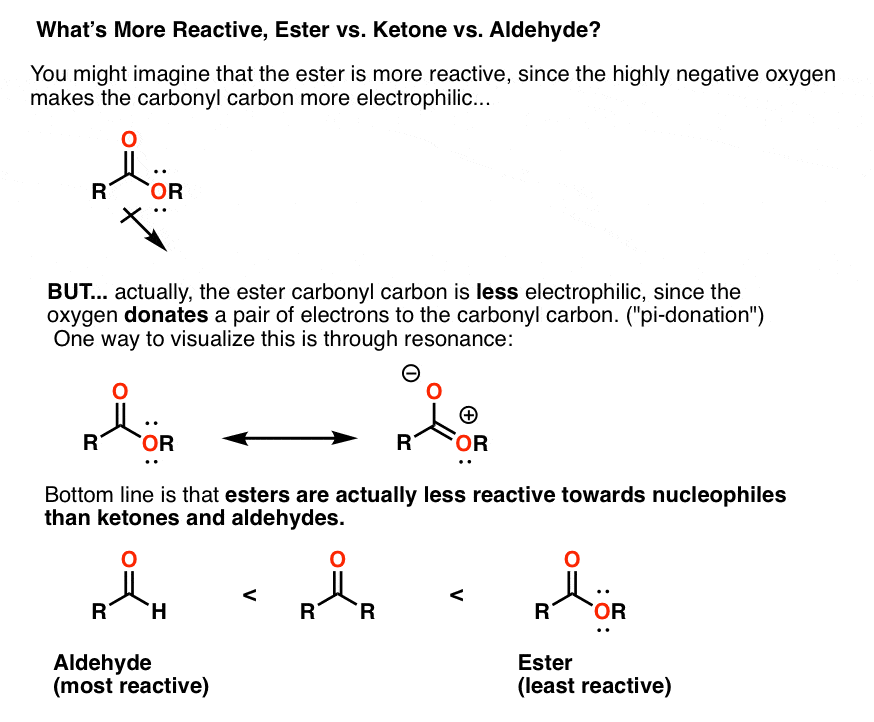
Note 3. Alas, no. Using 1 equivalent of Grignard will result in 0.5 equivalents of a tertiary alcohol and 0.5 equivalents of the