Reactions of Aromatic Molecules
Why are halogens ortho- para- directors?
Last updated: May 28th, 2026 |
Why are halogens ortho-, para- directors?
- All activating groups are also ortho-, para- directors.
- Halogens (F, Cl, Br, I) are notable in that they are deactivating ortho-, para- directors. Why?
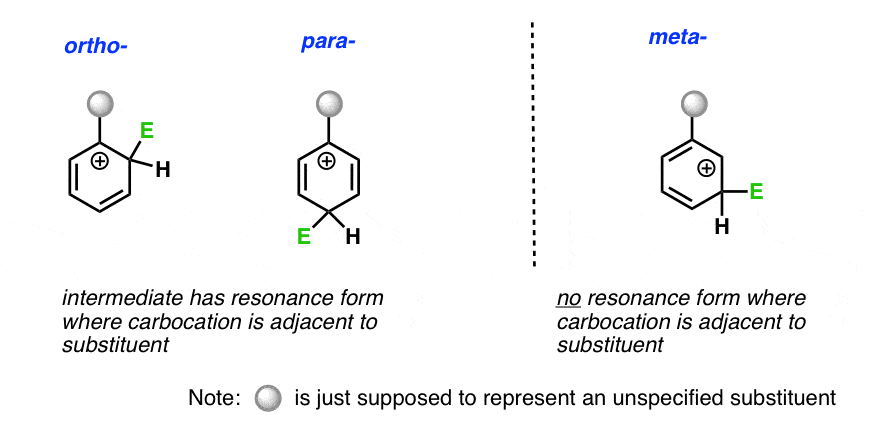
- In electrophilic aromatic substitution (EAS), addition at the ortho- or para– position results in a carbocation intermediate with a resonance form containing a carbocation directly adjacent to the directing group.
- Halogens have a lone pair that can form a pi-bond with the adjacent carbocation.
- Even though halogens are deactivating overall, this “pi donation” helps to stabilize the transition state leading to ortho– or para- products, which is why they are ortho-, para- directors.
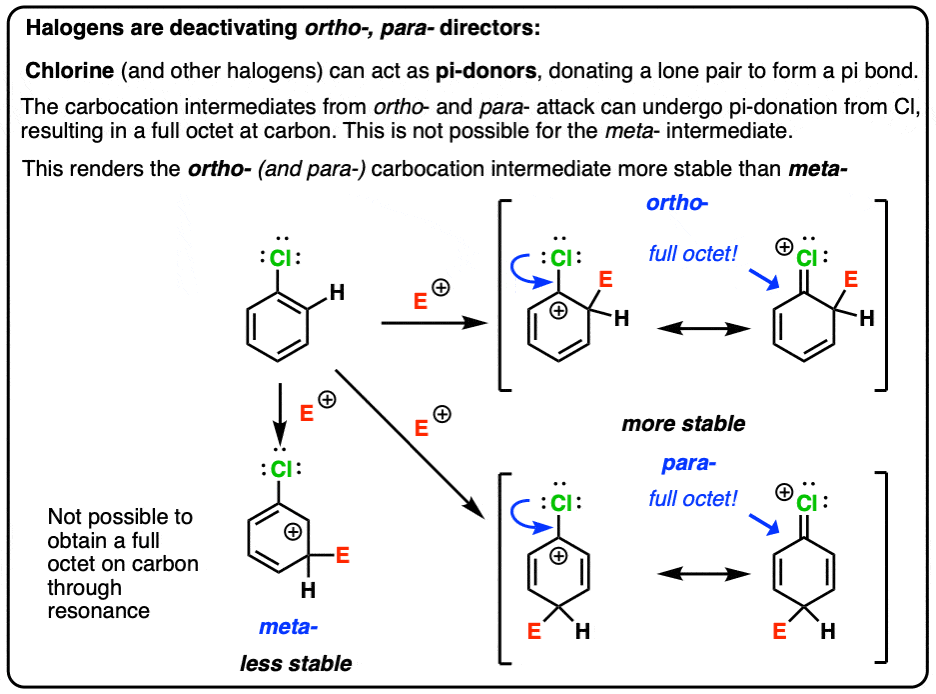
Table of Contents
- All “Activating Groups” Are ortho-, para- Directors
- Most “Deactivating Groups” Are meta- Directors
- So Why Are Halogens Ortho-, Para- Directors?
- The Lone Pair Of Halogens Stabilizes Adjacent Carbocations Formed In The ortho- And para- Intermediates
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. All “Activating Groups” Are ortho-, para- Directors
The previous post in this series tried to show that the key to understanding why a substituent is an ortho-, para- director or meta– director lies in understanding how it influences the stability of the ortho-, para- and meta- carbocation intermediates.
In this post, we’ll show why halogens are ortho-, para- directors even though they are deactivating.
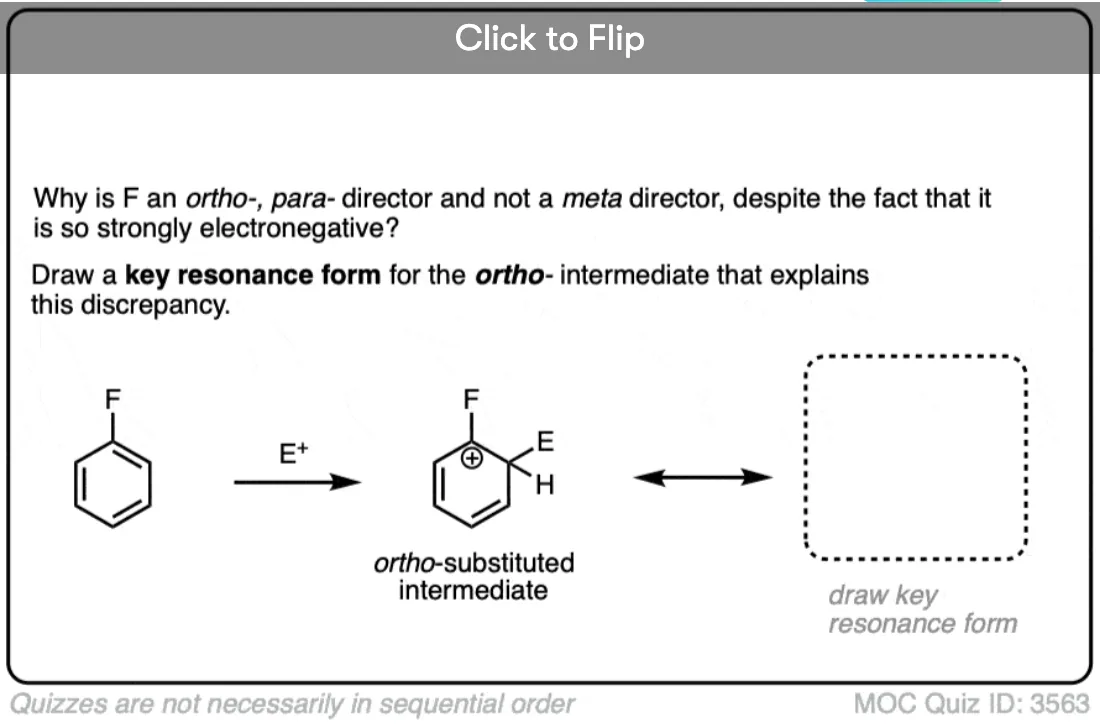
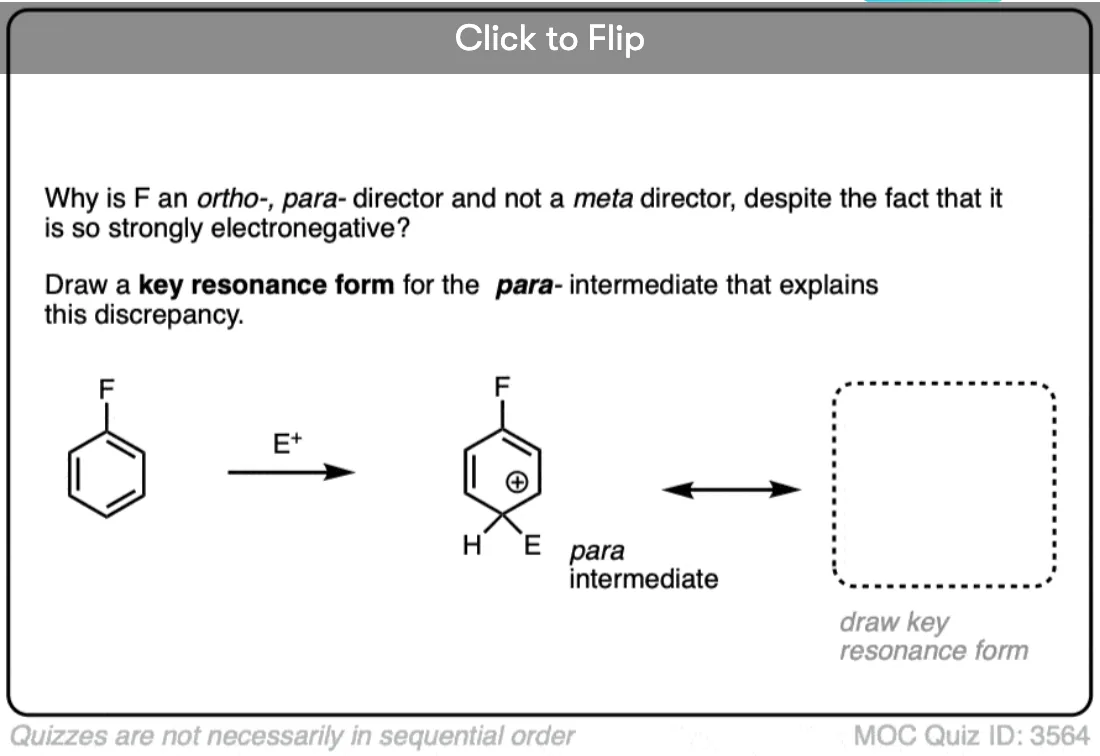
In ortho- and para- addition, there’s a resonance form where the carbocation ends up directly bonded to the substituent.
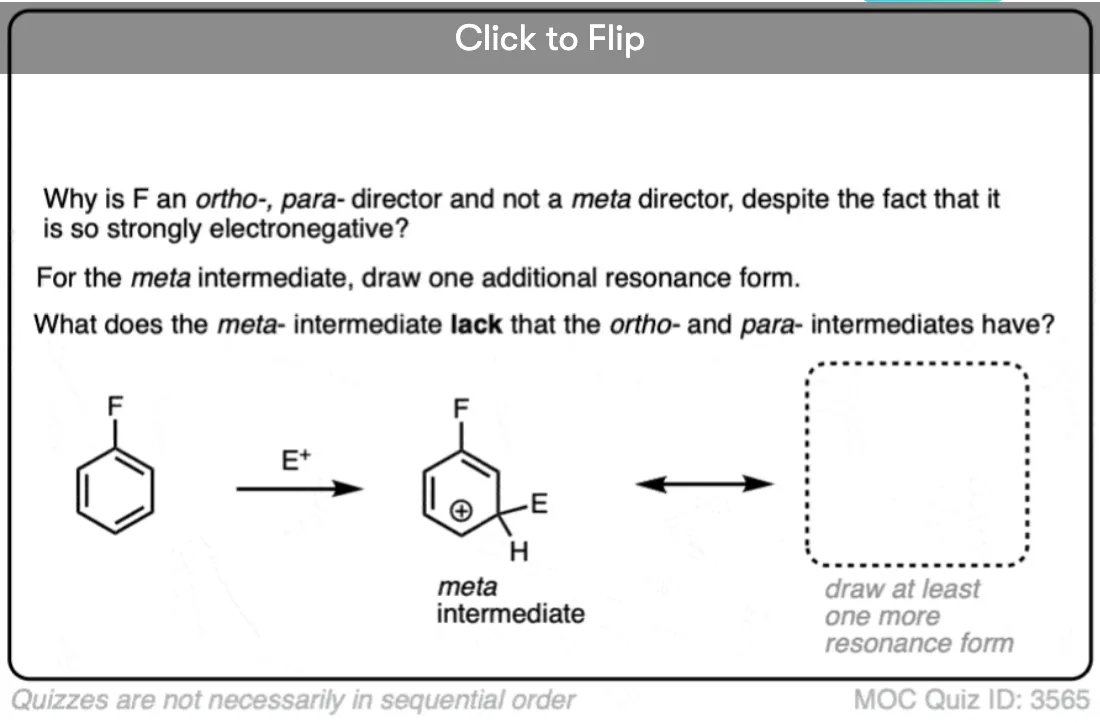
In meta– addition, the carbocation ends up on the carbon adjacent to the carbon bonded to the substituent.