starting ester. The reason why is that Step 2 [elimination] is quite fast!
Once elimination occurs, we will have ketone in the presence of an ester. For interesting reasons [see Note 2] ketones are more reactive than esters toward Grignard reagents, which means they will be consumed more quickly.
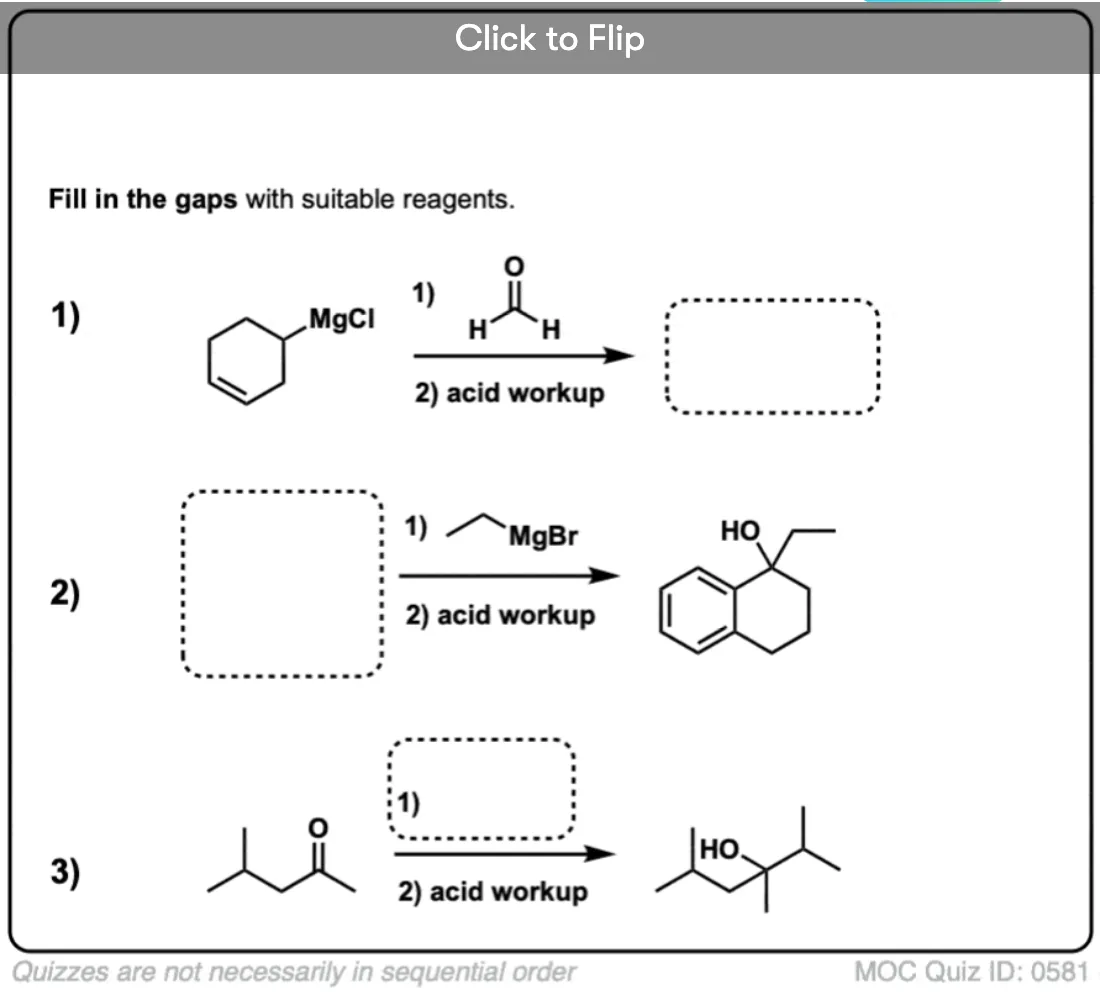