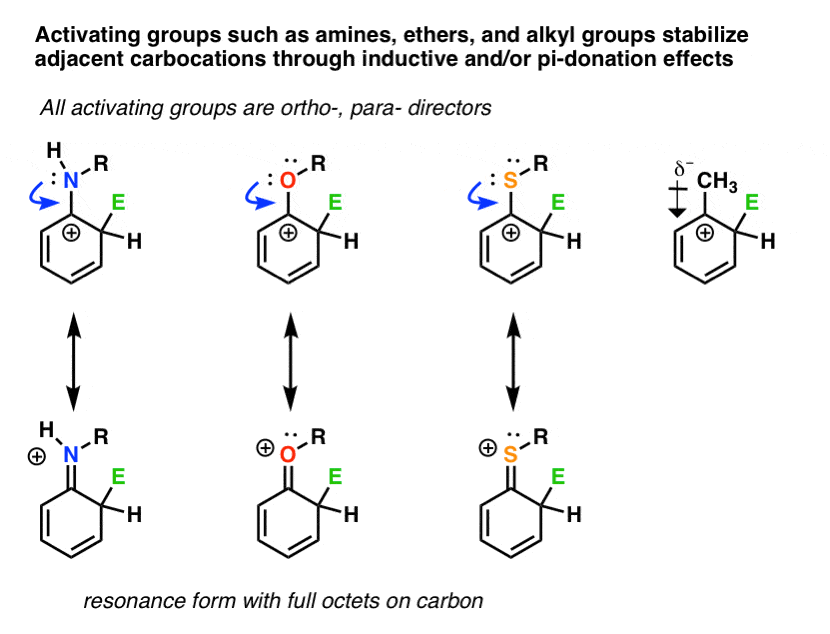
Any group that can donate electron density to a carbocation will be an ortho- , para- director.
We’ve seen that all activating groups (such as amines, ethers, and alkyl groups) are ortho-, para– directors.
These groups can donate electron-density to an adjacent carbocation through inductive effects (a.k.a. “sigma-donation”, as with alkyl groups) and/or pi-donation, where donation of a lone pair from an attached oxygen or nitrogen provides a key resonance form where all carbons have a full octet. Carbocations, being electron-poor, are stabilized by electron-rich neighbors.
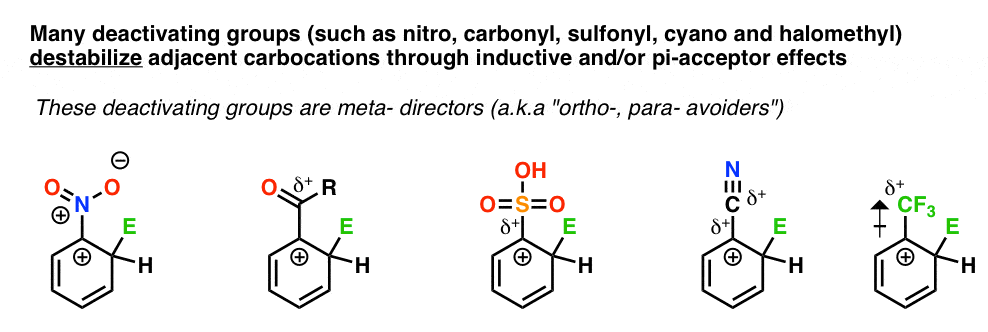
2. Most Deactivating Groups Are meta- Directors
Most deactivating groups are meta- directors.
They withdraw electron-density through an adjacent carbocation through being “sigma-acceptors” (such as the electron-withdrawing CF3 group, or the ammonium [–NR3+] group) and/or “pi acceptors”, such as nitro, carbonyl, or sulfonyl groups.
Carbocations are destabilized by electron-poor neighbors.
Which brings us to the peculiar case of halogens.
3. So Why Are Halogens Ortho-, Para- Directors?
Halogens are deactivating substituents, which is to say that the rate of electrophilic aromatic substitution is lowered when a halogen replaces hydrogen (H) as a substituent. [See this earlier post on “activating vs. deactivating substituents“] . This reflects their high electronegativity, withdrawing electron density from the ring. [Note 1]
At first glance, this might seem to preclude them from being ortho-para directors. But lo, they are!