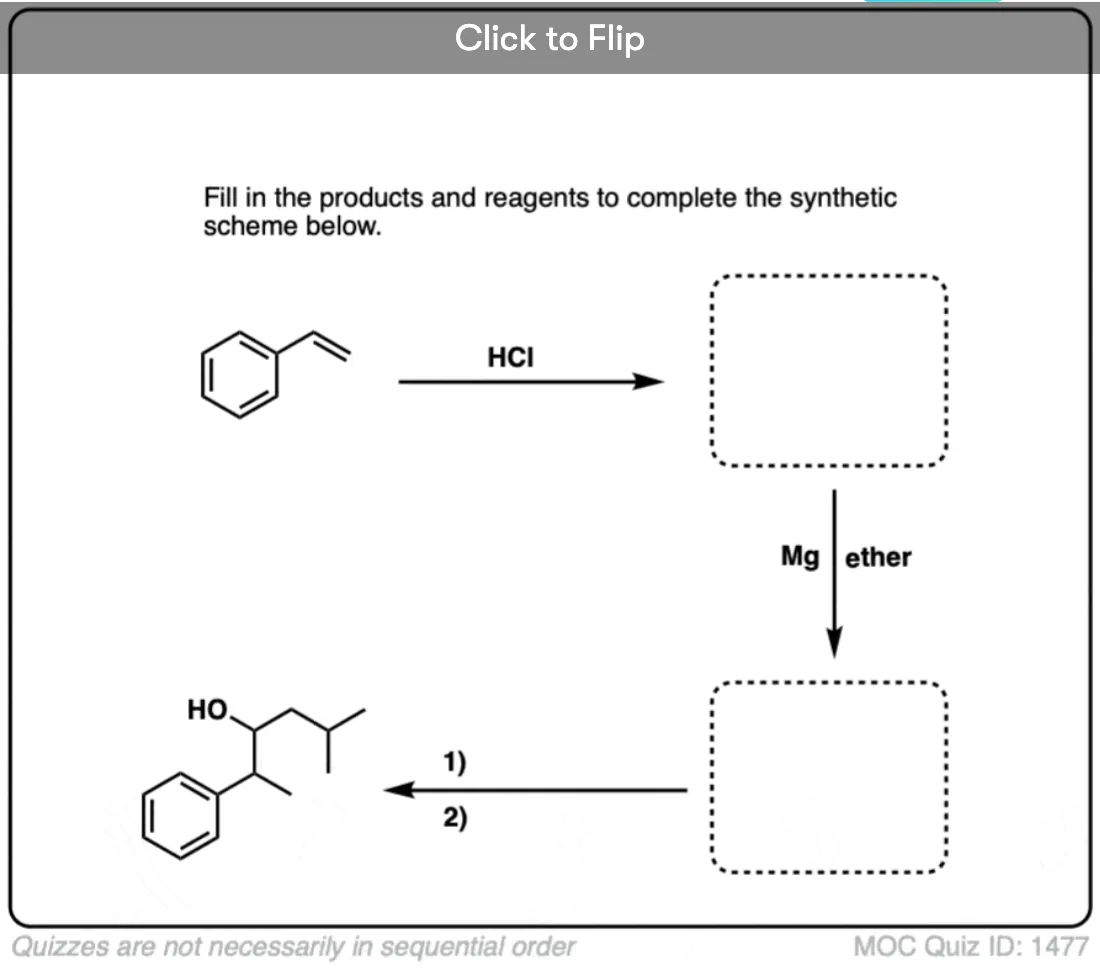
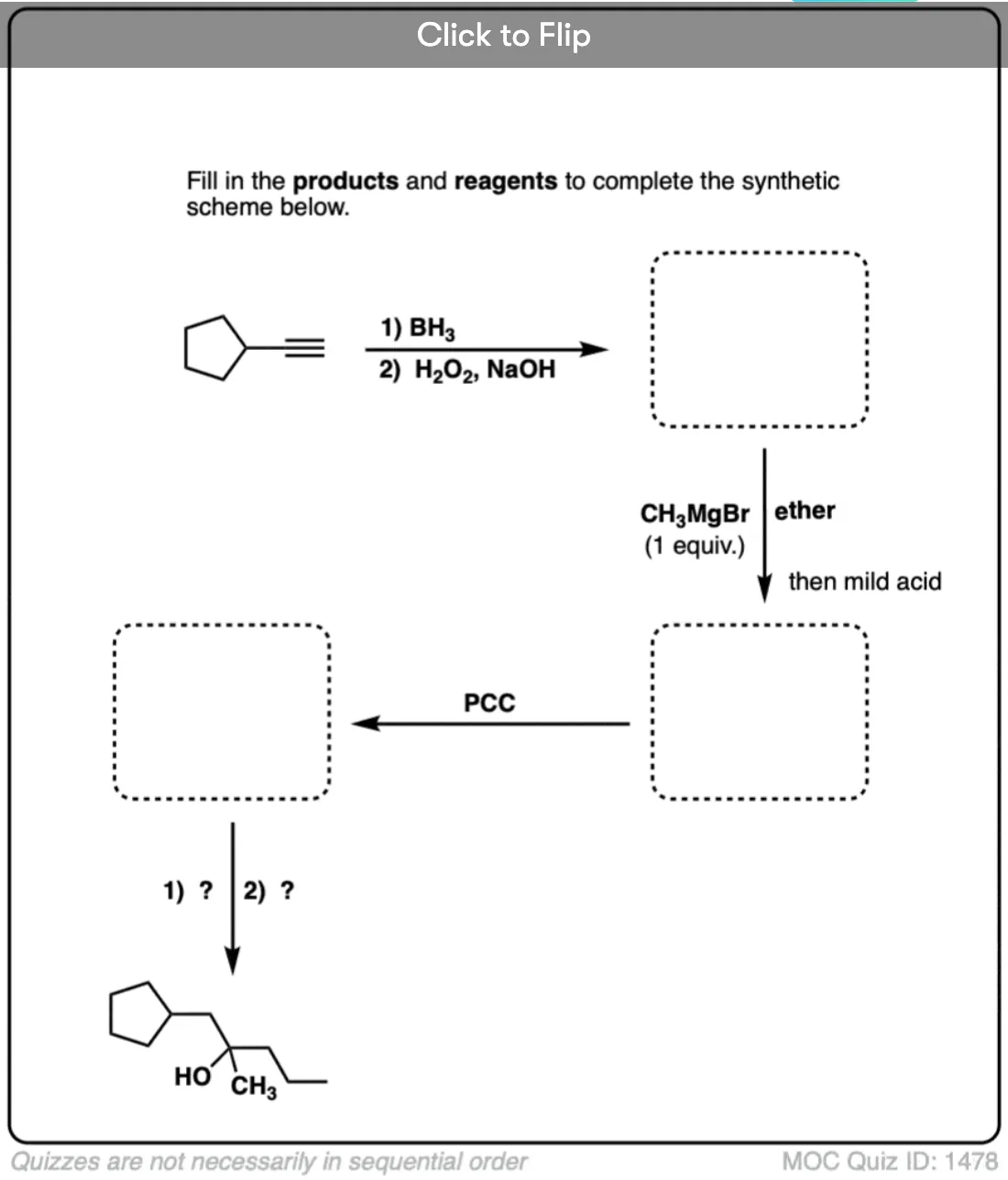
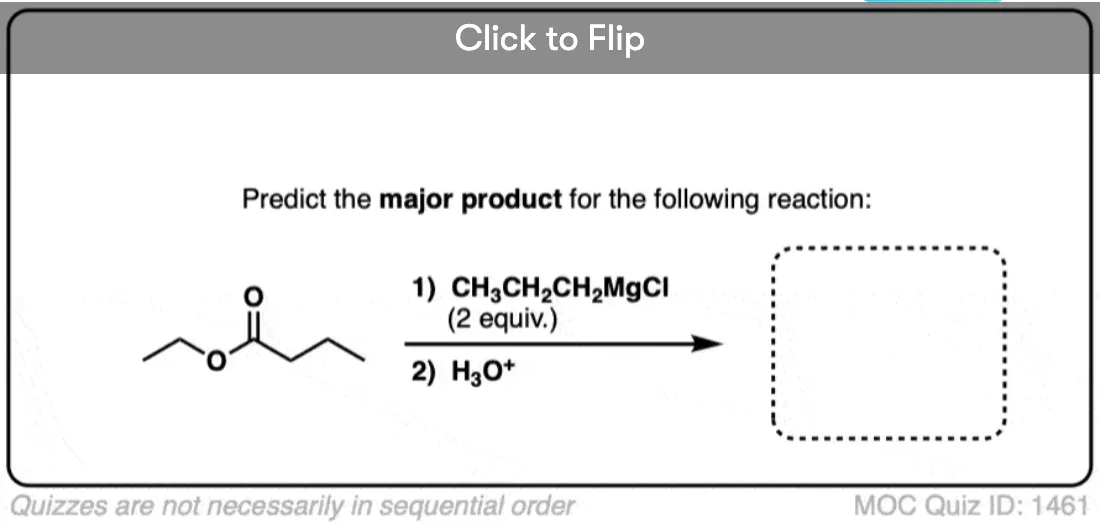
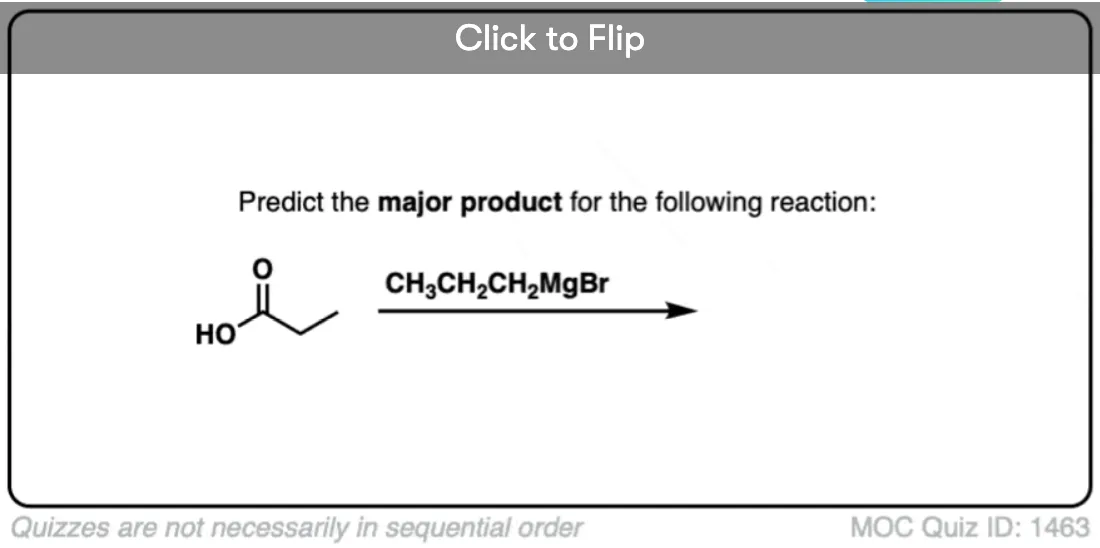
Quiz Yourself!
 Click to Flip
Click to Flip
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading:
- The Grignard Reagents
Dietmar Seyferth
Organometallics 2009 28 (6), 1598-1605
DOI: 10.1021/om900088z
A historical overview on Grignard reagents by the late Prof. Dietmar Seyferth (MIT), founding editor of the journal Organometallics. - Secondary and Tertiary Alkyllithium Compounds and Some Interconversion Reactions with Them
Henry Gilman, Fred W. Moore, and Ogden Baine
Journal of the American Chemical Society 1941, 63 (9), 2479-2482
DOI: 1021/ja01854a046
Prof. Henry Gilman (Iowa State) was a pioneer in organometallic chemistry in the first half of the 20th century. In this paper he describes the synthesis and reactivity of various alkyllithiums (n-butyllithium, s-butyllithium, isopropyllithium, and t-butyllithium). The synthesis is from the alkyl halide and lithium metal, as can be seen in the experimental section. - t-Butyllithium
Paul D. Bartlett, C. Gardner Swain, and Robert B. Woodward
Journal of the American Chemical Society 1941, 63 (11), 3229-3230
DOI: 1021/ja01856a501
This communication is from some legendary figures in organic chemistry and describes the preparation of t-butyllithium. - 2-PHENYLPYRIDINE
C. W. Evans and C. F. H. Allen
Org. Synth. 1938, 18, 70
DOI: 10.15227/orgsyn.018.0070
The first step in this procedure is a preparation of phenyllithium from bromobenzene and lithium metal. Organic Syntheses is a reputable source of reproducible and independently tested synthetic organic procedures. - The mechanism of the lithium – halogen Interchange reaction : a review of the literature
Bailey, W. F.; Patricia, J. J.
Organomet. Chem. 1988, 352 (1-2), 1-46
DOI: 10.1016/0022-328X(88)83017-1
In modern organic chemistry, organolithium reagents are rarely prepared from scratch (i.e. using Li metal), due to the ready availability of alkyllithium reagents from vendors (e.g. MeLi, the BuLi reagents, PhLi, etc.). Instead, these reagents can be used to form other organolithium species through a process known as lithium-halogen exchange. - What’s Going on with These Lithium Reagents?
Hans J. Reich
The Journal of Organic Chemistry 2012, 77 (13), 5471-5491
DOI: 1021/jo3005155
Prof. Hans Reich (U. Wisconsin-Madison) has spent his career studying the behavior of organolithium species, and this is an account of his research and the surprising findings he made. This is classic Physical Organic chemistry.
Why don’t grignard reagents do SN2 with alkyl halides?
I don’t have a great or satisfying answer for you. They just tend not to, or in cases where they do, the yields are low and are accompanied by other products.
The best substrates for SN2 reactions are alkyl halides, and generally what tends to happen is either no reaction, or the Grignard reagent reduces the alkyl halide to an alkane. Deprotonation can also happen.
Part of the reason is that the structure of Grignard reagents in solution is not simple – they form clusters – and this is not good from a steric hindrance standpoint. Also, what makes Grignards add so well to the C=O group is that the magnesium can act as a Lewis acid and activate the carbonyl towards nucleophilic attack. The halogen of alkyl halides are not activated (i.e. made into a better leaving group) by magnesium.
In order to do SN2 reactions, the best choice is to use a Gilman reagent (organocuprate). Those work well, especially with primary alkyl halides. They are less basic and perform fewer side reactions.
What happens when CH3MgBr reacts with Pent-2,4-dione?
How many equivalents of CH3MgBr?
Due to keto-enol tautomerism pent-2,4-dione has an acidic enol O-H that may be deprotonated faster than addition to the carbonyl can occur.
Can a Grignard reagent interact with molecular nitrogen, as in N2 gas?
No. Molecular N2 does not interact with Grignard reagents, but there are other metallic compounds which do (molybdenum compounds come to mind).
If we were to do an acid work up right after the first step of reaction of Grignard reagent with an ester, could a hemiacetal be formed? If not, why?
No, it’s not practical to quench the reaction fast enough to get single addition in most cases. There are ways to do mono-addition to form ketones from esters (e.g. using a Weinreb amide) but a fast quench of a Grignard is not it.
Why is acidic workup not done after the first step in case of reaction with ester?
There is no good way to time the workup so that it is done after the first addition occurs but before the second does.
After the first addition happens, the subsequent breakdown to the ketone and second addition occur very quickly.
If a ketone is desired, there are species known as “Weinreb amides” that form a stable tetrahedral intermediate after the first addition. This tetrahedral intermediate can be quenched and a ketone obtained.