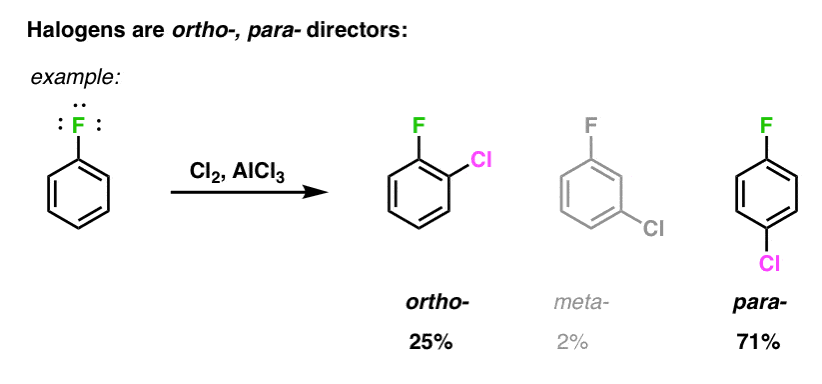
How can we rationalize this observation?
Recall that “activating” vs. “deactivating” just compares how well a substituent stabilizes a carbocation relative to hydrogen.
That’s not the right comparison here. Just like the old joke goes, it’s not about outrunning the bear – it’s about outrunning the other guy.
The key for a substituent being an ortho-, para- versus meta- director is the stability of the ortho- and para– carbocation intermediates versus the meta- carbocation intermediate.
4. The Lone Pair Of Halogens Stabilizes Adjacent Carbocations Formed In The ortho- And para- Intermediates
We can rationalize the ortho-, para- directing ability of halogens by noting that these atoms have attached lone pairs, and can (albeit poorly) act as pi-donors. This results in a resonance form where carbon has a full octet.
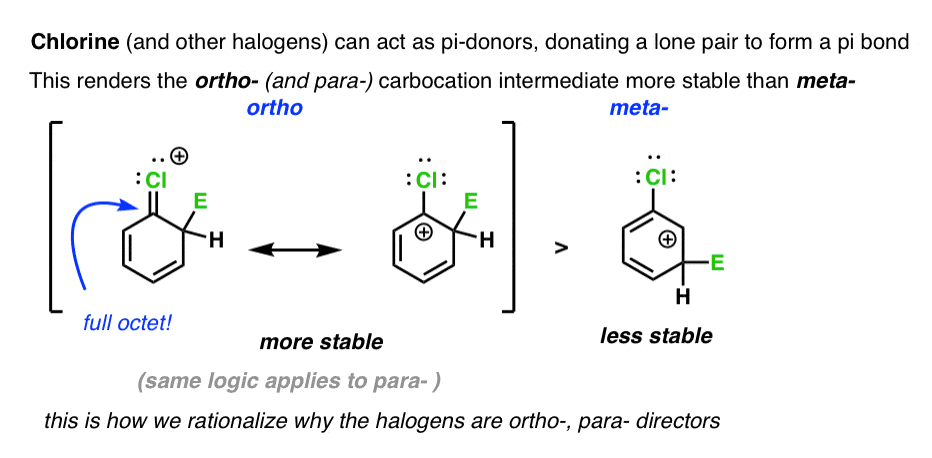
Note that I didn’t say “predict” – I said “rationalize” : – ) . Rationalization involves looking backward from a result and trying to understand why something might have happened. There are several variables at work here that tug in opposite directions, and predicting the magnitude of these individual effects in the absence of a strong computational model is a fool’s errand. That’s why we run experiments!
From these experiments, it seems that a carbocation intermediate which has a pi-donor is more important toward determining whether it is an ortho-, para- director than whether it is a strong electron withdrawing group.