Why grignard reaction need extremely cold conditions like -78°C
It does not, Grignard reactions happen fine at room temperature and above.
Hi James,
Thanks for creating this website. It has helped me a lot and I can’t thank you enough.
I have one doubt. Please if you can help me out. What happens when you take a very sterically hindered carbonyl or acid derivative substrate and equally sterically hindered grignard reagent? The mechanism in both case I believe is SN2 and SN(addition-elimination). Will the reaction proceed? Or not? Or if it does then how? And do you have any reference which I can go through? Actually back in the college, one of my professors had taught us about bulky G.R. giving out hydride , which attacks further on sterically hindered substrates. But since, I am not in college now, I can’t ask that professor. And I haven’t been able to find it in any standard textbooks. I checked clayden, klein and bruice.
You’ll probably have reduction of the carbonyl. I would check out March’s Advanced Organic Chemistry.
What happens if do not wash with saturated NaCl solution at the last step of Grignard reaction
A Grignard is generally quenched with aqueous solution and extracted with organic solvent. The purpose of the extraction is to remove any magnesium salts and other polar byproducts.
Can NaCl react with CH3MgBr?
One of the issues for NaCl would be getting it to dissolve in an appropriate solvent. But let’s just say we were able to get some to dissolve in ether, in the presence of CH3MgBr. About the only thing that might happen is some of the Br ions of the Grignard reagent might exchange with some of the Cl ions from the NaCl. This would not influence the reactivity of the Grignard reagent in any significant way. I hope this answers your question.
Can Grignard Reagent react with alkenes?
Thank You!
can we say what you explained in point 5 in another way that in esters there is a 4 member chelate formation due to ion-dipole interaction between oxygen and (MgBr)+ and as 4 member ring is not comfortable so it breaks and form MgBrOR and leaving us ketone
Hi, Will other organometallics react in the similar way as grignard reagent?
No, not exactly, the choice of metal can have a very large effect on reactivity. The reagents that are the most similar to Grignard reagents are organolithium reagents, which will also add to carbonyls (aldehydes, ketones, esters) as well as act as strong bases. Note that the electronegativity of lithium is very close to magnesium. Once you get toward the right half of the periodic table (such as copper or palladium) the reactivity becomes very different; the organometallic reagents are not nearly as basic, and there is much stronger bonding to “soft” species such as alkenes.
I had a doubt…in grignard synthesis of alcohols using carbonyl groups we usually get higher order(carbon wise) in the product so does this reaction exists:
R-Mg-X—>ROH i.e we get the same order
No, in that case you’re forming a C-OH bond, not a C-C bond, so you’d need to add a source of electrophilic oxygen.
In the reaction with esters, why does the nucleophile C2H5O- not attack in the second step, only grignard reagents attack?
It could attack, but attack is reversible. In contrast, the reaction with Grignards is irreversible since carbanions are such strong bases and poor leaving groups.
Hi
Can you please tell me grignard reagent reaction with 1 degree, 2 degree and 3 degree amines
No reaction, generally.
What will be the reaction of O2 with grignard reagent?
Grignards can react with O2 to give hydroperoxides, which can then be reduced to alcohols. See https://pubs.acs.org/doi/abs/10.1021/ja01627a069
What will be th action of RMgCl/RLi towards all type of amides? &also reply how many moles of Grignard reagents can be consumed by one mole of any type of amide(P/S/T)
For primary or secondary amides – deprotonation. For tertiary amides they do not generally give addition products. One exception is Weinreb amides. https://en.wikipedia.org/wiki/Weinreb_ketone_synthesis
Why does grignard reagent break cyclic ethers using only SN2 mechanism but not any SN1 ? A small question
I ask this because, during SN2 we should have bond breaking and making simultaneously. GR instead acts as a sp2 addition nucleophile…..
Kindly reply…plz
Generally Grignard reagents do not react with ethers… e.g. diethyl ether and THF are commonly used as solvents.
i get that, but what i wanted was to know a double bonds reactivity, like if in a compound both double bond(between 2 carbons) and a aldehyde is given, what will grignard reagent react with first.
Aldehyde. Grignards will add to carbonyls (1,2) before they do conjugate addition (1,4)
what is more reactive with grignard reagent, Aldehyde vs. Ketone vs. Ester vs. Alkene
Aldehyde > Ketone > Ester.
But here’s a question for you. Why?
Etheneone is C2H2O.Aliphatic stucture with a SP2 hybridisation to the carbon which has oxygen double bond.
Ah, I never heard “ketene” called etheneone before. Should still add 1,2.
What would be the reaction of Ethenone with Grignard reagent
I’m not sure what you’re referring to as ethenone, but if you mean methyl vinyl ketone, the answer is that Grignards add to the ketone.
Why alpha Carbon needs to be perpendicular to the carbonyl for deprotonation in ketones.
So that when the C-H bond breaks, the orbital (and its resulting lone pair) is aligned with p-orbitals in the C=O pi system, allowing for resonance stabilization.
Such a great job putting all this together
Thank you Philip – glad you find it useful.
I have worked on this Grignard addition to a Weinred amide:
2-ethyl hexyl MgBr + 2-ethyl hexyl-(C=O)-N(-Me)(-OMe)
The Grignard is from Sigma aldrich and the amide is lab-made.
A brief conclusion is that this reaction did not work. I recover the amide both by TLC and by 1H NMR.
Do you think this is caused by steric hindrance?
I doubt it is a steric hindrance issue. I wonder if it is a Grignard issue. I generally have seen better results using the organolithium reagents.
what is the reason behind this?
Do Grignards react with ethers? If yes then how?
No they do not. That is why ethers are useful protecting groups.
Another question!Is first step in addition of RMgX to ketones a SN2?is it a backside attack?what about stereochem?
It’s a carbonyl addition reaction and it could happen from either face of the ketone.
Hello! what about reaction of CO2 with grignard to form carboxilic acids?any post about it?
What is the stereochemistry of the product formed in the reaction of aldehyde or ketone with grignards reagent
Without a source of chirality, any addition that forms a chiral center will result in a mixture of stereoisomers.
What about the reaction of grignard reagent with nitriles and isonitriles?
Grignard reagents will give ketones, after hydrolysis of the intermediate imine. See https://pubs.acs.org/doi/abs/10.1021/ja00796a022
Relative reactivity order of grignard reagent with respect to alkyl group
On an absolute scale, it will correlate pretty well with basicity, but that’s not taking steric factors into account. The problem with Grignards is usually not that they aren’t “reactive enough”, but that the substrate is sterically hindered and side reactions result.
What happens when grignard reagent is reacted with 2° alcohol
It will be deprotonated to give the epoxide. An acid-base reaction, in other words. That’s it.
Ok… I get what grignard reagent would do with all the above-mentioned compounds… But I am stuck on this question…
Acetyl bromide reacting with excess of CH3MgI followed by treatment with a saturated solution of NH4Cl gives???
You’ve got an an acid chloride reacting with an excess of Grignard reagent. Acid chlorides react similarly to esters. The Grignard adds twice.
In many cases the workup step says, “H+”. However this can cause confusion because H+ in the presence of a tertiary alcohol can lead to formation of a carbocation (remember sn1 with alcohols). So instead of H+, NH4+ Cl- is sometimes used in the workup step. This is a strong enough acid (pKa 10) to protonate the negatively charged oxygen, but not a strong enough acid to protonate the neutral OH and thus go down the SN1/E1 pathway.
Does that help?
Hey, what will be the product for a three degree alcohols reacting with ‘traces of GR and H+’. Thankyou!
A tertiary alcohol reacting with “traces of grignard reagent” and H+. That doesn’t make any sense to me. Grignard reagents are destroyed by H+.
That was my conclusion too. This question was in my textbook. Perhaps a misprint. Thanks for all the help :)
Reactivity order of all oxygen contain compounds towards grignard reagent(aldehyde,ketone,ester,acid chloride,carboxylic acid,acid anhydrides,acid amide)?
aldehyde and acid chloride > ketone > ester >> amide. Carboxylic acids protonate Grignards and they don’t add to carboxylates.
Is it possible to form a ‘Grignard’ reagent with other Gp2 metals? Are the larger Gp2 metal ions not sufficiently polarising due to their lower charge density? I just ran an exam revision course and found this thread very useful for an unusual A level chem topic.
Hi Robert. Organocalcium species are known, but from my understanding they tend to give a lot of Wurtz-type coupling (much like organosodium reagents). I haven’t dug into this reference but it is covered here: J. Org. Chem. 1993 58, 1187. https://pubs.acs.org/doi/10.1021/jo00057a035
What is the stereochemistry of the final product?
is it Racemic?