Notes
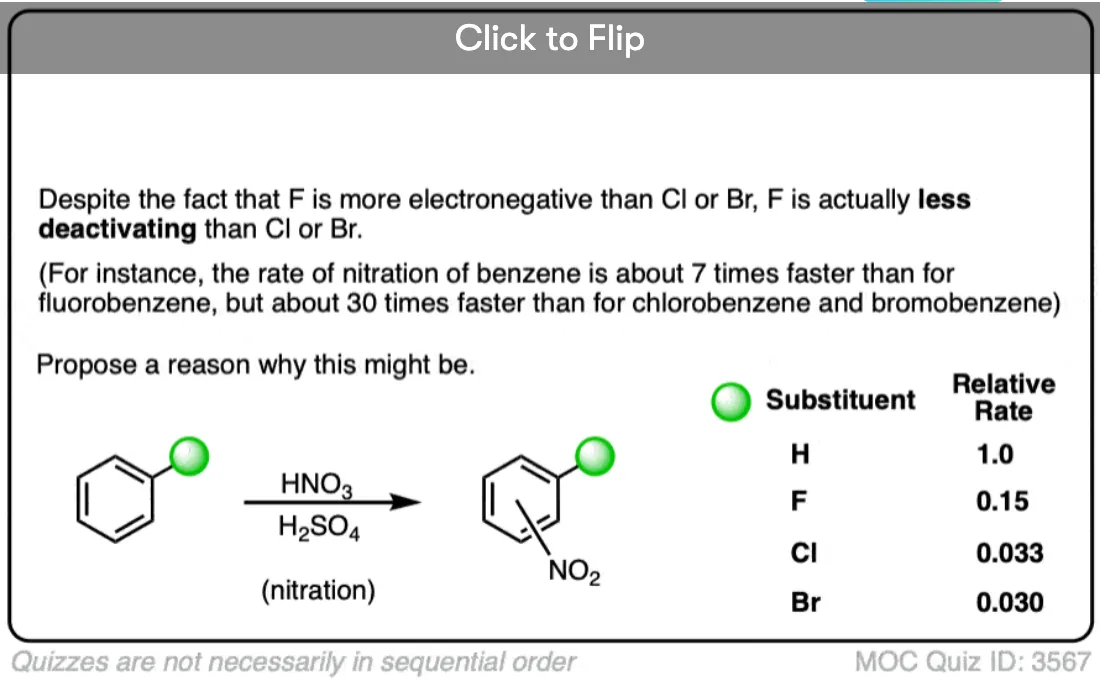
Note 1. It is interesting to note, however, that despite having the highest electronegativity, fluorine is actually the most activating of the halogens (the other halogens are relatively similar in their deactivating powers). This can be attributed to the better orbital overlap of the fluorine sp3 orbitals with the 2p orbitals of the pi system. [For similar reasons, BF3 is a worse Lewis acid than BCl3 and BBr3 , since the fluorine orbitals overlap much better with the empty boron 2p orbital].
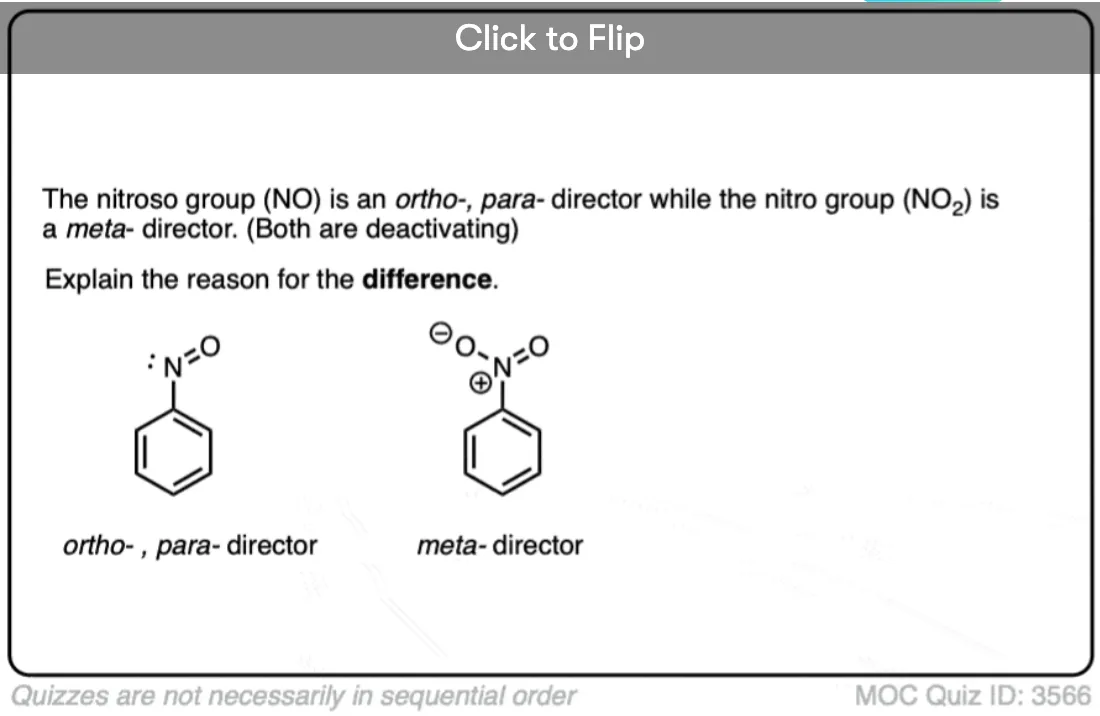
Are there any other deactivating ortho-, para- directors?
Yes. NO.
NO?
Yes, NO. Nitroso.
Knowing what we now know about halogens, what predictions would you make for the nitroso group, a group that is somewhat electron withdrawing, but also bears a lone pair on the nitrogen.
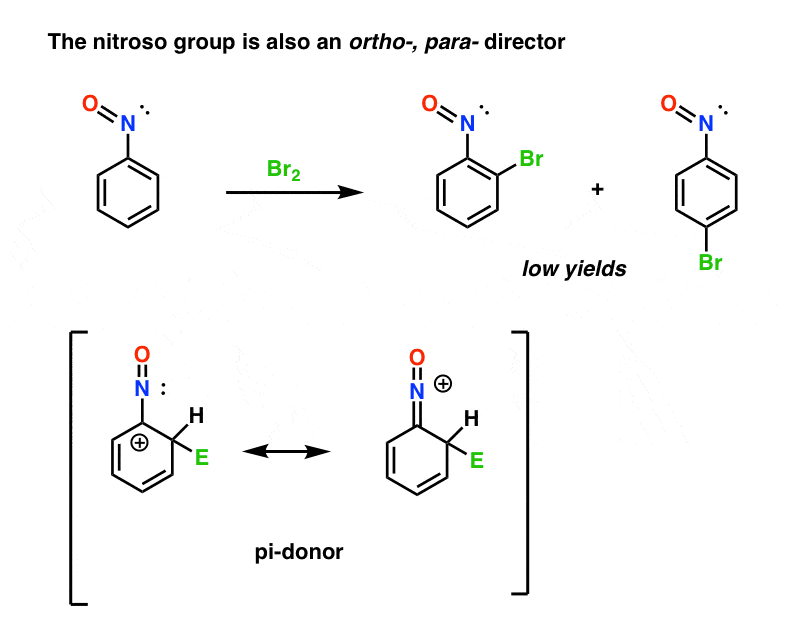
The yields aren’t great, but there you go.
Quiz Yourself!
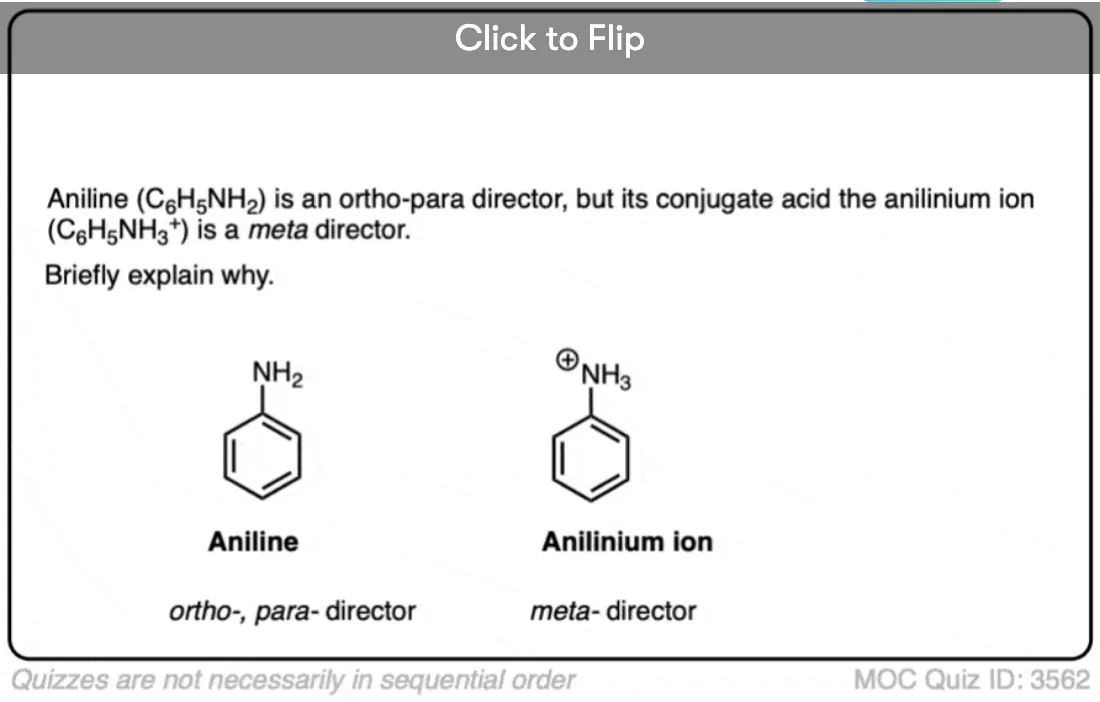
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
Become a MOC member to see the clickable quiz with answers on the back.