Hi Adam, it would depend on several factors. If you’re talking about epoxide opening, then if you start with a chiral, non-racemic epoxide, you’ll end up with a chiral, non racemic product because the stereocenter is unaffected. If you’re talking about addition to an achiral aldehyde or ketone, then in the absence of any chiral influence you’ll obtain a racemic product. If you’re talking about addition to a chiral, but racemic aldehyde or ketone then in the absence of any chiral influence you’ll end up with a mixture of diastereomers. This is a good question and deserves its own blog post.
How do you know when to use H3O+ v. NH4Cl?
That’s a great question. I put in both because you might see either. It’s very instructor dependent.
For introductory organic purposes, however, you can think of them as the same.
In practice NH4Cl is a little milder, especially with acid-sensitive molecules that might react with H3O+ . NH4Cl is a “safe choice” when you need a mild acid for workup.
Is the order of reactivity correct? Grignard reactivity order : chloride> anhydride> aldehyde> ketone> ester>amide?
Sounds about right. I wouldn’t want to try to run a competition experiment between an acid chloride and an aldehyde, but aldehydes are certainly more reactive than ketones.
Can a grignard reagent react with a molecule that does not have a functional group, such as methylcyclopropane?
Methylcyclopropane, no, because there’s no electron sink for the carbanion that would result. They can certainly react with acidic hydrocarbons like terminal alkynes and cyclopentadiene.
Hi, why can’t Grignards react with the alpha carbon on ketones and aldehydes since the pKa of that alpha carbon is around ~20 while the pKa of an alkane hydrogen is ~50?
Great question Jon. One of the tricky bits we don’t mention too much is that in order for the C-H bond to actually be acidic, it has to be aligned at 90 degrees to the carbonyl. So when the Grignard is in the vicinity of the carbonyl, two things can happen; it can add, or it can remove the C-H bond. However like I said the C-H bond has to be aligned perfectly for that to happen, whereas the carbonyl is under no such restraint.
The upshot is that addition tends to be faster than deprotonation in this case, so long as the carbonyl isn’t too sterically hindered.
Forgive me if I’m wrong, but for the illustration that appears just above the header “2. Reaction of Grignards With Aldehydes and Ketones,” did the ethyl group (going out of the plane) in the cycloheptane ring change to a methyl group by mistake?
Yes you are correct. Thanks for the spot. Fixed!
So I have a question, why does the reagent not get protonated in Ketones instead of attacking the carbonyl group?
The reagent can get protonated in cases where the Grignard or the ketone are sterically hindered. One complication of acidity of the alpha position of aldehydes or ketones is that the alpha C-H bond must be aligned with the C=O bond in order for it to be acidic; otherwise the resulting carbanion would not be resonance stabilized. There is no such barrier to addition to the carbonyl.
Grignard with amide?
Certainly not for primary or secondary amides. For intro organic chemistry puposes, generally amides considered to be unreactive…. but google “weinreb amide” for a more advanced treatment.
I was wondering about the reactivity of different grignards reagents, with the same R group. Meaning I, Cl, Br. If I where to have two methyl halogens in the solution, which one would be more readily to form the grignard?
The order of halide activity is I > Br > Cl. See March’s advanced organic chemistry 5th edition page 805. Under “Metallo-de-halogenation”.
Does presence of NH4Cl or H3O+ effect the sn2 mechanism of epoxide with GR?
NH4Cl or H3O+ will destroy Grignard reagent. Some Lewis acids (e.g. CeCl3) are compatible with Grignards however.
Esters are less reactive than ketones for addition, what about carbonyl halides?
Esters are certainly less reactive than ketones. I don’t know of a good competition experiment between a carbonyl halide and a ketone or aldehyde. All I would say is that in these three cases the first addition is very fast.
A question:If one equivalent of grignard reagent is made to reacts with a compound that contains a double bond and a carbonyl group(C=O),grignard will attack which one?
The carbonyl group. Addition to the double bond can be effected if you add a catalytic amount of a copper(I) salt such as CuBr.
reactions of grignard reagent will be affected or not by the change of solvent from protic to aprotic.
Grignard reagents will be destroyed if added to a protic solvent such as an alcohol or water.
Hey what if 1,3 dibromopropane reacts with grignard?
You will get a most interesting product with chemical formula C3H6. Can you figure out what it is?
What is the reaction between grignard reagent and ammonia?
I have never seen the use of a Grignard reagent in ammonia, although sometimes tertiary amines are added to Grignards to help them dissolve in benzene or toluene.