(Advanced) References and Further Reading
- A. F. Holleman, Die direkte Einführung von Substituenten in den Benzolkern
Rec. Trav. Chim. Pays-Bas 1910, 12, 455-456
DOI: 10.1002/recl.19100291205
A.F Holleman from 1910 said that ortho–para orientation is associated with activation and meta orientation with deactivation. - —The nature of the alternating effect in carbon chains. Part XXII. An attempt further to define the probable mechanism of orientation in aromatic substitution
Christopher Kelk Ingold and Florence Ruth Shaw
J. Chem. Soc. 1927, 2918-2926
DOI: 10.1039/JR9270002918
An early paper by the influential Physical Organic Chemist, Prof. C. K. Ingold, stating that halogenobenzenes are inductively electron-withdrawing but simultaneously resonance-stabilizing. - Influence of directing groups on nuclear reactivity in oriented aromatic substitutions. Part IV. Nitration of the halogenobenzenes
Marjorie L. Bird and Christopher K. Ingold
J. Chem. Soc. 1938, 918-929
DOI: 10.1039/JR9380000918
The relative rates of nitration for the halobenzenes are determined here, and it is seen that the order of reactivity is PhF>PhI>PhCl, PhBr - The Anomalous Reactivity of Fluorobenzene in Electrophilic Aromatic Substitution and Related Phenomena
Joel Rosenthal and David I. Schuster
Journal of Chemical Education 2003, 80 (6), 679
DOI: 1021/ed080p679
A very interesting paper, suitable for curious undergrads, and discusses something that most practicing organic chemists will know empirically – fluorobenzene is almost as reactive as benzene in EAS or Friedel-Crafts reactions, which is counterintuitive when one considers electronic effects. - —A new orientation rule and the anomaly of the nitroso-group
Dalziel Llewellyn Hammick and Walter S. Illingworth
J. Chem. Soc. 1930, 2358-2364
DOI: 10.1039/JR9300002358 - 93. The orienting power of the nitroso-group
Dalziel Ll. Hammick, Randal G. A. New, and Leslie E. Sutton
J. Chem. Soc. 1932, 742-748
DOI: 10.1039/JR9320000742
These two papers discuss the electronics of the nitroso substituent. Both papers refer to C. K. Ingold’s experiment where he observed p-substitution of nitrosobenzene from bromination in CS2. The authors attempt to explain this by suggesting that in certain solvents nitrosobenzene dimerizes, and the dimer prefers o,p-substitution. This is worth reevaluating with modern methods (hint, hint)!
Sir, will you do inorganic reaction mechanisms aswell?
Perhaps one day.
Thank you 💕
Nitroso group are not exactly deactivating group they are segregated as chamelian group which behave as activating as well as deactivating groups
Nitroso group can able exbiht both type of mesmeric effects.
In the fifth figure from top, chlorine is shown to suffer positive charge. With 3 electron pairs as well as a double bond, how chlorine can become electron deficient?
It’s a positive “formal” charge. Chlorine is still more electronegative than carbon, and is still partially negative. Here’s a post I wrote on why not to trust formal charge: https://www.masterorganicchemistry.com/2011/11/15/how_to_use_electronegativity/
The left structure in the figure with the Cl acting as an e-donor on the benzene ring during EArS has too many e around the Cl. One of the lone pairs has been used in the resonance interaction….
shoot. Thank you.
Here is a fun blog post here with some related discussion of the nitroso group, but sadly no experimental data: https://www.ch.imperial.ac.uk/rzepa/blog/?p=7234
It is mostly an article about the first curved arrows ever drawn. But near the end there is an interesting discussion about how geometry of the molecule changes between ground state and transition state. You alluded to it in your pictures.